

Abstract
The human gut is home to complex microbial populations that change dynamically in response to various internal and external stimuli. The gut microbiota provides numerous functional benefits that are crucial for human health but in the setting of a disturbed equilibrium, the microbial community can cause deleterious outcomes such as diseases and cancers. Characterization of the functional activities of human gut microbiota is fundamental to understand their roles in human health and disease. Metaproteomics, which refers to the study of the entire protein collection of the microbial community in a given sample is an emerging area of research that provides informative details concerning functional aspects of the microbiota. In this mini review, we present a summary of the progress of metaproteomic analysis for studying the functional role of gut microbiota. This is followed by an overview of the experimental approaches focusing on fecal specimen for metaproteomics and is concluded by a discussion on the challenges and future directions of metaproteomic research.
Keywords: Metaproteomics, Human, Gut Microbiota, Multi-omics, Technologies
Background
The human gastrointestinal (GI) tract is colonized by a highly diverse population of microbial community collectively known as the gut microbiota that play vital roles in maintaining human health [1]. Although relatively stable, alterations in the microbial consortium may occur due to factors such as aging, genetic mutations, inflammation and dietary change [2, 3]. Accumulating evidence indicates that imbalances in the microbial community or dysbiosis have potential adverse effects on human health, whereby such alterations are implicated in the development of numerous diseases, including metabolic disorders, inflammatory diseases and cancers [4].
Given the importance of gut microbiota in human health and disease development, it has been the subject of extensive investigations in recent years. The completion of a large-scale initiative known as the Human Microbiome Project in 2012 has marked an important milestone in the characterization of human microbiome in healthy individuals, which led to the establishment of a reference microbial genome database [5]. MetaHIT (Metagenomics of the Human Intestinal Tract) is another collaborative effort that aims to provide a reference catalog of gut microbiome in association with obesity and inflammatory bowel disease (IBD) [6]. Over the years, much effort has been devoted to determine microbiome composition in various diseased patients using metagenomic analysis to identify potential links between gut microbiota and diseases [7].
To date, the growing number of metagenomic studies have provided valuable insights into the structure and diversity of the gut microbial community and their genetic composition. However, it is important to note that there are several limitations in these studies. Firstly, these studies only uncovered gene sequences that were present but do not provide any clues regarding their actual gene or protein expression levels. Besides, metagenomic analysis does not discriminate between microbiota that are active, dormant or dead, as all microbial cells will be sequenced. Consequently, the precise functions of the gut microbiota are still largely unknown. Hence, other complementary approaches are needed to elucidate the functional capacity of human gut microbiota.
Over the past decade, metaproteomics which is defined as the large-scale profiling of the whole protein complement expressed by a complex microbial ecosystem [8], has been applied to analyze human gut microbiota. In comparison to metagenomics, metaproteomics is capable to reveal functional traits relevant to the underlying physiological states, thereby providing detailed insights into the connection between microbial diversity, functions and the impact on host biology. In this mini review, we summarize the recent progress of metaproteomic study in the context of human gut microbiota. Further, we discuss experimental approaches for metaproteomics and conclude by providing an outlook on the challenges and future research direction for metaproteomics.
Metaproteomics of human gut microbiota
In a pioneering metaproteomic study of human gut microbiota, an initial input into the establishment and functional role of gut microbiota during early growth was obtained from the analysis of fecal microbiota from two infants [9]. Undoubtedly, the study is limited in the depth of analysis due to the absence of a suitable database. Despite the relatively simple fecal protein profile, only Bifidobacterial transaldolases protein was successfully identified by de novo sequencing back then. A few years along the road, with the likes of more powerful analytical tools and metagenome data availability, Young et al. recently provided a more detailed fecal metaproteome profile of a preterm infant, shedding light on the functional clues and host-microbiota interactions during early development [10].
As for the more complex human adult, an initial comprehensive fecal metaproteome analysis was performed on a healthy monozygotic twin pair [11] and subsequently followed by a high-throughput temporal analysis of intestinal metaproteomes between three female adults [12]. Both studies identified a common core proteome that was mainly mapped to metabolic pathways and detected a distinctive but relatively stable metaproteome for each individual. Notably, both studies also demonstrated discrepancies between protein levels predicted from the metagenomes and phylogenetic data with their actual abundances, hence further emphasize the essentiality of metaproteomics in understanding the protein expression dynamics.
As noted from prior study, the composition of mucosa-associated microbiota varies from those residing inside the lumen [13]. This finding was further corroborated in an animal study where dissimilar metaproteome profiles of mucus, gut content and feces were reported [14]. Moreover, different intestinal sites have been found to affect host protein diversity more profoundly than microbial colonization states [15]. By applying a different sampling approach, Li et al. analyzed mucosal lavage samples from different intestinal locations to assess metaproteome of mucosa-associated microbiota [16]. Significant differences in the mucosal metaproteome were noted between the proximal and distal colon, implying distinct functionality within specific intestinal niches. This approach would be useful to investigate spatial distribution and activities at the mucosa-lumen interface, but might not be practically feasible as being invasive and costly.
