

Abstract
Bicuspid aortic valve (BAV) is the most common congenital heart defect. Although many BAV patients remain asymptomatic, at least 20% develop thoracic aortic aneurysm (TAA). Historically, BAV-related TAA was considered as a hemodynamic consequence of the valve defect. Multiple lines of evidence currently suggest that genetic determinants contribute to the pathogenesis of both BAV and TAA in affected individuals. Despite high heritability, only very few genes have been linked to BAV or BAV/TAA, such as NOTCH1, SMAD6, and MAT2A. Moreover, they only explain a minority of patients. Other candidate genes have been suggested based on the presence of BAV in knockout mouse models (e.g., GATA5, NOS3) or in syndromic (e.g., TGFBR1/2, TGFB2/3) or non-syndromic (e.g., ACTA2) TAA forms. We hypothesized that rare genetic variants in these genes may be enriched in patients presenting with both BAV and TAA. We performed targeted resequencing of 22 candidate genes using Haloplex target enrichment in a strictly defined BAV/TAA cohort (n = 441; BAV in addition to an aortic root or ascendens diameter ≥ 4.0 cm in adults, or a Z-score ≥ 3 in children) and in a collection of healthy controls with normal echocardiographic evaluation (n = 183). After additional burden analysis against the Exome Aggregation Consortium database, the strongest candidate susceptibility gene was SMAD6 (p = 0.002), with 2.5% (n = 11) of BAV/TAA patients harboring causal variants, including two nonsense, one in-frame deletion and two frameshift mutations. All six missense mutations were located in the functionally important MH1 and MH2 domains. In conclusion, we report a significant contribution of SMAD6 mutations to the etiology of the BAV/TAA phenotype.
Keywords: bicuspid aortic valve, thoracic aortic aneurysm, SMAD6, targeted gene panel, variant burden test
Introduction
With a prevalence of 1–2% in the general population, bicuspid aortic valve (BAV) is the most common congenital heart defect. It has a 3:1 male preponderance and is characterized by an aortic valve with two cusps instead of the normal three. BAV often coincides with aortic manifestations such as coarctation of the aorta and thoracic aortic aneurysm (TAA) (Verstraeten et al., 2016). The latter can lead to lethal dissections if left untreated. Although first described over 400 years ago and high heritability (89%) (Cripe et al., 2004), the genetic etiology of BAV, with or without TAA, remains largely elusive. It was initially suggested that TAA results from altered blood flow dynamics imposed by the abnormal bicuspid valve. Changes in shear stress were presumed to weaken the aortic wall, resulting in dilatation and rupture. At present, common genetic risk factors for BAV and TAA are proposed (Hinton, 2012), based on the following observations: (i) the aortic valve and the aorta share common embryologic origins [i.e., the cardiac neural crest (CNC) and the second heart field] (Martin et al., 2015), (ii) family members of BAV/TAA probands show TAA without valve abnormalities and/or BAV without aneurysmal disease (Loscalzo et al., 2007), and (iii) TAA formation in BAV probands that previously underwent valve replacement has been reported (Braverman et al., 2005).
Transmission of BAV/TAA mostly complies with an autosomal dominant inheritance pattern, displaying reduced penetrance and variable expressivity (Clementi et al., 1996; Huntington et al., 1997). Few genes have been robustly linked to the BAV phenotype to date. NOTCH1 is often considered the sole established BAV gene, either as an isolated finding or in association with early onset valve calcification, TAA, or other left-sided heart defects (Mohamed et al., 1797; Garg et al., 2005; McKellar et al., 2007; Foffa et al., 2013; Kent et al., 2013; Bonachea et al., 2014; Freylikhman et al., 2014; Kerstjens-Frederikse et al., 2016). SMAD6 (Tan et al., 2012) and MAT2A (Guo et al., 2015) have also been implicated in BAV, but only in a very limited number of patients. A dozen candidate genes emanated from knockout mouse models with increased BAV occurrence (Biben et al., 2000; Lee et al., 2000; Laforest and Nemer, 2011; Laforest et al., 2011; Thomas et al., 2012; Mommersteeg et al., 2015; Quintero-Rivera et al., 2015). The prevalence of BAV in these knockout models is often low (range: 2–42% in single knockouts) (Table 1), probably due to reduced penetrance and/or activation of compensatory mechanisms. Mutations in some syndromic (Attias et al., 2009; Callewaert et al., 2011; Lindsay et al., 2012; Nistri et al., 2012; van de Laar et al., 2012; Pepe et al., 2014) or non-syndromic (Guo et al., 2007) TAA genes also associate with increased BAV occurrence (Table 1).
Table 1.
Genes included in the targeted gene panel and the criteria on which their selection was based.
| Context | Gene | Incidence | References |
|---|---|---|---|
| BAV in humans | NOTCH1 | Mutations found in 27 BAV patients | Mohamed et al., 1797; Garg et al., 2005; McKellar et al., 2007; Foffa et al., 2013; Kent et al., 2013; Bonachea et al., 2014; Freylikhman et al., 2014; Kerstjens-Frederikse et al., 2016 |
| SMAD6 | Mutations found in 2 BAV patients | Tan et al., 2012 | |
| MAT2A | Mutations found in 1 BAV patient | Guo et al., 2015 | |
| BAV in mice | ACVR1 | BAV in 78–83% of Alk2FXKO/Gata5−Cre+ mice | Thomas et al., 2012 |
| GATA4 | BAV in 43% of Gata4+/−;Gata5+/− mice | Laforest and Nemer, 2011 | |
| GATA5 | BAV in 25% of Gata5−/− mice | Laforest et al., 2011 | |
| GATA6 | BAV in 25% Gata5+/−;Gata6+/− mice | Laforest and Nemer, 2011 | |
| MATR3 | BAV in 12% in Matr3+/− mice | Quintero-Rivera et al., 2015 | |
| NKX2-5 | BAV in 2–20% of Nkx2-5+/− mice | Biben et al., 2000 | |
| NOS3 | BAV in 42% of Nos3−/− mice | Lee et al., 2000 | |
| ROBO1 | BAV in 100% of Robo1−/−;Robo2−/− mice | Mommersteeg et al., 2015 | |
| ROBO2 | BAV in 100% of Robo1−/−;Robo2−/− mice | Mommersteeg et al., 2015 | |
| BAV in (non)syndromic TAA cases | FBN1 | Occasional BAV in Marfan syndrome | Attias et al., 2009; Nistri et al., 2012; Pepe et al., 2014 |
| ACTA2 | 7% BAV in non-syndromic TAA | Guo et al., 2007 | |
| ELN | Occasional BAV in cutis laxa | Callewaert et al., 2011 | |
| FLNA | Occasional BAV in X-linked valve disease | Jefferies et al., 2010 | |
| MYH11 | Occasional BAV in non-syndromic TAA | Personal observation | |
| SMAD3 | 3–11% BAV in Loeys-Dietz syndrome | van de Laar et al., 2012 | |
| TGFB2 | 8–13% BAV in Loeys-Dietz syndrome | Lindsay et al., 2012 | |
| TGFB3 | 4% BAV in Loeys-Dietz syndrome | Personal observation | |
| TGFBR1 | 8–12% BAV in Loeys-Dietz syndrome | Personal observation | |
| TGFBR2 | 8–12% BAV in Loeys-Dietz syndrome | Personal observation |
BAV, Bicuspid aortic valve; TAA, Thoracic aortic aneurysm.
