

Abstract
Cytosolic Ca2+ is a universal second messenger that is involved in many processes throughout the body, including the regulation of cell growth/cell division, apoptosis, and the secretion of both ions, and macromolecules. Tobacco smoke exerts multiple effects on airway epithelia and we have previously shown that Kentucky reference cigarette smoke exposure elevated the second messenger Ca2+, leading to dysfunctional ion secretion. In this study, we tested whether little cigar and commercial cigarette smoke exposure exerts similar effects on intracellular Ca2+ levels. Indeed, Swisher Sweets, Captain Black, and Cheyenne little cigars, as well as Camel, Marlboro, and Newport cigarettes, triggered a comparable increase in intracellular Ca2+ as seen with Kentucky reference cigarettes in human bronchial epithelia. We also found that Kentucky reference cigarette smoke exposure caused increases in Ca2+ in HEK293T cells and that similar increases in Ca2+ were seen with the tobacco smoke metabolites 1-NH2-naphthalene, formaldehyde, nicotine, and nicotine-derived nitrosamine ketone. Given the large number of physiological processes governed by changes in cytosolic Ca2+, our data suggest that Ca2+ signaling is a useful and reproducible assay that can be used to probe the propensity of tobacco products and their constituents to cause toxicity.
Keywords: : cigarette toxicity, cytosolic calcium release, tobacco constituents
Introduction
Tobacco smoking is considered one of the major risk factors for mortality and morbidity. Despite health warnings on the packages, ban on advertising, and increased tax on tobacco products, there are 1.3 billion smokers worldwide. In the United States alone, tobacco has accounted for about 20 million premature deaths since the publication of Surgeon General's report in 1964. Tobacco can be smoked in many different forms, including cigarettes, cigars, pipes, and hookah. Cigars are defined as “any roll of tobacco wrapped in leaf tobacco or any substance containing tobacco,” whereas cigarettes are tobacco wrapped in paper. Cigars that weigh less than three pounds per one thousand units are identified as “Little Cigars.” Although the deleterious effects of cigarette smoking have been documented widely, little is known about the health effects of related tobacco products such as little cigars on the lung.
Tobacco exposure triggers inflammatory reactions in the airways.1 Furthermore, smoker's airways are characterized by dehydrated mucus.2,3 Cystic fibrosis transmembrane conductance regulator (CFTR)-mediated regulation of airway surface liquid (ASL) volume plays a critical role in the maintenance of mucus hydration, and CFTR function is required to preserve the sterility of the lungs.4 Impaired CFTR function has been linked with mucus dehydration, hindered mucociliary clearance, and increased mortality.5 Indeed, in cystic fibrosis patients carrying the G551D mutation, addition of the CFTR potentiator drug ivacaftor (Vx770) that rapidly restores CFTRG551D function, leads to improvements in lung function.6 We and others have demonstrated that both acute and chronic exposure to cigarette smoke cause CFTR dysfunction in humans.7–9 We have also shown that tobacco exposure leads to increases in cytosolic Ca2+ levels, which in turn mediate CFTR internalization and ASL depletion.10 Smoke from other similar tobacco products is expected to exert similar effects causing impaired airway surface hydration resulting in pulmonary disease initiation and progression. In this study, we have looked at whether little cigars and commercial cigarettes can elicit the increases in cytoplasmic Ca2+ seen with Kentucky reference cigarettes and tested whether 14 common tobacco constituents can also induce increases in cytosolic Ca2+.
Materials and Methods
Human bronchial epithelial isolation and culture
Human bronchial epithelial cells (HBECs) were isolated from main stem/lobar bronchi of human excess donor and excised recipient normal lung by enzymatic digestion following protocols approved by the University of North Carolina Committee on the Protection of the Rights of Human Subjects.11 Cells were plated on 12 mm Transwell clear-collagen coated membrane inserts (Corning Costar) and transferred to air–liquid interface conditions 5–7 days after seeding when cells were confluent. Only primary cells were used for the study. Cells were maintained in air–liquid interface media which is a modified bronchial epithelial growth medium with 5% CO2 at 37°C described in details previously11 and used after 2–6 weeks of seeding as described.8,12 Cells were imaged in HEPES-buffered Ringer solution.