Apart from cataloguing gut microbial metaproteome in healthy individuals, comparative analyses to characterize differential protein profiles under altered physiological conditions have been performed. Antibiotics are known to cause disturbance in the microbiota composition and functions, which in turn will have potential consequences on health and disease [17]. A comprehensive multi-omics study revealed drastic changes in the protein profiles of the gut-associated microbiota following β-lactam therapy [18], which reflect functional adaptation of gut microbiota in response to the drug. Clearly, more studies are needed to understand how different antibiotics can shape the gut microbiota and the resulting effect on the host.
There is increasing evidence linking gut microbiota to diseases such as Inflammatory Bowel Disease (IBD), including Crohn's Disease (CD) and ulcerative colitis (UC), but the exact role of the microbiota is still unclear [19]. Erickson et al. studied alterations of gut microbiota in CD patients and found differentially expressed proteins that could be linked to the disease [20]. In another metaproteomic study that focused on mucosal-luminal interface in IBD, changes in the bacterial phylotypes were reported to be associated with host immune response and inflammation [21]. These findings provide an insight into host-microbiota interactions that may be correlated to the disease etiology. The disease-associated features were further corroborated by the discovery of distinct protein modules associated with IBD in the mucosal metaproteome of IBD patients, which were verified in an independent cohort [22].
Besides, altered intestinal microbiota has been implicated in the development of obesity but the mechanistic link remains obscure [23]. Striking enrichment of gut microbiota proteins involved in cell motility and vitamin B12 synthesis was reported in an obese adolescent subject, whereas the lean adolescent showed more active vitamin B6 synthesis [24]. Nevertheless, the results are rather preliminary as only one subject from each group was analyzed. In a recent fecal metaproteome study involving a larger group of individuals, Kolmeder et al. reported that the phylum Bacteroidetes was biologically more active in the obese group [25]. The authors have identified a subset of bacterial and human proteins that could be used to classify the subjects into their corresponding groups and unveiled functional shifts that could be correlated to obesity.
Functional associations of microbial imbalances with liver cirrhosis have also been characterized using metaproteomics. Remarkably, unique metaproteome and functional pathways were reported for the patients, highlighting distinct functional characteristics of the gut microbiota that could be linked to liver cirrhosis [26]. The results provide new insights into the host-microbe relationships in liver cirrhosis that warrant further investigation. Metaproteomics also has been applied to study the correlation between dysbiosis with cystic fibrosis. Debyser et al. detected significant differences in the gut microbial diversity and protein profiles, along with a strong increase in host acute phase proteins in the patients, which reflect the ongoing inflammatory condition [27]. The study also reported a set of host and microbial proteins that might serve as candidate biomarkers for cystic fibrosis.
In the trending research area of microbiota and probiotic, no conclusive data is observed regarding probiotic intervention on the host and microbial functionalities. Probiotic consumption did not cause significant alteration on the overall fecal protein profiles nor the functional pathways despite a reduction of fecal host proteins and concomitant increase in bacterial proteins [28]. In spite of some evidence for therapeutic effect shown by several probiotic studies in diseases, there are conflicting findings reported on the implications of probiotic supplementation in the healthy population [29].
Overall, metaproteomic study is gradually gaining momentum to unravel the functionality of the complex microbial consortium. From understanding the role of microbiota in healthy individuals, the field has progressed to explore the functional profiles of dysbiosis in various diseases, as summarized in Table 1. Metaproteomics has the potential to dissect microbial functionality, which could help to understand the underlying pathophysiology and pave the way for targeted approach to improve health and disease (Fig. 1). Yet, we are only just beginning to decipher such associations and in addition to the diseases mentioned above, it would be interesting to explore other disease manifestations such as neurological disorders [30] and colorectal cancer [31].
Table 1.