To date, no major BAV/TAA gene has emerged. The described genes have been associated with BAV, but their contribution to the etiology of BAV/TAA has never been examined systematically. Here, we evaluate this contribution in 22 BAV-associated genes (Table 1) using a targeted gene panel and variant burden approach.
Materials and methods
Study cohort
Genomic DNA (gDNA) of 441 BAV/TAA patients was collected through a collaborative effort involving 8 different centers (Supplementary Table 1). Patients were selected based on the presence of BAV and either an aortic diameter at the sinus of Valsalva or the ascending aorta of at least 4.0 cm in adults, or a Z-score exceeding 3 in children. Aortic diameter dimensions were determined using echocardiography, computed tomography or magnetic resonance imaging. A positive family history was defined as having at least one first- or second-degree relative with BAV and/or TAA. Control gDNA was obtained from 183 cancer patients who presented at the SickKids Hospital, Toronto, Canada. None of the controls showed structural heart disease upon examination with echocardiography. All study participants or their legal guardians gave informed consent at the respective sample-contributing centers.
Targeted enrichment
Genes (n = 22) were selected for targeted resequencing based on the following criteria: (i) mutations occur in human BAV cases (n = 3), (ii) knockout mouse models present with incomplete penetrance of BAV (n = 9), and (iii) occasional or increased BAV manifestation occurs in patients with mutations in known TAA genes (n = 10) (Table 1). Enrichment of all exons of these candidate genes, including ±10 nucleotides of adjacent intronic sequence, was performed with a custom Haloplex target enrichment kit per instructions of the manufacturer (Agilent Technologies, USA). Probe design covered a theoretical 99.7% of the complete target region (560 kb). Pooled samples were sequenced either on a HiSeq 2500 (Illumina, USA) with 2 × 150 bp reads or on a HiSeq 1500 (Illumina, USA) with 2 × 100 bp reads.
Data analysis and filtering
The raw data were processed using an in-house-developed Galaxy-based pipeline, followed by variant calling with the Genome Analysis Toolkit Unified Genotyper (DePristo et al., 2011). Variants were subsequently annotated and filtered with the in-house developed database VariantDB (Vandeweyer et al., 2014), which uses ANNOVAR. Heterozygous coding or splice site (±2 bp from exon-intron boundaries for nucleotide substitution, and ±5 bp for multi-bp deletions or insertions) variants with an allelic balance between 0.25 and 0.85 (FLNA in males: 0.75–1) and a minimum coverage of 10 reads were selected. Finally, we included variants that fitted within at least one of the following three categories; unique variants [absent in the Exome Aggregation Consortium (ExAC) database (Lek et al., 2016)], variants with an ExAC Minor Allele Frequency (MAF) lower than 0.01% or variants with an ExAC MAF between 0.01% and 0.1% that had a Combined Annotation Dependent Depletion (CADD) (Kircher et al., 2014) score above 20. All splice region variants underwent splice site effect prediction using ALAMUT (Interactive Biosoftware, France). Synonymous variants outside of splicing regions were not taken into account.
The ExAC database was used as an independent control dataset. The raw data of variants (~all ExAC datasets) fulfilling ExAC's quality control parameters (“PASS”) were extracted from the offline version of ExAC v0.3.1. Since the ExAC variants were annotated using VEP, whereas our patient variant annotation was ANNOVAR-based, we re-annotated the ExAC variants with ANNOVAR. The same variant filtering strategy as described for the patient cohort was subsequently applied. For each selected ExAC variant, the allele frequency was determined by computing the ratio of the Mutant Allele Count (mAC) and Total Allele Count (tAC). Next, we re-scaled each variant's mAC by multiplying its computed allele frequency by its respective tAC_Adj, i.e., the tAC average of all variants in that specific gene. Finally, the variant counts for each panel gene were obtained by summing up the re-scaled mACs.
Validation by sanger sequencing
Variants discussed in the results section were confirmed with Sanger sequencing. Primers were designed using Primer3 software (Untergasser et al., 2012) v4.0.0 and polymerase chain reaction (PCR) products were purified with Calf Intestinal Alkaline Phosphatase (Sigma-Aldrich, USA). Sequencing reactions were performed using the BigDye Terminator Cycle Sequencing kit (Applied Biosystems, Life Technologies, USA), followed by capillary electrophoresis on an ABI3130XL (Applied Biosystems, Life Technologies, USA). The obtained sequences were analyzed with CLC DNA Workbench v5.0.2 (CLC bio, Denmark).
Segregation analysis
When family members were available, Sanger sequencing of the SMAD6 variants identified in the proband was performed in additional relatives to check if the phenotype segregated with the variant.
Statistical analysis
We performed burden analyses comparing frequencies of the variants fulfilling the three criteria that were mentioned in “Section Data Analysis and Filtering” between patients and controls. Whereas the Fisher's Exact Test was used to statistically compare variant frequencies in the patient cohort to those in the study control cohort, the Chi-Square Test with Yates' correction was used for the patient-ExAC comparison. No p-values were calculated if the number of variants in patients and/or controls was zero. Fisher's Exact statistics were also used to determine if significant variant type enrichment and/or domain clustering of variants occurs in patients. Statistical significance was considered when p < 0.05.
Results
The patient cohort consisted of 441 BAV/TAA patients (75% males and 25% females) with an average age at inclusion of 63.5 ± 14.4 years. For these patients, the most common associated feature was coarctation of the aorta (2.9%, n = 13). About 3% (n = 14) had other additional findings such as mitral valve prolaps, aortic stenosis, dilated cardiomyopathy, aortic insufficiency, patent ductus arteriosus or intracranial aneurysm. 46.7% (n = 206) had a left-right leaflet BAV orientation, 15.9% (n = 70) had a right-non-coronary leaflet BAV orientation and for 37.4% (n = 165) of the patients the subtype of valve leaflet morphology was not specified. A positive family history was known for 9.3% of the patients, whereas for the remainder the family history was negative or unknown. The study control cohort (n = 183) consisted of 58% males and 42% females. The average age at inclusion of this control cohort was 13.1 ± 5.1 years.
Targeted gene panel sequencing reached an overall coverage at 10x of 99.13% of the targeted regions. In total, 169 variants passed our selection criteria in our patient and control group (Supplementary Table 2). Of these, 112 variants were identified in 441 patients. They included 101 missense, 2 nonsense, 2 splice-site, 5 in-frame indel, and 2 frameshift variants. The 183 study controls contained 57 variants including 53 missense, 1 nonsense, 2 splice-site, and 1 frameshift variant. After applying the identical filtering criteria to the ExAC control cohort, 15,660 variants were retained in on average 54,940 individuals: i.e., 14,931 missense, 190 splice-site, 72 nonsense, 10 no-stop, 204 frameshift, and 253 in-frame indel variants.
To validate our control cohort, we compared its variant frequencies for the 22 selected candidate genes to those of the ExAC cohort. No significant differences were observed (Figure 1). We then performed a variant burden analysis equating the numbers of patient variants per gene to the numbers found in the control cohort (Table 2). Results are graphically presented in Figure 1, showing the proportion of variants per gene in the three different cohorts. Although a few genes (e.g., FLNA) showed trends toward significance when comparing our study patient and control cohort, we decided to focus on the patient-ExAC comparison because of the larger number of controls in the ExAC cohort and hence, higher power. Only SMAD6 reached significance (p = 0.002) in the patient-ExAC comparison. Remarkably, a protective effect for NOS3 and NOTCH1 variants was suggested (p = 0.06 and p = 0.05, respectively).