HEK293T cell culture
HEK293T cells were cultured in 384-well plates as described.13 HEPES-buffered Ringer solution was used for imaging.8
Tobacco smoke exposure of HBECs
All tobacco products were stored at ambient laboratory temperature for 24 hours before use. Ten × 35 mL filtered puffs at a rate of one puff per 30 seconds without covering the ventilation holes were used per exposure. Since HBECs were imaged during the exposure on an inverted epifluorescent microscope, tobacco smoke was generated manually using a 50 mL syringe. As a control, we generated smoke from Kentucky Research Cigarettes (CODE 3R4F, Class A cigarettes) and tested six other commercial tobacco products: Marlboro (Class A cigarettes), Newport Nonmenthol cigarettes, Camel (Blue) cigarettes, Swisher Sweets Little Cigars (Filtered Little Cigars), Captain Black Little Cigars, and Cheyenne Cigars (Full Flavor). For air exposure, cells were exposed to 10 × 35 mL puffs of air. Cultures were placed in sterile Ringer's solution during imaging.
Fluorescent Ca2+ Imaging of HBECs
Epifluorescence measurements on individual cultures were performed using a Nikon Ti-S microscope with Hamamatsu Flash 4.0 Camera, Ludl Filter wheels, and a 40 × 1.2 NA plan fluor oil immersion lens. HBECs were basolaterally loaded with 5 μM Fluo-8 AM (Abcam ab142773), 1× PowerLoad™ concentrate (Molecular Probes P10020), and 2.5 mM probenecid at 37°C for 30 minutes in culture media for smoking experiments. Fura-2 (Sigma, F0888) was used in the nicotine experiments at a concentration of 4 μM. Cultures were washed with phosphate-buffered saline after an additional 30 minutes; tobacco smoke exposure was carried out in a custom-made chamber on the stage of the epifluorescent microscope. While within the chamber, cultures placed in Ringer's solution and the mucosal surface, which was under thin film conditions, were exposed to filtered smoke from the tobacco products. Fluo-8 fluorescence (excitation 490 nm, emission 520 nm) was measured at timed intervals before and after tobacco smoke. Fura-2 fluorescence (excitation 340/380 nm, 510 emission nm) was used to measure nicotine effects as per manufacturer's protocol. Data are presented as fold change of fluorescence after 15 minutes of start of smoke exposure compared to initial fluorescence intensity after subtracting background fluorescence from unstained cultures.
Fluorescent Ca2+ imaging of HEK293T
HEK293T cells were cultured in 384-well plates at a density of 10,000 cells/well and incubated overnight at 37°C and 5% CO2 to allow cells to recuperate. On the following day, media was replaced with 20 μL of 1× Fluo-4 Direct, and cells were incubated at 37°C and 5% CO2 for 1 hour. Various cigarette constituents were then added onto the cells at 10 mM and immediately imaged using a Tecan Infinite M1000 multiplate reader at 37°C. Changes in Fluo-4 Direct fluorescence (excitation 494 nm, emission 516 nm) were measured every 30 seconds for 8 minutes after chemical exposure.
Calcein-AM and propidium iodide staining of HEK293T
HEK293T cells were cultured in a 384-well plate at a density of 10,000 cells/well and incubated overnight at 37°C and 5% CO2 to allow cells to recuperate. Twenty-four hours later, various cigarette constituents were added to the cells and incubated at 37°C. After 1 hour of chemical exposure, chemicals and media were replaced, and cells were incubated with 2 μM of calcein-AM (Thermo Fisher Scientific) and 2 mg/mL μM of propidium Iodide (Sigma) to stain the live and dead cells, respectively, for 30 minutes at 37°C. Fluorescence was captured using an Infinite M1000 multiplate reader (Tecan).