Summary of metaproteomic studies of human gut microbiota
| Sample type | Subject | Separation method | Mass spectrometer | Database | No. of proteins identified | Reference |
|---|---|---|---|---|---|---|
| Feces | 2 infants | 2-D PAGE | MALDI-TOF | Public database | >200 spots, 1 protein identified | Klaassens et al., 2007 |
| Feces | 1 infant | 2D-LC | LTQ Orbitrap | Databases from metagenomic sequences, microbial species, human and common contaminants | 4,031 proteins | Young et al., 2015 |
| Feces | 1 monozygotic twin pair (adult) | 2D-LC | LTQ Orbitrap | Databases from two human metagenomes, human database and common contaminants | 446 human proteins 1,368 microbial proteins |
Verberkmoes et al., 2009 |
| Feces | 3 healthy adults | 1-DE and LC | LTQ Orbitrap | Databases from intestinal microbes, two metagenomes, human protein and food database | 1,790 proteins | Kolmeder et al., 2012 |
| Mucosal lavage | 38 healthy adults | LC WCX magnetic beads |
LTQ Orbitrap MALDI-TOF |
Swiss-Prot | 300 human proteins | Li et al., 2011 |
| Feces | 1 adult with antibiotic treatment | 1-DE and LC | LTQ Orbitrap | Matched metagenome | 3,011 proteins | Pérez-Cobas et al., 2013 |
| Feces | 6 monozygotic twin pairs (1 set healthy, 3 sets concordant twins with CD, 2 sets discordant twins) | 2D-LC | LTQ Orbitrap | Matched metagenome and human microbial isolate reference genome database | 2,904 proteins | Erickson et al., 2012 |
| Mucosal lavage | First cohort: 3 healthy, 6 UC patients Second cohort: 13 healthy, 14 CD, 15 UC |
WCX Protein Chip arrays | SELDI-TOF | Uniprot | 589 proteins | Presley et al., 2012 |
| Mucosal lavage | Discovery cohort: 51 (17 healthy, 13 UC, 21 CD) Validation cohort: 38 healthy |
WCX magnetic beads LC |
MALDI-TOF LTQ Orbitrap |
Swiss-Prot | 599 protein/peptide peaks | Li et al., 2016 |
| Feces | 1 lean and 1 obese teenager | 1-DE and LC | LTQ Orbitrap | Matched and unmatched metagenomes | 613 proteins | Ferrer et al., 2013 |
| Feces | 9 normal, 4 overweight, 16 obese | 1-DE and LC | Orbitrap | In-house metaproteome database | 893,007 MS/MS spectra | Kolmeder et al., 2015 |
| Feces | 3 liver cirrhosis patients and their corresponding spouse | 1-DE and LC | Quadrupole-Orbitrap | UniProt | 5,020 proteins | Wei et al., 2016 |
| Feces | 15 children with cystic fibrosis and their unaffected siblings | 1-DE and LC | Ion trap-FTICR | NCBI | 1,676 proteins | Debyser et al., 2016 |
| Feces | 16 healthy adults (8 probiotics, 8 placebo) | 1-DE and LC | LTQ Orbitrap | In-house metaproteome database | 4,966 peptides | Kolmeder et al., 2016 |
| Feces | 1 healthy adult | LC | LTQ Orbitrap | UniProtKB Swiss-Prot |
3,911 proteins (not centrifuged) 4,587 proteins (centrifuged) |
Tanca et al., 2015 |
| Feces | 2 premature infants | 2D-LC | LTQ Orbitrap | In-house metaproteome database | 807 and 342 proteins (without filtering) 1264 and 1012 proteins (with double filtering) |
Xiong et al., 2015b |
| Feces | 2 healthy adults | 1-DE and LC | LTQ Orbitrap | Matched metagenomes Synthetic metagenomes (iterative search) |
2,331 and 1,870 peptides 5,010 and 3,542 peptides |
Rooijers et al., 2011 |
| Feces | 4 healthy volunteers | LC | LTQ Orbitrap | Metagenome databases UniProt-based databases All bacterial sequences |
>10,000 peptides | Tanca et al., 2016 |
| Feces Mucosal lavage |
8 mice 8 pediatric volunteers |
LC | Q Exactive | Microbial gene catalog databases Target-decoy database |
30,749 mouse protein groups 19,011 human protein groups |
Zhang et al., 2016b |
| Mucosal lavage | 5 teenagers | LC | LTQ Orbitrap | In-house Human Intestinal Microbial Protein Database Target-decoy database |
4,014 protein groups quantified | Zhang et al., 2016a |
1-DE one-dimensional electrophoresis, 2-D PAGE two-dimensional polyacrylamide gel electrophoresis, 2-D LC two-dimensional liquid chromatography, FTICR Fourier transform ion cyclotron resonance, LC liquid chromatography, LTQ linear trap quadrupole, MALDI-TOF matrix-assisted laser desorption/ionization time-of-flight, NCBI National Center for Biotechnology Information, SELDI-TOF surface-enhanced laser desorption/ionization time-of-flight, WCX weak cation exchange
Fig. 1.