Figure 1.

Proportion of variant alleles per gene in the patient group, control group and ExAC cohort. Variants were selected as follows: First, we selected heterozygous coding or splice site variants with an allelic balance between 0.25 and 0.85 (FLNA in males: 0.75–1) and a minimum coverage of 10x. Next, we made three variant groups based on their frequency in the ExAC database; that is, variants that are absent from the ExAC control dataset (blue), variants with an ExAC MAF lower than 0.01% (orange) and variants with an ExAC MAF between 0.01% and 0.1% that had a CADD score above 20 (gray). Only statistics of the patient-ExAC comparison are shown (**p ≤ 0.01). No statistically significant differences in allele frequencies were observed between our control cohort and the ExAC controls. Abbreviations: ExAC, Exome Aggregation Consortium; MAF, Minor Allele frequency; CADD, Combined Annotation Dependent Depletion.
Table 2.
Variant burden comparisons per gene between patients and either study controls or ExAC controls.
| Gene | Number of variants in 882 patient alleles | Number of variants in 366 control alleles | Number of variants in ExAC alleles | p-value patients-controls | p-value patients-ExAC |
|---|---|---|---|---|---|
| ACTA2 | 2 | 1 | 109 in 120,631 | 1.00 | 0.44 |
| ACVR1 | 2 | 1 | 202 in 120,994 | 1.00 | 0.98 |
| ELN | 4 | 2 | 728 in 113,954 | 1.00 | 0.63 |
| FBN1 | 16 | 5 | 1,740 in 120,988 | 0.81 | 0.43 |
| FLNA | 3* | 6* | 1,133 in 84,359* | 0.03 | 0.15 |
| GATA4 | 5 | 1 | 260 in 105,980 | 0.68 | 0.11 |
| GATA5 | 2 | 3 | 259 in 86,819 | 0.15 | 0.94 |
| GATA6 | 5 | 3 | 240 in 95,775 | 0.70 | 0.13 |
| MAT2A | 0 | 0 | 74 in 116,667 | / | / |
| MATR3 | 1 | 0 | 382 in 119,089 | / | 0.43 |
| MYH11 | 17 | 8 | 2,513 in 119,001 | 0.82 | 0.79 |
| NKX2-5 | 5 | 0 | 360 in 98,978 | / | 0.47 |
| NOS3 | 5 | 7 | 1,390 in 102,070 | 0.05 | 0.06 |
| NOTCH1 | 10 | 7 | 2,181 in 101,245 | 0.29 | 0.05 |
| ROBO1 | 12 | 5 | 1,354 in 113,390 | 1.00 | 0.77 |
| ROBO2 | 9 | 5 | 1,245 in 119,282 | 0.57 | 0.95 |
| SMAD3 | 0 | 1 | 95 in 111,500 | / | / |
| SMAD6 | 11 | 1 | 450 in 94,779 | 0.20 | 0.002 |
| TGFB2 | 1 | 0 | 192 in 117,070 | / | 0.71 |
| TGFB3 | 0 | 0 | 205 in 121,315 | / | / |
| TGFBR1 | 2 | 0 | 181 in 118,320 | / | 0.90 |
| TGFBR2 | 0 | 1 | 366 in 115,147 | / | / |
Variant burden analyses were performed comparing frequencies of the variants fulfilling the three criteria that were mentioned in “Section Data Analysis and Filtering” between patients and controls. Whereas, the Fisher's Exact Test was used to statistically compare variant frequencies in the patient cohort to those in the study control cohort, the Chi-Square Test with Yates' correction was used for the patient-ExAC comparison. No p-values were calculated if the number of variants in patients and/or controls was zero. Statistical significance was considered when p < 0.05. The asterisks denote that in these cases the number of alleles is consistent with the number of X-chromosomes, i.e., 553 patient alleles and 260 control alleles were checked for variants. Statistically significant p-values are represented in bold.
We identified 11 SMAD6 variants in 441 patients (2.5%). These included two frameshift deletions, two nonsense mutations, one in-frame deletion, and six missense variants (Figure 2). Only a single individual (0.55%) in the study control cohort harbored a SMAD6 missense variant. The ExAC database harbored 450 SMAD6 variants in 47,389 individuals (0.9%). Whereas 36.4% (n = 4/11) of the SMAD6 mutations in the patient cohort were loss of function (LOF; frameshift, nonsense or splice site) mutations, truncating SMAD6 mutations were found in only 4.0% (n = 18/450) of the ExAC individuals, demonstrating a clear enrichment in BAV/TAA patients compared to controls (p = 0.001).
Figure 2.
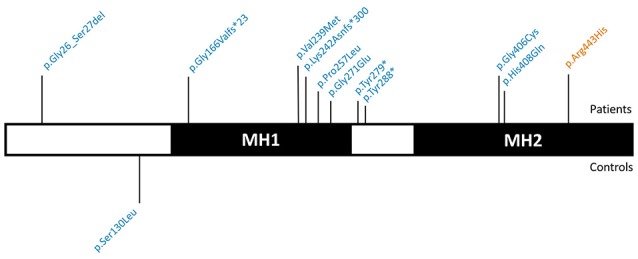
Graphical representation of the identified SMAD6 variants. SMAD6 has two major protein domains, a DNA-binding MH1 domain and a MH2 domain that interacts with components of the TGF-β and BMP signaling pathways. Variants above the protein have been found in patients, while those below the protein occurred in control individuals. Variants in blue are absent from the ExAC database, variants in orange have an ExAC MAF below 0.01%. Abbreviations: TGF-β, Transforming growth factor-β; BMP, Bone morphogenetic protein; ExAC, Exome Aggregation Consortium; MAF, Minor Allele frequency.
The SMAD6 c.726del variant leads to a frameshift (p.Lys242Asnfs*300) and a predicted protein with a C-terminal extension due to loss of the intended stop codon. The c.454_461del frameshift variant (p.Gly166Valfs*23) causes the introduction of a premature stop codon, most likely resulting in haploinsufficiency due to nonsense-mediated mRNA decay (NMD). Also the two nonsense variants (p.Tyr279* and p.Tyr288*) are predicted to lead to NMD. All of the missense variants cluster in the functionally important MH1 and MH2 domains (Makkar et al., 2009) (amino acids 148–275 and 331–496, respectively), which is not the case for the sole missense variant (p.Ser130Leu) found in a control individual (Figure 2). All but one (p.Arg443His) of the identified variants were absent in the ExAC control cohort (v0.3.1; Supplementary Table 2). Moreover, the missense variants in the patient cohort (7/7) are enriched in the MH1 and MH2 domains when compared to ExAC controls (n = 228/430; p = 0.02).