Solutions and chemicals
All chemicals were purchased from Sigma-Aldrich, with the exception of calcein-AM, Fura2-AM, and Fluo-4 Direct, which were purchased from Thermo Fisher Scientific. Ringer solution for all experiments was (in mM) as follows: 120 NaCl, 5.2 KCl, 1.2 MgCl2, 1.2 CaCl2 · 2H2O, 12 NaHCO3, 24 HEPES, 10 glucose, pH 7.4.
Statistical analysis
Statistical analysis was performed using GraphPad Prism 5.0 with p < 0.05 considered as significant. All values are shown as mean ± standard error of the mean. Data were analyzed by analysis of variance. All experiments were performed using HBECs from three donors or on HEK293T cells tested on three separate occasions.
Results
Previously, we have demonstrated that the cytosolic calcium concentration is increased following exposure to cigarette smoke, which leads to CFTR internalization and ASL dehydration.14 To test whether tobacco smoke from little cigars and commercial cigarettes also affected Ca2+ signaling, we exposed polarized HBECs to 10 puffs from different tobacco products and monitored changes in Fluo-8 emission, as an indicator of cytosolic Ca2+ levels. All the tobacco products tested elicited a significant rise in cytosolic Ca2+ (Fig. 1), which typically peaked at ∼15 minutes and is shown as the increase in fluorescence emission divided by baseline emission (F/F0). We next tested the effects of nicotine on polarized HBECs. Nicotine induced a small but significant response that was slightly less than what we observed with the purinergic agonist ATP (Fig. 2).
FIG. 1.

Tobacco smoke exposure elicits increases in cytosolic Ca2+ in HBECs. Changes in Fluo-8 levels were measured by epifluorescence as an indicator of cytosolic Ca2+ levels following acute exposure of 10 filtered puffs of air/smoke for Kentucky reference cigarettes, three brands of little cigars, and three brands of commercial cigarettes (n = 8/group). Data are shown as peak changes (F/F0). (A) Time trace of fluorescence changes for air (open black circle), Kentucky (grey square), and Cheyenne (black triangle) cigarette exposure. Cells were exposed to air or smoke for 10 minutes, and fluorescence was measured for 17 minutes at 30-second intervals. (B) Fluorescence measured after 16 minutes of exposure for air, Kentucky, Swisher Sweets, Captain Black, Cheyenne, Marlboro, Newport, and Camel cigarettes. *p < 0.05. HBEC, human bronchial epithelial cell.
FIG. 2.
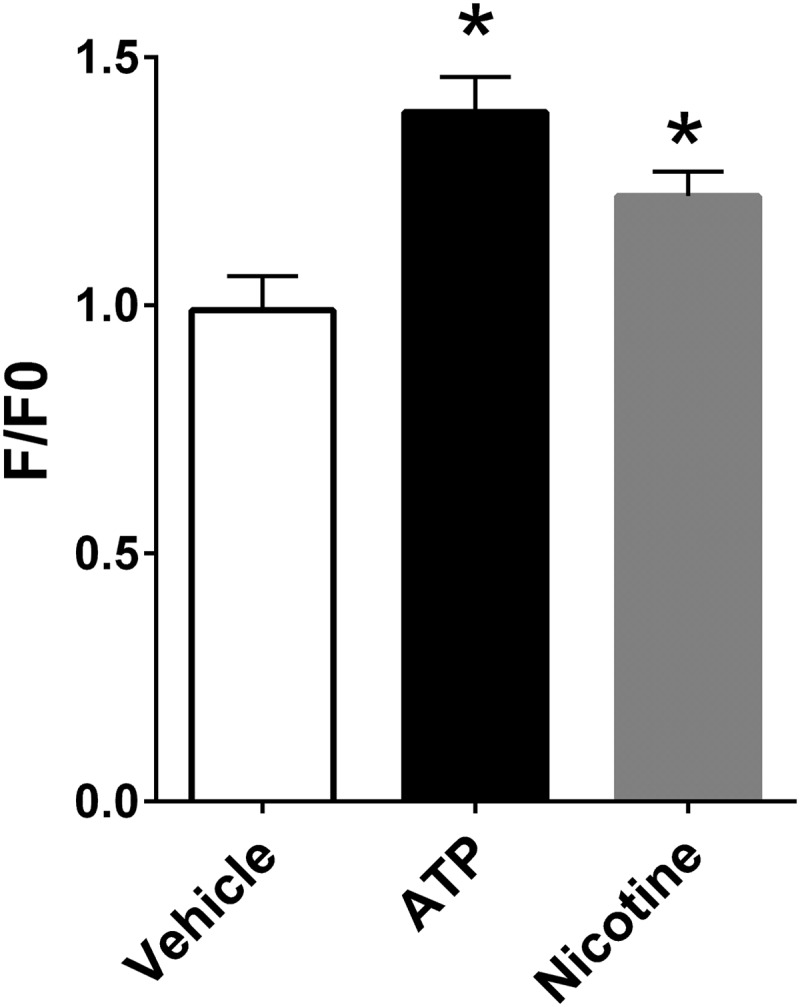
Nicotine increases Fura-2 fluorescence in polarized HBECs. Intracellular Fura-2 levels were measured in polarized HBECs as an indicator of cytoplasmic Ca2+ levels. Data are shown as peak changes in Fura-2 emission. Nicotine and ATP were added at 100 μM. All n = 6/group. *p < 0.05.