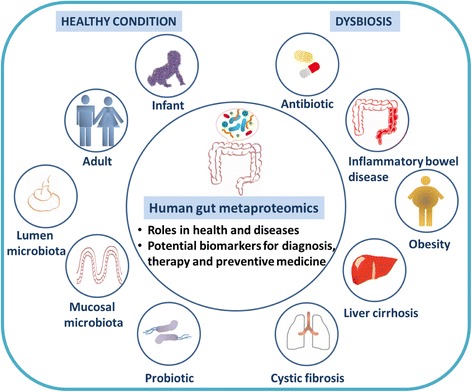
Applications of human gut metaproteomic study. Metaproteomic analysis has been employed to characterize functional roles of gut microbiota in healthy and disease conditions, which help to unravel the molecular mechanism underlying homeostasis and disease pathogenesis. Knowledge gained from metaproteomic study could be useful to devise strategies in disease prevention and management for improved human health
Experimental considerations for metaproteomics
Metaproteomics workflow typically includes sample collection, protein extraction, fractionation, mass spectrometry (MS) analysis and database searches [32]. For human gut microbiota study, fecal and mucosal lavage samples are commonly employed to characterize global proteome of the entire gut and the mucosa interface, respectively. This mini review will focus on fecal sample as it is more widely used for metaproteomics. Sample storage is a crucial yet sometimes overlooked step in metaproteomics. Several independent studies have revealed that different storage temperatures may introduce considerable alterations to the microbial profiles and highlighted that proper storage is critical to maintain sample stability [33–35]. Moreover, it was found that frozen intact fecal material was more stable than frozen extracted proteins, hence is recommended for long-term storage [36].
Apart from storage, sample processing is another key step in metaproteomics. Sample preparation protocol primarily depends on the research questions, which isolate either host or microbial proteins or both. Most previous studies have focused on proteins of microbial origin and employed centrifugation to remove other interfering substances. However, it was observed that despite greater microbial protein identifications, the centrifugation step caused considerable protein loss due to non-specific removal of microbial cells, which led to bias in the analysis [37]. Conversely, stool without pretreatment provides a better representation of the microbial proteins and allows concurrent analysis of human proteins. This highlights the importance of careful consideration in selecting a suitable approach for sample processing. Alternatively, a double filtering separation step has been shown useful to deplete human proteins for selective enrichment of microbial proteins, which was demonstrated to enhance proteome coverage by facilitating the identification of low-abundance proteins [38].
Next, efficient protein extraction from the complex microbial samples is critical to allow accurate representation of the intracellular protein content. In the metaproteomic analysis of environmental samples, different protein extraction methods have been shown to isolate different subset of proteins with only minimal overlap, which underlines the importance of selecting appropriate protocol to obtain optimal protein sample [39]. For gut microbiota study, several studies have indicated that mechanical disruption by bead beating was an efficient protein extraction method, particularly for lysing Gram-positive bacteria [40, 41]. Thus far, there is a major gap in the characterization of extracellular proteins that may serve as major mediators of host-microbiota interactions. The challenge to capture the secreted proteins from a complex ecosystem is huge, as consideration for intracellular protein removal either from the host or microbiota must be taken into account. Fecal samples may provide sufficient protein yield for this kind of secretome study but protein loss is inevitable given the necessity of an extensive clean-up procedure that follows due to the nature of the sample itself. Fecal proteins may also undergo some alteration along the intestine. Lichtman et al. described the enrichment of secreted gut luminal proteins from feces that can be applied to facilitate analysis of secreted host proteome [42]. Other than that, targeted analysis of specific subcellular fraction such as membrane proteins and post-translational modifications are also likely to provide additional functional insights.
To date, MS remains as the analytical platform of choice for metaproteomics. Prior to MS analysis, extensive fractionation using multidimensional LC separations (GeLC-MS/MS or 2D-LC-MS/MS) is particularly useful to reduce sample complexity and improve protein identification. The final and fairly demanding stage for metaproteomics is data analysis. Several software tools such as Pipasic [43], MetaProteomeAnalyzer [44] and Unipept [45] have been developed to facilitate metaproteomic data analysis. One of the key elements for a successful metaproteomic study is the availability of a relevant database for mass spectra searching. Strategy using either matched or unmatched metagenomes has been successfully employed for metaproteomic protein identifications [24, 46]. Furthermore, iterative workflow using synthetic metagenome generated from known gut microbiota has been shown successful to enhance protein identifications [47].
The choice of database is a critical factor in data analysis. Parallel use of multiple databases to improve protein yields may be the way forward as demonstrated by Tanca et al. in which the use of different databases in gut microbial metaproteome data analysis has led to complementary identification of unique peptides [46]. More recently, a data analysis pipeline coupling publicly accessible gene catalog databases with iterative database searching known as MetaPro-IQ was introduced by Zhang et al. [48]. The pipeline enabled efficient identification and quantification of over 120,000 peptides corresponding to >30,000 protein groups from human and mouse gut microbial metaproteome. To date, it represents the most extensive metaproteome coverage and appears to be a promising approach for future metaproteomic study.
Challenges and future directions
Despite the great potential of metaproteomics to decipher the diverse roles of microbial members within the human host, there are many obstacles that need to be surmounted. First and foremost, inherent sample complexity associated with the highly diverse microbial community is among the major hurdles for metaproteomic study. The vast protein dynamic range often hinders the detection of low-abundant proteins from the minor species. This problem could be partly alleviated by applying different fractionation and enrichment strategies, such as capillary and microchip methods [49] to reduce sample complexity and increase protein detection, but at the expense of increased cost and analysis time.