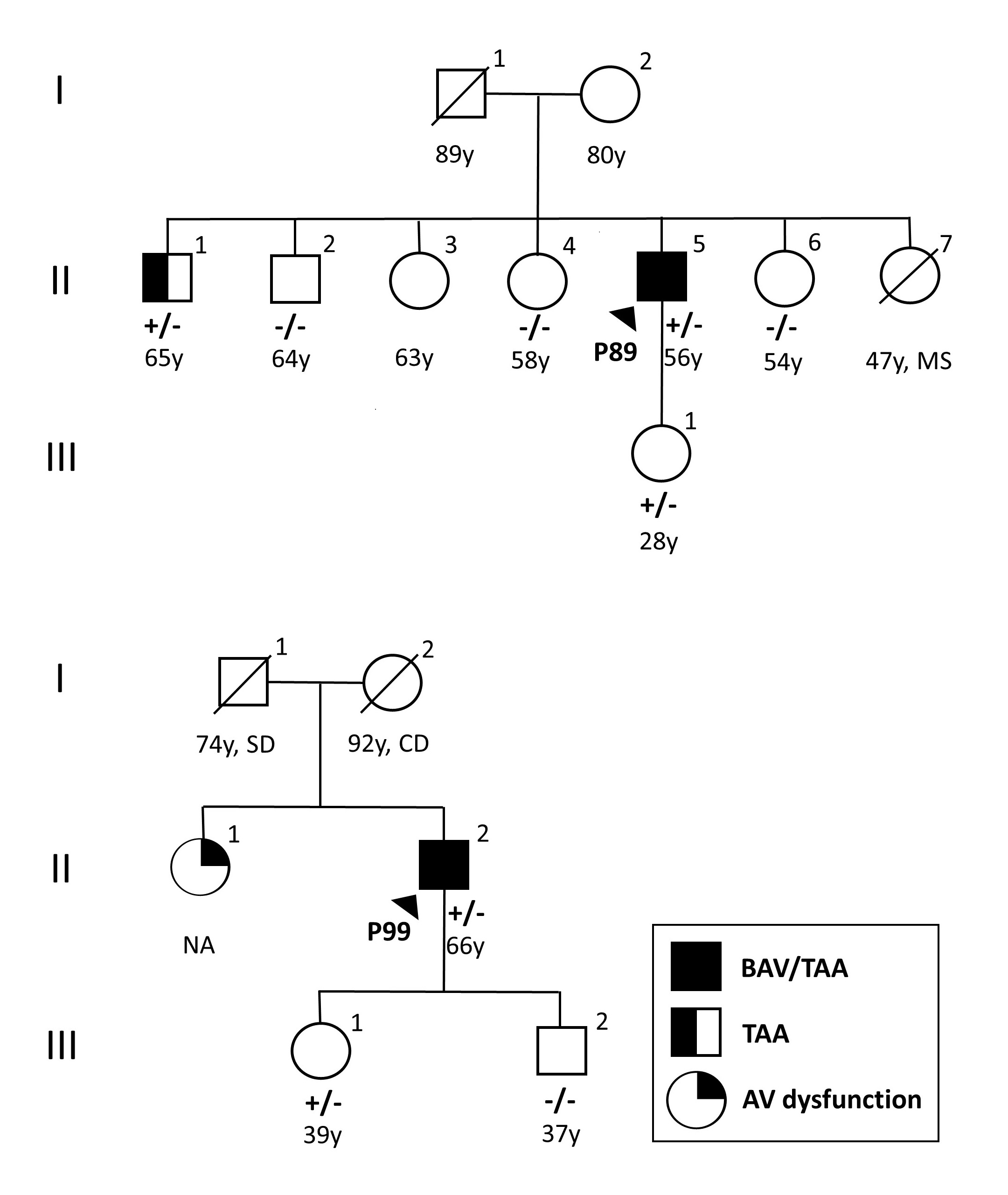
For two SMAD6 mutation carriers (P89, p.Gly271Glu; P99, p.Gly166Valfs*23), gDNA of family members was available for segregation analysis (Supplementary Figure 1). Although neither of these probands had a documented family history of BAV/TAA, a brother of P89 has been diagnosed with a sinus of Valsalva aneurysm (45 mm) and carried the SMAD6 mutation. The mutation was also observed in an unaffected daughter (age 28) of the proband (Supplementary Figure 1). Three unaffected siblings at ages 54, 58, and 64 did not carry the mutation. No gDNA was available from a sister of P99 with unspecified aortic valve problems. The p.Gly166Valfs*23 mutation was found in an unaffected daughter (age 39) of P99 but was absent in his 39 year-old unaffected son (Supplementary Figure 1).
Intriguingly, two genes (NOTCH1 and NOS3) that previously had been associated with increased BAV risk in humans (Mohamed et al., 1797; Garg et al., 2005; McKellar et al., 2007; Foffa et al., 2013) and/or mice (Lee et al., 2000; Bosse et al., 2013) revealed borderline significance for protection from BAV/TAA (p = 0.05 and p = 0.06, respectively). Analysis of NOTCH1 identified 10 variants in patients (2.3%), including two splice-site variants, vs. seven variants (all missense) in controls (3.8%) and 2,181 (4.3%) variants in ExAC. One variant in the patient cohort (c.5167+3_5167+6del) leads to complete loss of the 5' donor splice site of intron 27, predicted to result in skipping of exon 27 (149 bp) and hence a frameshift. For the second variant (p.S784S), the predicted effect on splicing is more ambiguous. If loss of the 5' donor splice site of intron 14 would occur, skipping of exon 14 (146 bp) would again lead to a frameshift event. Unfortunately, cDNA to reliably determine the precise effect of these mutations on splicing is not available. None of the NOTCH1 variants that we identified in BAV/TAA patients has previously been reported in the literature. We did not observe any variant-domain clustering or significant differences in CADD scores when comparing the patient and control NOTCH1 variants. Similarly, for NOS3 a total of five missense variants (1.1%) was found in patients, whereas the control cohort harbored seven variants (3.8%), including one out-of-frame mutation (p.Leu927Hisfs*32). In the ExAC control cohort, 1,390 NOS3 variants (2.7%) were found in 51,035 individuals.
Based on statistical analyses of BAV/TAA heritability and the fact that BAV/TAA shows prominent gender bias, oligogenic inheritance of BAV/TAA is an emerging concept (Andelfinger et al., 2016; Verstraeten et al., 2016). To test for such oligogenic patterns, we determined the number of patients and controls in our study cohort with variants in at least two out of the 22 analyzed genes. In the patient cohort, 10 patients presented with two variants (2.3%), while the control group harbored 7 individuals that carried two variants (3.8%). Based on these data, there is no evidence for a digenic or multigenic model in the analyzed genes (p = 0.29).
Discussion
So far, no gene with a contribution of more than 1% to BAV or BAV/TAA has been identified in humans. Gene identification has been hampered by low penetrance, variable clinical expressivity, the likelihood of BAV-phenocopies within individual families and, most likely, substantial locus heterogeneity (Verstraeten et al., 2016). NOTCH1 has been suggested as a BAV(/TAA) gene, but does not contribute greatly to disease etiology. About 20 other genes have been associated with BAV in humans and mice (Table 1), but few of them also showed association with TAA. This suggests that whereas some disease genes might be linked to both BAV and TAA, others increase risk for only one of the component phenotypes. In this study, we used a targeted gene panel approach to study the prevalence of mutations in genes that previously have been associated with BAV and/or TAA in people or mice in a cohort of BAV/TAA patients. In total, 22 genes were sequenced in 441 BAV/TAA patients and 183 controls. SMAD6 was identified as the most important known gene in the etiology of BAV with associated TAA. With 11 mutation-carrying probands, SMAD6 offers a molecular explanation for 2.5% of our study population. For two of the variants segregation analysis in relatives could be performed, revealing the presence of one of the respective SMAD6 mutations in a TAA patient and two rather young individuals (age 28 & 39) that might still develop TAA later in life. Four unaffected individuals (age 37, 54, 58, 64) did not carry a SMAD6 mutation. As two nonsense and two frameshift SMAD6 variants in our cohort are predicted to lead to haploinsufficiency, LOF is the most likely mechanism. All the patient-specific missense variants (n = 7) are in the functionally important MH1 and MH2 domains of SMAD6 (Makkar et al., 2009). LOF missense mutations in SMAD2 and SMAD3 causing Loeys-Dietz syndrome, another syndromic TAA form, are also located in the MH1 and MH2 domains (van de Laar et al., 2011; Micha et al., 2015). The MH1 domain of SMAD6 binds DNA (Bai and Cao, 2002), while the MH2 domain interacts with key components of the transforming growth factor (TGF)-β and bone morphogenetic protein (BMP) signaling cascades (Hanyu et al., 2001; Lin et al., 2003; Jung et al., 2013). In 2012, two missense variants in the MH2 domain of SMAD6 were identified in two patients with BAV in association with mild to moderate aortic stenosis (Tan et al., 2012). Interestingly, in our cohort, one SMAD6 patient (p.Tyr288*) presented with coarctation in addition to BAV and TAA. Moreover, mice lacking expression of the murine orthologue of SMAD6, i.e., Madh6−/− mice, also present with cardiovascular pathologies, including abnormal vascular smooth muscle cell relaxation, thickening of the cardiac valves and misplaced septation and ossification of the outflow tract (OFT) (Galvin et al., 2000). As such, our findings confirm a role for SMAD6 mutations in the etiology of BAV and expand the spectrum of SMAD6-related cardiovascular manifestations with BAV-related TAA.