Since we saw only a moderate stimulation of Ca2+ release with nicotine, we next looked for other chemicals. Over 7000 different chemicals are present in whole tobacco smoke.15 These chemicals have been grouped according to their propensity for causing respiratory disease, cardiovascular disease, or cancer, although these groupings are not mutually exclusive. Examples of chemicals thought to induce respiratory disease include formaldehyde, acetaldehyde, acrolein, and benzene.15 To determine which constituents in tobacco smoke could have caused the observed increases in Ca2+, we tested 13 individual tobacco components by exposing HEK293T cells loaded with the Ca2+-indicator Fluo-4. We used HEK293T cells since they have previously been shown to elicit a similar increase in Ca2+ and a similar internalization of CFTR in HBECs after tobacco smoke exposure.14 HEK293T cells cultured in 384-well plates elicited similar increases in Ca2+ to vehicle, thapsigargin, and tobacco smoke constituents in Fig. 3.14 In contrast, of the 13 constituents tested, only 1-NH2-naphtalene, formaldehyde, nicotine, and nicotine-derived nitrosamine ketone (NNK) elicited increases in intracellular Ca2+ (Fig. 3).
FIG. 3.

Tobacco smoke constituents increase the fluorescence of the Ca2+-sensitive dye Fluo-4. HEK293T cells were cultured in 384-well plates and exposed to vehicle (− control), 1 μM thapsigargin (+ control), or 10 mM of the constituents as indicated. Data are shown as normalized peak changes in Fluo-4 emission (compared to vehicle). All n = 9/group. *p < 0.05.
To confirm that these increases in Ca2+ were not due to acute cell death, we performed viability assays on HEK293T cells grown in 384-well plates using calcein-AM and propidium iodide as live and dead stains, respectively. After 24 hours of culture in 384-well plates, HEK293T cells exposed to vehicle (media) yielded a F490/F530 ratio of ∼0.8, which we took as our baseline for a predominantly healthy culture. At higher doses, 1-NH2-naphtalene, acrolein, crotonaldehyde, and formaldehyde caused significant cell death (i.e., a decrease in the F495/F530 ratio). Ethanol was included as a positive control, which also killed cells (Fig. 4). In contrast, the other compounds tested (4ABP, acetaldehyde, acrylonitrile, benzene, isoprene, nicotine, NNK, N-Nitrosonornicotine, and toluene) had little effect on cell viability (Fig. 4A–C). A summary bar graph of all compounds and controls at 2.86 mM is shown in Fig. 4D.