Beyond protein identification, quantitative analysis is important to determine key microbial players that contribute to metabolic functions [50]. However, given the enormous species and metabolic diversity, robust approaches for quantitative metaproteomic are still lacking. Protein-based stable isotope probing (protein-SIP) has been reported as a powerful technique in environmental studies to decipher metabolic activity of distinct microbial members [51]. Concerning gut microbiota, label-free quantification is the most common strategy adopted in the aforementioned metaproteomic studies, but has limited accuracy. The most recent take on quantitative metaproteomics of human gut microbiota is the application of metabolic labeling for improved peptide quantification [52]. This newly introduced method hold the potential to facilitate future metaproteomic study.
There are also technical limitations implicated in metaproteomic analysis. Standardized procedures for metaproteomics are yet to be established, which might lead to suboptimal findings and hinder inter-laboratory comparison. Moreover, existing analytical platforms are also limited in terms of their sensitivity to analyze protein sample with such a wide dynamic range. With the emerging technology advancement in mass spectrometry such as the data-independent acquisition strategy (MSE and SWATH), reproducibility of protein quantification and the depth of proteome analysis were significantly improved [53]. Apart from that, identification of proteins from the complex microbial consortium, which may harbor up to hundreds or thousands of species, has also proven to be a difficult task. Lack of complete genome sequences for the highly heterogeneous microbial community, particularly the poorly characterized and uncultivable species poses a big challenge for researchers. Nonetheless, the availability of sequence data from the blooming metagenomic studies and new analysis softwares are expected to counteract these issues.
Additionally, it is evident that sample complexity and inter-individual variation in gut microbiota are extensive [16]. It is also important to note that host and microbiota interactions involve delicate interplay between factors such as age, genetics, immunity and dietary habits, thus these could act as potential confounding variables. Clearly, studies involving a larger set of well-defined subjects are neccesary to capture a more accurate functional input of the gut microbial ecosystem. Rodent and gnotobiotic animals can be custom-designed to circumvent obstacles related to human study, thus represent valuable models for studying gut microbiota and have been employed in several metaproteomic studies [14, 42, 54].
Despite all of the limitations, metaproteomic research has already led to some remarkable discoveries on the functional features of gut microbiota. With the ongoing development to address those challenges, we envision that the field will further advance in the future. Furthermore, integration of metaproteomic with other omics approaches is expected to provide a more comprehensive and meaningful elucidation of the microbial ecosystem.
Conclusion
With the growing interest to understand the link between gut microbiota in health and diseases, metaproteomic analysis is instrumental to characterize the activity and functional pathways of the microbial community. Although challenging, further advances in sample preparation methods, development of more sophisticated analytical tools alongside with the availability of relevant software and databases are expected to facilitate the progress of metaproteomics in the coming years. By harnessing the power of the emerging technologies, it is anticipated that more details on the microbial functionality and their connection with human host will be uncovered in the future.
Acknowledgments
Not applicable.
Funding
Dr. Siok-Fong Chin is supported by grants from Trans-Disciplinary Research Grant Scheme and Fundamental Research Grant Scheme funded by Ministry of Higher Education, Malaysia (TRGS/2/2014/UKM/02/3/2, FRGS/2/2013/SKK01/UKM/03/5). Dr. Hui-min Neoh is also supported by Trans-Disciplinary and Fundamental Research Grant Scheme from Ministry of Higher Education Malaysia (TRGS/2/2014/UKM/02/3/1, FRGS/1/2014/SKK04/UKM/03/1).
Availability of data and materials
Not applicable.
Authors’ contributions
All authors conceived the content of the manuscript, P-YL and S-FC wrote the manuscript, and all authors reviewed and approved the manuscript.
Competing interests
The authors declare that they have no competing interest.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Abbreviations
- CD
Crohn's Disease
- GI
Gastrointestinal
- IBD
Inflammatory Bowel Disease
- MetaHIT
Metagenomics of the Human Intestinal Tract
- MS
Mass spectrometry
- protein-SIP
Protein-based stable isotope probing
- UC
Ulcerative colitis
Contributor Information
Pey Yee Lee, Email: leepeyyee@yahoo.com.
Siok-Fong Chin, Email: chinsiokfong@ppukm.ukm.edu.my.
Hui-min Neoh, Email: hui-min@ppukm.ukm.edu.my.
Rahman Jamal, Email: rahmanj@ppukm.ukm.edu.my.