SMAD6 is highly expressed in the cardiac valves and OFT of the embryonic heart, in the late-embryonic, and adult vascular endothelium as well as in the vascular smooth muscle cells of the adult aortic root (Galvin et al., 2000; Dickel et al., 2016). Upregulation in response to laminar shear stress has been reported (Topper et al., 1997). SMAD6 encodes an inhibitory SMAD protein which negatively regulates BMP signaling by binding to BMP type I receptors or by establishing competitive interactions for SMAD4 (Imamura et al., 1997; Hata et al., 1998). In doing so, SMAD1/5/8 phosphorylation and/or nuclear translocation are prevented. Additionally, SMAD6 cooperates with SMURF E3 ubiquitin ligases to prime ubiquitin-mediated proteasomal degradation of BMP receptors and SMAD effector proteins (Murakami et al., 2003), including SMAD1 and 5. BMP signaling has previously been independently implicated in BAV- and TAA-related processes (Cai et al., 2012; Garside et al., 2013). In addition to mediating CNC cell migration into the cardiac cushions and differentiation to smooth muscle cells, BMP signaling promotes endothelial-to-mesenchymal transition and instigates mesenchymal cell invasion (Kaartinen et al., 2004; Garside et al., 2013). While SMAD6 and SMAD7 are thought to have a predominant negative regulatory effect on BMP and TGF-β signaling, respectively, there is strong evidence that this specificity is not absolute and that SMAD6 can directly suppress the TGF-β signaling cascade. Important crosstalk between BMP, TGF-β and NOTCH signaling has been reported (Garside et al., 2013). Many syndromic forms of TAA are caused by mutations in genes encoding effectors or regulators of the TGF-β signaling pathway (including TGFB2/3, TGFBR1/2, SMAD2/3, SKI) (Loeys et al., 2005; van de Laar et al., 2011; Boileau et al., 2012; Carmignac et al., 2012; Doyle et al., 2012; Lindsay et al., 2012; Bertoli-Avella et al., 2015; Micha et al., 2015), with increased activity observed in aortic specimens from people and mice with these conditions. An increased prevalence of BAV has been observed in patients carrying mutations in these genes (Table 1). Overall, these results imply that mutations in SMAD6 likely cause BAV/TAA through impaired negative regulation of BMP and/or TGF-β signaling.
Multiple studies have previously reported a link between NOTCH1 mutations and BAV (Mohamed et al., 1797; Garg et al., 2005; McKellar et al., 2007; Foffa et al., 2013). In 2005, a nonsense and a frameshift NOTCH1 mutation were found to segregate with BAV associated with early onset valve calcification in the respective families (Garg et al., 2005). Since the initial report, multiple NOTCH1, mostly missense, variants have been associated with BAV, BAV/TAA, aortic valve stenosis, coarctation, and hypoplastic left heart (Mohamed et al., 1797; McKellar et al., 2007; Iascone et al., 2012; Foffa et al., 2013; Freylikhman et al., 2014; Preuss et al., 2016; Irtyuga et al., 2017). In addition to these mutations in association with left-sided heart defects, frameshift and nonsense mutations were also identified in patients with right-sided heart defects affecting the pulmonary valve and conotruncal disease including pulmonary atresia with intact ventricular septum, tetralogy of Fallot, and truncus arteriosus, and other congenital heart diseases, such as anomalous pulmonary venous return, atrial septal defect, and ventricular septal defect (Kerstjens-Frederikse et al., 2016). Mouse models have confirmed a role for Notch1 in the development of the aortic valve and the cardiac OFT (Koenig et al., 2016). Unexpectedly, in our dataset NOTCH1 did not stand out as a prominent BAV/TAA gene, with the suggestion that NOTCH1 variants might even be protective. Sample selection bias might contribute to this observation as NOTCH1 variants appear to associate with early and severe valve calcification and seem to be enriched in families with highly penetrant BAV but far lower penetrance of TAA (Kent et al., 2013). Given that our study did not select for valve calcification and prioritized the BAV/TAA phenotype, it is understandable that NOTCH1 variants would be underrepresented. It also seems notable that only missense variants were seen in controls, while multiple variants in the patient cohort are predicted to have a more overt impact on protein expression and function.
Similarly, our variant burden test suggested that NOS3 variants might be protective for BAV/TAA development. NOS3, the endothelial specific nitric oxide (NO) synthase, is important in balancing NO production and in the reduction of oxidative stress (Forstermann and Munzel, 2006). Its role in cardiac development is demonstrated by the formation of BAV in Nos3-targeted mice (Table 1). Furthermore, it has already been shown that specific NOS3 polymorphisms can affect NO production (Oliveira-Paula et al., 2016), and increased NO levels have been found in a MFS mouse model and in Adamts1-deficient mice that develop TAA (Oller et al., 2017). Pharmacological inhibition of NOS2 in mice led to a protective effect in aortic aneurysm development (Oller et al., 2017). This supports the importance of NO levels and nitric oxide synthases in aneurysm pathology. The variants in NOS3 identified in the current study may lead to less active NOS3 and as such may protect against development of aortic aneurysm.
Our study has several methodological limitations: (i) The small number of genes included in our study, as well as the patient cohort size, precludes the ability to detect oligogenic inheritance or gene-gene interactions involved in BAV/TAA. An extended experiment in a larger BAV/TAA cohort, including BAV-related pathways instead of selected genes, could give us more insight regarding how genes work together in BAV and/or TAA development; (ii) The size of the patient and study/ExAC control cohort only allows us to detect BAV/TAA genes with a fairly large contribution (variant burden in patients: ≥3% & ≥2%, respectively); (iii) The control cohort consists of younger, adolescent patients that did not show cardiac complications at the time of investigation but may still develop complications such as TAA later-on in life. Therefore, the ExAC database was used as an additional dataset for allele frequencies in a cohort without gross developmental defects.
Our study specifically assesses the presence of pathogenic variants in BAV-associated genes in a large BAV/TAA cohort. We conclude that SMAD6 is currently the most important contributor to the genetic architecture of BAV/TAA. More research and larger cohorts will be needed to fully elucidate the genetic architecture of this common but complex cardiovascular pathology.