FIG. 4.

The effects of tobacco smoke constituents on HEK293T cell viability. (A–C) HEK293T cells were cultured in 384-well plates for 24 hours, exposed to the chemicals of interest for 1 hour, and then stained with calcein-AM and propidium iodide to measure live and dead cells, respectively. Data are shown as the ratio calcein/propidium iodide emission (F495/F540). (D) Summary bar graph of data from (A–C) taken at 2.86 mM. All bars are n = 9 wells from three separate experiments. *p < 0.05.
Discussion
We have previously shown that acute tobacco exposure rapidly dehydrates the ASL, due to a tobacco-induced decrease in CFTR-mediated Cl− secretion.8 We also found that a variety of common inhibitors of cell signaling, including inhibitors of protein kinase A and PI3-kinase, had no effect on tobacco-induced CFTR diminution.10 However, chelation of Ca2+ with BAPTA-AM was the most potent maneuver that prevented loss of CFTR.10 Additional investigation revealed that tobacco exposure caused a sustained increase in intracellular Ca2+ (Ca2+i). Furthermore, tobacco induced increases in cytoplasmic Ca2+i were not inhibited by (i) the removal of extracellular Ca2+, (ii) endoplasmic reticulum Ca2+ depletion with thapsigargin, or (iii) addition of the mitochondrial uncoupler carbonyl cyanide 3-cholophenyl hydrazone (CCCP) indicating that Ca2+ influx, endoplasmic reticulum, and mitochondrial Ca2+ stores are not involved in tobacco-induced Ca2+ release.10 However, this increase in cytosolic Ca2+ was bafilomycin sensitive, suggesting that the Ca2+ came from lysosomes and persisted even with chronic tobacco exposure.10 In this study, we show that Kentucky, Camel, Marlboro, and Newport cigarettes, as well as little cigars (Swisher sweets, Captain Black, and Cheyenne), were equally efficacious at eliciting a rise in Ca2+i, suggesting that this is a common phenomenon of tobacco exposure (Fig. 1).
Intracellular Ca2+ is an important second messenger that regulates critical functions ranging from cell growth to differentiation to cell death.16,17 In the airways, Ca2+ also regulates ciliary beating,18,19 Cl−/ASL secretion,20 cytokine secretion, and mucin secretion.21 The majority of cytoplasmic Ca2+ emanates from the endoplasmic reticulum and excess cytoplasmic Ca2+ are usually rapidly taken up into this store soon after release. This is desirable because Ca2+ is usually only transiently elevated as a cytoplasmic second messenger.22 Our previously published data indicated that tobacco induced Ca2+ was not released from the endoplasmic reticulum or mitochondria and instead most likely came from lysosomes.10 While we do not know the efflux pathway present in lysosomes, two pore channels and transient receptor potential cation channels (TRPMLs) are candidate Ca2+ channels that may have been activated by tobacco smoke to trigger lysosomal Ca2+ release.23–25
Prolonged elevation of Ca2+ is associated with stress responses and a transition to an apoptotic state, regardless of whether the cells actually undergo cell death or not.26 Thus, beyond the effects that we see on CFTR, tobacco induced elevations in intracellular Ca2+ may exert additional effects on the epithelia, as described above and could contribute to the inflammation/mucus secretion seen with tobacco exposure by triggering cytokine release and mucin secretion. Furthermore, deranged Ca2+ homeostasis has been linked to autoimmune disease, asthma, and cancer, further suggesting that tobacco-induced Ca2+ release may be an important biomarker of exposure.27–29