References
- 1.Backhed F, Ley RE, Sonnenburg JL, Peterson DA, Gordon JI. Host-bacterial mutualism in the human intestine. Science. 2005;307:1915–1920. doi: 10.1126/science.1104816. [DOI] [PubMed] [Google Scholar]
- 2.Costello EK, Lauber CL, Hamady M, Fierer N, Gordon JI, Knight R. Bacterial community variation in human body habitats across space and time. Science. 2009;326:1694–1697. doi: 10.1126/science.1177486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Holmes E, Li JV, Marchesi JR, Nicholson JK. Gut microbiota composition and activity in relation to host metabolic phenotype and disease risk. Cell Metab. 2012;16:559–564. doi: 10.1016/j.cmet.2012.10.007. [DOI] [PubMed] [Google Scholar]
- 4.Carding S, Verbeke K, Vipond DT, Corfe BM, Owen LJ. Dysbiosis of the gut microbiota in disease. Microb. Ecol. Health Dis. 2015;26:26191. doi: 10.3402/mehd.v26.26191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Human Microbiome Project Consortium Structure, function and diversity of the healthy human microbiome. Nature. 2012;486:207–214. doi: 10.1038/nature11234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li J, Jia H, Cai X, Zhong H, Feng Q, Sunagawa S, et al. An integrated catalog of reference genes in the human gut microbiome. Nat. Biotechnol. 2014;32:834–841. doi: 10.1038/nbt.2942. [DOI] [PubMed] [Google Scholar]
- 7.Wang W-L, Xu S-Y, Ren Z-G, Tao L, Jiang J-W, Zheng S-S. Application of metagenomics in the human gut microbiome. World J Gastroenterol. 2015;21:803–814. doi: 10.3748/wjg.v21.i3.803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wilmes P, Bond PL. The application of two-dimensional polyacrylamide gel electrophoresis and downstream analyses to a mixed community of prokaryotic microorganisms. Environ. Microbiol. 2004;6:911–920. doi: 10.1111/j.1462-2920.2004.00687.x. [DOI] [PubMed] [Google Scholar]
- 9.Klaassens ES, de Vos WM, Vaughan EE. Metaproteomics approach to study the functionality of the microbiota in the human infant gastrointestinal tract. Appl. Environ. Microbiol. 2007;73:1388–1392. doi: 10.1128/AEM.01921-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Young JC, Pan C, Adams RM, Brooks B, Banfield JF, Morowitz MJ, et al. Metaproteomics reveals functional shifts in microbial and human proteins during a preterm infant gut colonization case. Proteomics. 2015;15:3463–3473. doi: 10.1002/pmic.201400563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Verberkmoes NC, Russell AL, Shah M, Godzik A, Rosenquist M, Halfvarson J, et al. Shotgun metaproteomics of the human distal gut microbiota. ISME J. 2009;3:179–189. doi: 10.1038/ismej.2008.108. [DOI] [PubMed] [Google Scholar]
- 12.Kolmeder CA, de Been M, Nikkilä J, Ritamo I, Mättö J, Valmu L, et al. Comparative metaproteomics and diversity analysis of human intestinal microbiota testifies for its temporal stability and expression of core functions. PLoS ONE. 2012;7:e29913. doi: 10.1371/journal.pone.0029913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Eckburg PB, Bik EM, Bernstein CN, Purdom E, Dethlefsen L, Sargent M, et al. Diversity of the human intestinal microbial flora. Science. 2005;308:1635–1638. doi: 10.1126/science.1110591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Haange SB, Oberbach A, Schlichting N, Hugenholtz F, Smidt H, von Bergen M, et al. Metaproteome analysis and molecular genetics of rat intestinal microbiota reveals section and localization resolved species distribution and enzymatic functionalities. J. Proteome Res. 2012;11:5406–5417. doi: 10.1021/pr3006364. [DOI] [PubMed] [Google Scholar]
- 15.Lichtman JS, Alsentzer E, Jaffe M, Sprockett D, Masutani E, Ikwa E, et al. The effect of microbial colonization on the host proteome varies by gastrointestinal location. ISME J. 2016;10:1170–1181. doi: 10.1038/ismej.2015.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li X, LeBlanc J, Truong A, Vuthoori R, Chen SS, Lustgarten JL, et al. A metaproteomic approach to study human-microbial ecosystems at the mucosal luminal interface. PLoS ONE. 2011;6:e26542. doi: 10.1371/journal.pone.0026542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Francino MP. Antibiotics and the human gut microbiome: dysbioses and accumulation of resistances. Front. Microbiol. 2015;6:1543. doi: 10.3389/fmicb.2015.01543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pérez-Cobas AE, Gosalbes MJ, Friedrichs A, Knecht H, Artacho A, Eismann K, et al. Gut microbiota disturbance during antibiotic therapy: a multi-omic approach. Gut. 2013;62:1591–1601. doi: 10.1136/gutjnl-2012-303184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Matsuoka K, Kanai T. The gut microbiota and inflammatory bowel disease. Semin. Immunopathol. 