Ethics statement
This study was carried out in accordance with written informed consent from all subjects. All subjects gave written informed consent in accordance with the Declaration of Helsinki. The protocol was approved by the Ethics Committee of the Antwerp University Hospital and all participating centers.
Author contributions
All authors revised the work critically. All authors provided final approval of the version for publication. All authors agreed to be accountable for all aspects of the work and ensure that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. More specific contributions to the work are: EG, AAK, IL, CP, EC, NB, FW, RG, LV, SAM, SM, LM, HB, AF, AM, PE, GA, HD, AV, and BL contributed to conception and design of the work. EG, AAK, IL, EC, MA, NB, GvdB, BW, GV, JM, RZ, DZ, SAM, SM, LM, JV, IV, MW, EM, GG, MN, AK, MK, SS, TD, XJ, JA, PE, AV, and BL contributed to acquisition of the data. EG, AAK, CP, FW, GA, LV, HD, AV, and BL contributed to analysis of the data. EG, IL, CP, EC, MA, FW, RG, RZ, DZ, SAM, SM, LM, HB, AF, AM, LV, JV, IV, MW, EM, GG, MN, AK, MK, SS, TD, XJ, JA, PE, HD, AV, and BL contributed to interpretation of the data.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Footnotes
Funding. This research was supported by funding from the University of Antwerp (Lanceringsproject), the Fund for Scientific Research, Flanders (FWO, Belgium, G.0221.12), The Dutch Heart Foundation (2013T093), the Foundation Leducq (MIBAVA—Leducq 12CVD03). BL is senior clinical investigator of the Fund for Scientific Research, Flanders (FWO). AV and GV are FWO postdoctoral researchers and JM (FWO) and IL (FWO-SB) hold a PhD mandate. BL holds a starting grant from the European Research Council (ERC- StG-2012-30972-BRAVE).
Supplementary material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fphys.2017.00400/full#supplementary-material
{kind=link}
References
- Andelfinger G., Loeys B., Dietz H. (2016). A decade of discovery in the genetic understanding of thoracic aortic disease. Can. J. Cardiol. 32, 13–25. 10.1016/j.cjca.2015.10.017 [DOI] [PubMed] [Google Scholar]
- Attias D., Stheneur C., Roy C., Collod-Beroud G., Detaint D., Faivre L., et al. (2009). Comparison of clinical presentations and outcomes between patients with TGFBR2 and FBN1 mutations in Marfan syndrome and related disorders. Circulation 120, 2541–2549. 10.1161/CIRCULATIONAHA.109.887042 [DOI] [PubMed] [Google Scholar]
- Bai S., Cao X. (2002). A nuclear antagonistic mechanism of inhibitory Smads in transforming growth factor-beta signaling. J. Biol. Chem. 277, 4176–4182. 10.1074/jbc.M105105200 [DOI] [PubMed] [Google Scholar]
- Bertoli-Avella A. M., Gillis E., Morisaki H., Verhagen J. M., de Graaf B. M., van de Beek G., et al. (2015). Mutations in a TGF-β ligand, TGFB3, cause syndromic aortic aneurysms and dissections. J. Am. Coll. Cardiol. 65, 1324–1336. 10.1016/j.jacc.2015.01.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biben C., Weber R., Kesteven S., Stanley E., McDonald L., Elliott D. A., et al. (2000). Cardiac septal and valvular dysmorphogenesis in mice heterozygous for mutations in the homeobox gene Nkx2-5. Circ. Res. 87, 888–895. 10.1161/01.RES.87.10.888 [DOI] [PubMed] [Google Scholar]
- Boileau C., Guo D. C., Hanna N., Regalado E. S., Detaint D., Gong L., et al. (2012). TGFB2 mutations cause familial thoracic aortic aneurysms and dissections associated with mild systemic features of Marfan syndrome. Nat. Genet. 44, 916–921. 10.1038/ng.2348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonachea E. M., Zender G., White P., Corsmeier D., Newsom D., Fitzgerald-Butt S., et al. (2014). Use of a targeted, combinatorial next-generation sequencing approach for the study of bicuspid aortic valve. BMC Med. Genomics 7:56. 10.1186/1755-8794-7-56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosse K., Hans C. P., Zhao N., Koenig S. N., Huang N., Guggilam A., et al. (2013). Endothelial nitric oxide signaling regulates Notch1 in aortic valve disease. J. Mol. Cell. Cardiol. 60, 27–35. 10.1016/j.yjmcc.2013.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braverman A. C., Guven H., Beardslee M. A., Makan M., Kates A. M., Moon M. R. (2005). The bicuspid aortic valve. Curr. Probl. Cardiol. 30, 470–522. 10.1016/j.cpcardiol.2005.06.002 [DOI] [PubMed] [Google Scholar]
- Cai J., Pardali E., Sanchez-Duffhues G., ten Dijke P. (2012). BMP signaling in vascular diseases. FEBS Lett. 586, 1993–2002. 10.1016/j.febslet.2012.04.030 [DOI] [PubMed] [Google Scholar]
- Callewaert B., Renard M., Hucthagowder V., Albrecht B., Hausser I., Blair E., et al. (2011). New insights into the pathogenesis of autosomal-dominant cutis laxa with report of five ELN mutations. Hum. Mutat. 32, 445–455. 10.1002/humu.21462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmignac V., Thevenon J., Ades L., Callewaert B., Julia S., Thauvin-Robinet C., et al. (2012). In-frame mutations in exon 1 of SKI cause dominant Shprintzen-Goldberg syndrome. Am. J. Hum. Genet. 91, 950–957. 10.1016/j.ajhg.2012.10.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clementi M., Notari L., Borghi A., Tenconi R. (1996). Familial congenital bicuspid aortic valve: a disorder of uncertain inheritance. Am. J. Med. Genet. 62, 336–338. 10.1002/(SICI)1096-8628(19960424)62:4<336::AID-AJMG2>3.0.CO;2-P [DOI] [PubMed] [Google Scholar]
- Cripe L., Andelfinger G., Martin L. J., Shooner K., Benson D. W. (2004). Bicuspid aortic valve is heritable. J. Am. Coll. Cardiol. 44, 138–143. 10.1016/j.jacc.2004.03.050 [DOI] [PubMed] [Google Scholar]
- DePristo M. A., Banks E., Poplin R., Garimella K. V., Maguire J. R., Hartl C., et al. (2011). A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 43, 491–498. 10.1038/ng.806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickel D. E., Barozzi I., Zhu Y., Fukuda-Yuzawa Y., Osterwalder M., Mannion B. J., et al. (2016). Genome-wide compendium and functional assessment of in vivo heart enhancers. Nat. Commun. 7:12923. 10.1038/ncomms12923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle A. J., Doyle J. J., Bessling S. L., Maragh S., Lindsay M. E., Schepers D., et al. (2012). Mutations in the TGF-beta repressor SKI cause Shprintzen-Goldberg syndrome with aortic aneurysm. Nat. Genet. 44, 1249–1254. 10.1038/ng.2421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foffa I., Ait Ali L., Panesi P., Mariani M., Festa P., Botto N., et al. (2013). Sequencing of NOTCH1, GATA5, TGFBR1 and TGFBR2 genes in familial cases of bicuspid aortic valve. BMC Med. Genet. 14:44. 10.1186/1471-2350-14-44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forstermann U., Munzel T. (2006). Endothelial nitric oxide synthase in vascular disease: from marvel to menace. Circulation 113, 1708–1714. 10.1161/CIRCULATIONAHA.105.602532 [DOI] [PubMed] [Google Scholar]