In addition to observing an increase in Ca2+ with common tobacco products (Fig. 1), we also observed increases in Fluo-4 fluorescence following 1-NH2-naphthalene, formaldehyde, nicotine, or NNK (Fig. 3). While there are many more chemicals present in smoke than the 13 compounds that we tested, these 13 compounds represent many of those thought to be responsible for respiratory disease.15 Thus, our data suggest that 1-NH2-naphthalene, formaldehyde, nicotine, and NNK are capable of causing the increases in cytosolic Ca2+ that can trigger the signaling cascade that leads to CFTR internalization and airway dehydration. However, the ability of either of these compounds to induce changes in intracellular Ca2+ has not, to the best of our knowledge, previously been described. In addition to the increase in Ca2+ shown in HEK293T, we observed a small but significant increase in Ca2+ when we added nicotine to HBECs (Fig. 2), suggesting that nicotine may contribute to the increase in cytoplasmic Ca2+ seen with tobacco exposure. However, the increase in Fura-2 emission with nicotine was less than what we observed in HBECs following exposure to cigarettes or little cigars, suggesting that other components of tobacco smoke other than nicotine are responsible for the increase seen in Fig. 1. That said, activation of nicotinic-acetylcholine receptors has previously been associated with CFTR dysfunction.30 Thus, there may be dual inhibitory effects of nicotine and other metabolites on Ca2+ signaling and, hence, CFTR function.
In conclusion, we have shown that cigarettes and little cigars induce increases in intracellular Ca2+. Increases in intracellular Ca2+ can also be induced by 1-NH2-naphthalene, formaldehyde, nicotine, and NNK. Our data suggest that tobacco or chemical-induced increases in Ca2+ are a robust assay to use to search for potential tobacco-induced toxicity in vitro.
Acknowledgments
The support of the UNC CF Center Tissue core is gratefully acknowledged. This work was funded by NIH/FDA HL120100. Research reported in this publication was, in part, supported by NIH and the FDA Center for Tobacco Products (CTP). The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH or the Food and Drug Administration.
Author Disclosure Statement
No competing financial interests exist.
References
- 1.Rom O, Avezov K, Aizenbud D, et al. Cigarette smoking and inflammation revisited. Respir Physiol Neurobiol 2013:187;5–10 [DOI] [PubMed] [Google Scholar]
- 2.Ghosh A, Boucher RC, Tarran R. Airway hydration and copd. Cell Mol Life Sci 2015:72;3637–3652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rab A, Rowe SM, Raju SV, et al. Cigarette smoke and CFTR: implications in the pathogenesis of copd. Am J Physiol Lung Cell Mol Physiol 2013:305;L530–L541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Boucher RC. Evidence for airway surface dehydration as the initiating event in cf airway disease. J Intern Med 2007:261;5–16 [DOI] [PubMed] [Google Scholar]
- 5.Chmiel JF, Davis PB. State of the art: why do the lungs of patients with cystic fibrosis become infected and why can't they clear the infection? Respir Res 2003:4;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ramsey BW, Davies J, McElvaney NG, et al. A CFTR potentiator in patients with cystic fibrosis and the g551d mutation. N Engl J Med 2011:365;1663–1672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cantin AM, Hanrahan JW, Bilodeau G, et al. Cystic fibrosis transmembrane conductance regulator function is suppressed in cigarette smokers. Am J Respir Crit Care Med 2006:173;1139–1144 [DOI] [PubMed] [Google Scholar]
- 8.Clunes LA, Davies CM, Coakley RD, et al. Cigarette smoke exposure induces CFTR internalization and insolubility, leading to airway surface liquid dehydration. FASEB J 2012:26;533–545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dransfield MT, Wilhelm AM, Flanagan B, et al. Acquired cystic fibrosis transmembrane conductance regulator dysfunction in the lower airways in copd. Chest 2013:144;498–506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rasmussen JE, Sheridan JT, Polk W, et al. Cigarette smoke-induced Ca2+ release leads to cystic fibrosis transmembrane conductance regulator (CFTR) dysfunction. J Biol Chem 2014:289:7671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Randell SH, Fulcher ML, O'Neal W, et al. Primary epithelial cell models for cystic fibrosis research. Methods Mol Biol 2011:742;285–310 [DOI] [PubMed] [Google Scholar]