2015;37:47–55. doi: 10.1007/s00281-014-0454-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Erickson AR, Cantarel BL, Lamendella R, Darzi Y, Mongodin EF, Pan C, et al. Integrated metagenomics/metaproteomics reveals human host-microbiota signatures of Crohn's disease. PLoS ONE. 2012;7:e49138. doi: 10.1371/journal.pone.0049138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Presley LL, Ye J, Li X, Leblanc J, Zhang Z, Ruegger PM, et al. Host-microbe relationships in inflammatory bowel disease detected by bacterial and metaproteomic analysis of the mucosal-luminal interface. Inflamm. Bowel Dis. 2012;18:409–417. doi: 10.1002/ibd.21793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li X, LeBlanc J, Elashoff D, McHardy I, Tong M, Roth B, et al. Microgeographic proteomic networks of the human colonic mucosa and their association with inflammatory bowel disease. Cell. Mol. Gastroenterol. Hepatol. 2016;2:567–583. doi: 10.1016/j.jcmgh.2016.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gérard P. Gut microbiota and obesity. Cell. Mol. Life Sci. 2016;73:147. doi: 10.1007/s00018-015-2061-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ferrer M, Ruiz A, Lanza F, Haange SB, Oberbach A, Till H, et al. Microbiota from the distal guts of lean and obese adolescents exhibit partial functional redundancy besides clear differences in community structure. Environ. Microbiol. 2013;15:211–226. doi: 10.1111/j.1462-2920.2012.02845.x. [DOI] [PubMed] [Google Scholar]
- 25.Kolmeder CA, Ritari J, Verdam FJ, Muth T, Keskitalo S, Varjosalo M, et al. Colonic metaproteomic signatures of active bacteria and the host in obesity. Proteomics. 2015;15:3544–3552. doi: 10.1002/pmic.201500049. [DOI] [PubMed] [Google Scholar]
- 26.Wei X, Jiang S, Chen Y, Zhao X, Li H, Lin W, et al. Cirrhosis related functionality characteristic of the fecal microbiota as revealed by a metaproteomic approach. BMC Gastroenterol. 2016;16:121. doi: 10.1186/s12876-016-0534-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Debyser G, Mesuere B, Clement L, Van de Weygaert J, Van Hecke P, Duytschaever G, et al. Faecal proteomics: A tool to investigate dysbiosis and inflammation in patients with cystic fibrosis. J. Cyst. Fibros. 2016;15:242–250. doi: 10.1016/j.jcf.2015.08.003. [DOI] [PubMed] [Google Scholar]
- 28.Kolmeder CA, Salojärvi J, Ritari J, de Been M, Raes J, Falony G, et al. Faecal metaproteomic analysis reveals a personalized and stable functional microbiome and limited effects of a probiotic intervention in adults. PLoS ONE. 2016;11:e0153294. doi: 10.1371/journal.pone.0153294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kristensen NB, Bryrup T, Allin KH, Nielsen T, Hansen TH, Pedersen O. Alterations in fecal microbiota composition by probiotic supplementation in healthy adults: a systematic review of randomized controlled trials. Genome Med. 2016;8:52. doi: 10.1186/s13073-016-0300-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Carabotti M, Scirocco A, Maselli MA, Severi C. The gut-brain axis: interactions between enteric microbiota, central and enteric nervous systems. Ann. Gastroenterol. 2015;28:203–209. [PMC free article] [PubMed] [Google Scholar]
- 31.Gagnière J, Raisch J, Veziant J, Barnich N, Bonnet R, Buc E, et al. Gut microbiota imbalance and colorectal cancer. World J. Gastroenterol. 2016;22:501–518. doi: 10.3748/wjg.v22.i2.501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kolmeder CA, de Vos WM. Metaproteomics of our microbiome - developing insight in function and activity in man and model systems. J. Proteomics. 2014;97:3–16. doi: 10.1016/j.jprot.2013.05.018. [DOI] [PubMed] [Google Scholar]
- 33.Choo JM, Leong LE, Rogers GB. Sample storage conditions significantly influence faecal microbiome profiles. Sci. Rep. 2015;5:16350. doi: 10.1038/srep16350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fouhy F, Deane J, Rea MC, O'Sullivan Ó, Ross RP, O'Callaghan G, et al. The effects of freezing on faecal microbiota as determined using MiSeq sequencing and culture-based investigations. PLoS ONE. 2015;10:e0119355. doi: 10.1371/journal.pone.0119355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tedjo DI, Jonkers DM, Savelkoul PH, Masclee AA, van Best N, Pierik MJ, et al. The effect of sampling and storage on the fecal microbiota composition in healthy and diseased subjects. PLoS ONE. 2015;10:e0126685. doi: 10.1371/journal.pone.0126685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Morris LS, Marchesi JR. Assessing the impact of long term frozen storage of faecal samples on protein concentration and protease activity. J. Microbiol. Methods. 2016;123:31–38. doi: 10.1016/j.mimet.2016.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tanca A, Palomba A, Pisanu S, Addis MF, Uzzau S. Enrichment or depletion? The impact of stool pretreatment on metaproteomic characterization of the human gut microbiota. Proteomics. 2015;15:3474–3485. doi: 10.1002/pmic.201400573. [DOI] [PubMed] [Google Scholar]