- Freylikhman O., Tatarinova T., Smolina N., Zhuk S., Klyushina A., Kiselev A., et al. (2014). Variants in the NOTCH1 gene in patients with aortic coarctation. Congenit. Heart Dis. 9, 391–396. 10.1111/chd.12157 [DOI] [PubMed] [Google Scholar]
- Galvin K. M., Donovan M. J., Lynch C. A., Meyer R. I., Paul R. J., Lorenz J. N., et al. (2000). A role for smad6 in development and homeostasis of the cardiovascular system. Nat. Genet. 24, 171–174. 10.1038/72835 [DOI] [PubMed] [Google Scholar]
- Garg V., Muth A. N., Ransom J. F., Schluterman M. K., Barnes R., King I. N., et al. (2005). Mutations in NOTCH1 cause aortic valve disease. Nature. 437, 270–274. 10.1038/nature03940 [DOI] [PubMed] [Google Scholar]
- Garside V. C., Chang A. C., Karsan A., Hoodless P. A. (2013). Co-ordinating Notch BMP and TGF-β signaling during heart valve development. Cell. Mol. Life Sci. 70, 2899–2917. 10.1007/s00018-012-1197-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo D. C., Gong L., Regalado E. S., Santos-Cortez R. L., Zhao R., Cai B., et al. (2015). MAT2A mutations predispose individuals to thoracic aortic aneurysms. Am. J. Hum. Genet. 96, 170–177. 10.1016/j.ajhg.2014.11.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo D. C., Pannu H., Tran-Fadulu V., Papke C. L., Yu R. K., Avidan N., et al. (2007). Mutations in smooth muscle α-actin (ACTA2) lead to thoracic aortic aneurysms and dissections. Nat. Genet. 39, 1488–1493. 10.1038/ng.2007.6 [DOI] [PubMed] [Google Scholar]
- Hanyu A., Ishidou Y., Ebisawa T., Shimanuki T., Imamura T., Miyazono K. (2001). The N domain of Smad7 is essential for specific inhibition of transforming growth factor-beta signaling. J. Cell Biol. 155, 1017–1027. 10.1083/jcb.200106023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hata A., Lagna G., Massague J., Hemmati-Brivanlou A. (1998). Smad6 inhibits BMP/Smad1 signaling by specifically competing with the Smad4 tumor suppressor. Genes Dev. 12, 186–197. 10.1101/gad.12.2.186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinton R. B. (2012). Bicuspid aortic valve and thoracic aortic aneurysm: three patient populations, two disease phenotypes, and one shared genotype. Cardiol. Res. Pract. 2012:926975. 10.1155/2012/926975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huntington K., Hunter A. G., Chan K. L. (1997). A prospective study to assess the frequency of familial clustering of congenital bicuspid aortic valve. J. Am. Coll. Cardiol. 30, 1809–1812. 10.1016/S0735-1097(97)00372-0 [DOI] [PubMed] [Google Scholar]
- Iascone M., Ciccone R., Galletti L., Marchetti D., Seddio F., Lincesso A. R., et al. (2012). Identification of de novo mutations and rare variants in hypoplastic left heart syndrome. Clin. Genet. 81, 542–554. 10.1111/j.1399-0004.2011.01674.x [DOI] [PubMed] [Google Scholar]
- Imamura T., Takase M., Nishihara A., Oeda E., Hanai J., Kawabata M., et al. (1997). Smad6 inhibits signalling by the TGF-beta superfamily. Nature 389, 622–626. [DOI] [PubMed] [Google Scholar]
- Irtyuga O., Malashicheva A., Zhiduleva E., Freylikhman O., Rotar O., Back M., et al. (2017). NOTCH1 mutations in aortic stenosis: association with osteoprotegerin/RANK/RANKL. Biomed. Res. Int. 2017:6917907. 10.1155/2017/6917907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jefferies J. L., Taylor M. D., Rossano J., Belmont J. W., Craigen W. J. (2010). Novel cardiac findings in periventricular nodular heterotopia. Am. J. Med. Genet. A. 152A, 165–168. 10.1002/ajmg.a.33110 [DOI] [PubMed] [Google Scholar]
- Jung S. M., Lee J. H., Park J., Oh Y. S., Lee S. K., Park J. S., et al. (2013). Smad6 inhibits non-canonical TGF-β1 signalling by recruiting the deubiquitinase A20 to TRAF6. Nat. Commun. 4:2562. 10.1038/ncomms3562 [DOI] [PubMed] [Google Scholar]
- Kaartinen V., Dudas M., Nagy A., Sridurongrit S., Lu M. M., Epstein J. A. (2004). Cardiac outflow tract defects in mice lacking ALK2 in neural crest cells. Development 131, 3481–3490. 10.1242/dev.01214 [DOI] [PubMed] [Google Scholar]
- Kent K. C., Crenshaw M. L., Goh D. L., Dietz H. C. (2013). Genotype-phenotype correlation in patients with bicuspid aortic valve and aneurysm. J. Thorac. Cardiovasc. Surg. 146, 158–1565. 10.1016/j.jtcvs.2012.09.060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerstjens-Frederikse W. S., van de Laar I. M., Vos Y. J., Verhagen J. M., Berger R. M., Lichtenbelt K. D., et al. (2016). Cardiovascular malformations caused by NOTCH1 mutations do not keep left: data on 428 probands with left-sided CHD and their families. Genet. Med. 18, 914–923. 10.1038/gim.2015.193 [DOI] [PubMed] [Google Scholar]
- Kircher M., Witten D. M., Jain P., O'Roak B. J., Cooper G. M., Shendure J. (2014). A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 46, 310–315. 10.1038/ng.2892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koenig S. N., Bosse K., Majumdar U., Bonachea E. M., Radtke F., Garg V. (2016). Endothelial notch1 is required for proper development of the semilunar valves and cardiac outflow tract. J. Am. Heart Assoc. 5:e003075. 10.1161/JAHA.115.003075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laforest B., Andelfinger G., Nemer M. (2011). Loss of Gata5 in mice leads to bicuspid aortic valve. J. Clin. Invest. 121, 2876–2887. 10.1172/JCI44555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laforest B., Nemer M. (2011). GATA5 interacts with GATA4 and GATA6 in outflow tract development. Dev. Biol. 358, 368–378. 10.1016/j.ydbio.2011.07.037 [DOI] [PubMed] [Google Scholar]
- Lee T. C., Zhao Y. D., Courtman D. W., Stewart D. J. (2000). Abnormal aortic valve development in mice lacking endothelial nitric oxide synthase. Circulation 101, 2345–2348. 10.1161/01.CIR.101.20.2345 [DOI] [PubMed] [Google Scholar]
- Lek M., Karczewski K. J., Minikel E. V., Samocha K. E., Banks E., Fennell T., et al. (2016). Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285–291. 10.1038/nature19057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin X., Liang Y. Y., Sun B., Liang M., Shi Y., Brunicardi F. C., et al. (2003). Smad6 recruits transcription corepressor CtBP to repress bone morphogenetic protein-induced transcription. Mol. Cell. Biol. 23, 9081–9093. 10.1128/MCB.23.24.9081-9093.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsay M. E., Schepers D., Bolar N. A., Doyle J. J., Gallo E., Fert-Bober J., et al. (2012). Loss-of-function mutations in TGFB2 cause a syndromic presentation of thoracic aortic aneurysm. Nat. Genet. 44, 922–927. 10.1038/ng.2349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loeys B. L., Chen J., Neptune E. R., Judge D. P., Podowski M., Holm T., et al. (2005). A syndrome of altered cardiovascular, craniofacial, neurocognitive and skeletal development caused by mutations in TGFBR1 or TGFBR2. Nat. Genet. 37, 275–281. 10.1038/ng1511 [DOI] [PubMed] [Google Scholar]