- 12.Clunes LA, Bridges A, Alexis N, et al. In vivo versus in vitro airway surface liquid nicotine levels following cigarette smoke exposure. J Anal Toxicol 2008:32;201–207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Watson MJ, Lee SL, Marklew AJ, et al. The cystic fibrosis transmembrane conductance regulator (CFTR) uses its c-terminus to regulate the a2b adenosine receptor. Sci Rep 2016:6;27390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Monterisi S, Casavola V, Zaccolo M. Local modulation of cystic fibrosis conductance regulator: cytoskeleton and compartmentalized camp signalling. Br J Pharmacol 2013:169;1–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fowles J, Dybing E. Application of toxicological risk assessment principles to the chemical constituents of cigarette smoke. Tob Control 2003:12;424–430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Putney JW., Jr. Calcium signaling, second edition. In: Methods in Signal Transduction. Putney JW., Jr. (ed); pp. 1–536. Boca Raton, FL: Taylor & Francis Group; 2006 [Google Scholar]
- 17.Feske S. Orai1 and stim1 deficiency in human and mice: roles of store-operated Ca2+ entry in the immune system and beyond. Immunol Rev 2009:231;189–209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Korngreen A, Priel Z. Purinergic stimulation of rabbit ciliated airway epithelia: control by multiple calcium sources. J Physiol 1996:497;53–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lorenzo IM, Liedtke W, Sanderson MJ, et al. Trpv4 channel participates in receptor-operated calcium entry and ciliary beat frequency regulation in mouse airway epithelial cells. Proc Natl Acad Sci U S A 2008:105;12611–12616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tarran R, Loewen ME, Paradiso AM, et al. Regulation of murine airway surface liquid volume by CFTR and Ca2+-activated cl- conductances. J Gen Physiol 2002:120;407–418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schmid A, Clunes LA, Salathe M, et al. Nucleotide-mediated airway clearance. Subcell Biochem 2011:55;95–138 [DOI] [PubMed] [Google Scholar]
- 22.Prakriya M. Store-operated orai channels: structure and function. Curr Top Membr 2013:71;1–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kiselyov KK, Ahuja M, Rybalchenko V, et al. The intracellular ca(2)(+) channels of membrane traffic. Channels (Austin) 2012:6;344–351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Calcraft PJ, Ruas M, Pan Z, et al. Naadp mobilizes calcium from acidic organelles through two-pore channels. Nature 2009:459;596–600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chandra M, Zhou H, Li Q, et al. A role for the Ca2+ channel TRPML1 in gastric acid secretion, based on analysis of knockout mice. Gastroenterology 2011:140;857–867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.La Rovere RM, Roest G, Bultynck G, et al. Intracellular Ca(2+) signaling and Ca(2+) microdomains in the control of cell survival, apoptosis and autophagy. Cell Calcium 2016:60;74–87 [DOI] [PubMed] [Google Scholar]
- 27.Kaufmann U, Shaw PJ, Kozhaya L, et al. Selective orai1 inhibition ameliorates autoimmune central nervous system inflammation by suppressing effector but not regulatory t cell function. J Immunol 2016:196;573–585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stanisz H, Vultur A, Herlyn M, et al. The role of orai-stim calcium channels in melanocytes and melanoma. J Physiol 2016:594;2825–2835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yarova PL, Stewart AL, Sathish V, et al. Calcium-sensing receptor antagonists abrogate airway hyperresponsiveness and inflammation in allergic asthma. Sci Transl Med 2015:7;284ra260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Maouche K, Medjber K, Zahm JM, et al. Contribution of alpha7 nicotinic receptor to airway epithelium dysfunction under nicotine exposure. Proc Natl Acad Sci U S A 2013:110;4099–4104 [DOI] [PMC free article] [PubMed] [Google Scholar]