- 38.Xiong W, Giannone RJ, Morowitz MJ, Banfield JF, Hettich RL. Development of an enhanced metaproteomic approach for deepening the microbiome characterization of the human infant gut. J. Proteome Res. 2015;14:133–141. doi: 10.1021/pr500936p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Leary DH, Hervey WJ, Deschamps JR, Kusterbeck AW, Vora GJ. Which metaproteome? The impact of protein extraction bias on metaproteomic analyses. Mol. Cell. Probes. 2013;27:193–199. doi: 10.1016/j.mcp.2013.06.003. [DOI] [PubMed] [Google Scholar]
- 40.Santiago A, Panda S, Mengels G, Martinez X, Azpiroz F, Dore J, et al. Processing faecal samples: a step forward for standards in microbial community analysis. BMC Microbiol. 2014;14:112. doi: 10.1186/1471-2180-14-112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Smith B, Li N, Andersen AS, Slotved HC, Krogfelt KA. Optimising bacterial DNA extraction from faecal samples: comparison of three methods. Open Microbiol. J. 2011;5:14–17. doi: 10.2174/1874285801105010014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lichtman JS, Marcobal A, Sonnenburg JL, Elias JE. Host-centric proteomics of stool: a novel strategy focused on intestinal responses to the gut microbiota. Mol. Cell. Proteomics. 2013;12:3310–3318. doi: 10.1074/mcp.M113.029967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Penzlin A, Lindner MS, Doellinger J, Dabrowski PW, Nitsche A, Renard BY. Pipasic: similarity and expression correction for strain-level identification and quantification in metaproteomics. Bioinformatics. 2014;30:i149–156. doi: 10.1093/bioinformatics/btu267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Muth T, Behne A, Heyer R, Kohrs F, Benndorf D, Hoffmann M, et al. The MetaProteomeAnalyzer: a powerful open-source software suite for metaproteomics data analysis and interpretation. J. Proteome Res. 2015;14:1557–1565. doi: 10.1021/pr501246w. [DOI] [PubMed] [Google Scholar]
- 45.Mesuere B, Willems T, Van der Jeugt F, Devreese B, Vandamme P, Dawyndt P. Unipept web services for metaproteomics analysis. Bioinformatics. 2016;32:1746–1748. doi: 10.1093/bioinformatics/btw039. [DOI] [PubMed] [Google Scholar]
- 46.Tanca A, Palomba A, Fraumene C, Pagnozzi D, Manghina V, Deligios M, et al. The impact of sequence database choice on metaproteomic results in gut microbiota studies. Microbiome. 2016;4:51. doi: 10.1186/s40168-016-0196-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rooijers K, Kolmeder C, Juste C, Doré J, de Been M, Boeren S, et al. An iterative workflow for mining the human intestinal metaproteome. BMC Genomics. 2011;12:6. doi: 10.1186/1471-2164-12-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang X, Ning Z, Mayne J, Moore JI, Li J, Butcher J, et al. MetaPro-IQ: a universal metaproteomic approach to studying human and mouse gut microbiota. Microbiome. 2016;4:31. doi: 10.1186/s40168-016-0176-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Štěpánová S, Kašička V. Recent developments and applications of capillary and microchip electrophoresis in proteomic and peptidomic analyses. J Sep Sci. 2016;39:198–211. doi: 10.1002/jssc.201500973. [DOI] [PubMed] [Google Scholar]
- 50.von Bergen M, Jehmlich N, Taubert M, Vogt C, Bastida F, Herbst FA, et al. Insights from quantitative metaproteomics and protein-stable isotope probing into microbial ecology. ISME J. 2013;7:1877–1885. doi: 10.1038/ismej.2013.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jehmlich N, Vogt C, Lünsmann V, Richnow HH, von Bergen M. Protein-SIP in environmental studies. Curr. Opin. Biotechnol. 2016;41:26–33. doi: 10.1016/j.copbio.2016.04.010. [DOI] [PubMed] [Google Scholar]
- 52.Zhang X, Ning Z, Mayne Z, Deeke SA, Li J, Starr AE, et al. In vitro metabolic labeling of intestinal microbiota for quantitative metaproteomics. Anal. Chem. 2016;88:6120–6125. doi: 10.1021/acs.analchem.6b01412. [DOI] [PubMed] [Google Scholar]
- 53.Sajic T, Liu Y, Aebersold R. Using data-independent, high-resolution mass spectrometry in protein biomarker research: perspectives and clinical applications. Proteomics Clin. Appl. 2015;9:307–321. doi: 10.1002/prca.201400117. [DOI] [PubMed] [Google Scholar]
- 54.Lichtman JS, Ferreyra JA, Ng KM, Smits SA, Sonnenburg JL, Elias JE. Host-microbiota interactions in the pathogenesis of antibiotic-associated diseases. Cell Rep. 2016;14:1049–1061. doi: 10.1016/j.celrep.2016.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.