- Loscalzo M. L., Goh D. L., Loeys B., Kent K. C., Spevak P. J., Dietz H. C. (2007). Familial thoracic aortic dilation and bicommissural aortic valve: a prospective analysis of natural history and inheritance. Am. J. Med. Genet. A 143A, 1960–1967. 10.1002/ajmg.a.31872 [DOI] [PubMed] [Google Scholar]
- Makkar P., Metpally R. P., Sangadala S., Reddy B. V. (2009). Modeling and analysis of MH1 domain of Smads and their interaction with promoter DNA sequence motif. J. Mol. Graph. Model. 27, 803–812. 10.1016/j.jmgm.2008.12.003 [DOI] [PubMed] [Google Scholar]
- Martin P., Kloesel B., Norris R., Lindsay M., Milan D., Body S. (2015). Embryonic development of the bicuspid aortic valve. J. Cardiovasc. Dev. Dis. 2:248. 10.3390/jcdd2040248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKellar S. H., Tester D. J., Yagubyan M., Majumdar R., Ackerman M. J., Sundt T. M., et al. (2007). Novel NOTCH1 mutations in patients with bicuspid aortic valve disease and thoracic aortic aneurysms. J. Thorac. Cardiovasc. Surg. 134, 290–296. 10.1016/j.jtcvs.2007.02.041 [DOI] [PubMed] [Google Scholar]
- Micha D., Guo D. C., Hilhorst-Hofstee Y., van Kooten F., Atmaja D., Overwater E., et al. (2015). SMAD2 mutations are associated with arterial aneurysms and dissections. Hum. Mutat. 36, 1145–1149. 10.1002/humu.22854 [DOI] [PubMed] [Google Scholar]
- Mohamed S. A., Aherrahrou Z., Liptau H., Erasmi A. W., Hagemann C., Wrobel S., et al. (1797). Novel missense mutations (p.T596M and p.PH) in NOTCH1 in patients with bicuspid aortic valve. Biochem. Biophys. Res. Commun. 345, 1460–1465. [DOI] [PubMed] [Google Scholar]
- Mommersteeg M. T., Yeh M. L., Parnavelas J. G., Andrews W. D. (2015). Disrupted Slit-Robo signalling results in membranous ventricular septum defects and bicuspid aortic valves. Cardiovasc. Res. 106, 55–66. 10.1093/cvr/cvv040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami G., Watabe T., Takaoka K., Miyazono K., Imamura T. (2003). Cooperative inhibition of bone morphogenetic protein signaling by Smurf1 and inhibitory Smads. Mol. Biol. Cell. 14, 2809–2817. 10.1091/mbc.E02-07-0441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nistri S., Porciani M. C., Attanasio M., Abbate R., Gensini G. F., Pepe G. (2012). Association of Marfan syndrome and bicuspid aortic valve: frequency and outcome. Int. J. Cardiol. 155, 324–325. 10.1016/j.ijcard.2011.12.009 [DOI] [PubMed] [Google Scholar]
- Oliveira-Paula G. H., Lacchini R., Tanus-Santos J. E. (2016). Endothelial nitric oxide synthase: from biochemistry and gene structure to clinical implications of NOS3 polymorphisms. Gene 575(2 Pt 3), 584–99. 10.1016/j.gene.2015.09.061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oller J., Mendez-Barbero N., Ruiz E. J., Villahoz S., Renard M., Canelas L. I., et al. (2017). Nitric oxide mediates aortic disease in mice deficient in the metalloprotease Adamts1 and in a mouse model of Marfan syndrome. Nat. Med. 23, 200–212. 10.1038/nm.4266 [DOI] [PubMed] [Google Scholar]
- Pepe G., Nistri S., Giusti B., Sticchi E., Attanasio M., Porciani C., et al. (2014). Identification of fibrillin 1 gene mutations in patients with bicuspid aortic valve (BAV) without Marfan syndrome. BMC Med. Genet. 15:23. 10.1186/1471-2350-15-23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preuss C., Capredon M., Wunnemann F., Chetaille P., Prince A., Godard B., et al. (2016). Family based whole exome sequencing reveals the multifaceted role of notch signaling in congenital heart disease. PLoS Genet. 12:e1006335. 10.1371/journal.pgen.1006335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintero-Rivera F., Xi Q. J., Keppler-Noreuil K. M., Lee J. H., Higgins A. W., Anchan R. M., et al. (2015). MATR3 disruption in human and mouse associated with bicuspid aortic valve, aortic coarctation and patent ductus arteriosus. Hum. Mol. Genet. 24, 2375–2389. 10.1093/hmg/ddv004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan H. L., Glen E., Topf A., Hall D., O'Sullivan J. J., Sneddon L., et al. (2012). Nonsynonymous variants in the SMAD6 gene predispose to congenital cardiovascular malformation. Hum. Mutat. 33, 720–727. 10.1002/humu.22030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas P. S., Sridurongrit S., Ruiz-Lozano P., Kaartinen V. (2012). Deficient signaling via Alk2 (Acvr1) leads to bicuspid aortic valve development. PLoS ONE 7:e35539. 10.1371/journal.pone.0035539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topper J. N., Cai J., Qiu Y., Anderson K. R., Xu Y. Y., Deeds J. D., et al. (1997). Vascular MADs: two novel MAD-related genes selectively inducible by flow in human vascular endothelium. Proc. Natl. Acad. Sci. U.S.A. 94, 9314–9319. 10.1073/pnas.94.17.9314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Untergasser A., Cutcutache I., Koressaar T., Ye J., Faircloth B. C., Remm M., et al. (2012). Primer3–new capabilities and interfaces. Nucleic Acids Res. 40:e115. 10.1093/nar/gks596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de Laar I. M., Oldenburg R. A., Pals G., Roos-Hesselink J. W., de Graaf B. M., Verhagen J. M., et al. (2011). Mutations in SMAD3 cause a syndromic form of aortic aneurysms and dissections with early-onset osteoarthritis. Nat. Genet. 43, 121–126. 10.1038/ng.744 [DOI] [PubMed] [Google Scholar]
- van de Laar I. M., van der Linde D., Oei E. H., Bos P. K., Bessems J. H., Bierma-Zeinstra S. M., et al. (2012). Phenotypic spectrum of the SMAD3-related aneurysms-osteoarthritis syndrome. J. Med. Genet. 49, 47–57. 10.1136/jmedgenet-2011-100382 [DOI] [PubMed] [Google Scholar]
- Vandeweyer G., Van Laer L., Loeys B., Van den Bulcke T., Kooy R. F. (2014). VariantDB: a flexible annotation and filtering portal for next generation sequencing data. Genome Med. 6:746. 10.1186/s13073-014-0074-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verstraeten A., Roos-Hesselink J., Loeys B. (2016). Bicuspid aortic valve, in Clinical Cardiogenetics, eds Baars H. F., Doevendans P. A. F. M., Houweling A. C., van Tintelen J. P. (Cham: Springer International Publishing; ), 295–308. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.