

Abstract
Pediatric renal cell carcinoma (RCC) is a rare cancer that can be associated with inherited diseases including tuberous sclerosis complex (TSC) caused by germline mutations in TSC1 or TSC2. Somatic mutations in TSC1 and TSC2 have also been reported in adult RCC, which predict response to mTOR inhibitors. Here, we present the first case of RCC in a child with methylmalonic acidemia (MMA). Clinical whole exome sequencing of blood and tumor samples confirmed the diagnosis of MMA and revealed two somatic inactivating mutations in TSC2, suggesting the potential consideration of an mTOR inhibitor in the event of tumor recurrence.
Keywords: methylmalonic acidemia, renal cell carcinoma, TSC2
1 INTRODUCTION
Pediatric renal cell carcinoma (RCC) is a rare disease (comprising 2–6% of kidney tumors in children) that is known to have significant biological and clinical differences from adult RCC.1,2 Approximately, half of pediatric cases harbor translocations involving the microphthalmia transcription factor family genes, TFE or TFEB.1,3 Other genetic alterations, such as ALK rearrangements, are reported in a minority of tumors.4 Although most RCC appears sporadic, a number of inherited cancer syndromes have been associated with the development of RCC, primarily in adults, including von Hippel-Lindau, Birt-Hogg-Dube, hereditary papillary RCC, and tuberous sclerosis complex (TSC), as well as defects in the Krebs cycle enzyme succinate dehydrogenase.5–7 Additionally, there are case reports of secondary pediatric RCC after treatment of neuroblastoma and other malignancies.8,9 Here, we present the first reported case of RCC in a child with methylmalonic acidemia (MMA), an inborn error of amino acid metabolism not previously linked to kidney cancer.
2 CASE REPORT
A 6-year-old Hispanic female with MMA, renal tubular acidosis, and stage 4 chronic kidney disease was referred to the Texas Children’s Cancer Center following 1 year of hypercalcemia and an elevated and increasing parathyroid hormone related peptide (PTHrP). Decision of whether to proceed with liver and kidney transplantation was pending oncologic evaluation. Clinical examination revealed her to be at baseline mental status with no signs or symptoms concerning for cancer or TSC. No fever, night sweats, easy bruising, enlarged lymph nodes, vomiting, diarrhea, hematuria, or dysuria were reported. Her MMA was well controlled, with no metabolic crises for at least 6 months prior to her office visit. Laboratory results were notable for hypercalcemia (12.6 mg/dl, normal range 8.9 to 10.4 mg/dl), elevated creatinine (2.1 mg/dl, corresponding to eGFR 24 ml/1.73 m2/min by Schwartz equation), and elevated PTHrP (59 pg/ml, normal range 14–27 pg/ml). A subsequent positron emission tomography-computed tomography (PET/CT) scan revealed a solitary focus of intense hyper-metabolic uptake within the anterior aspect of the interpolar left kidney and an underlying 1.5 cm soft tissue lesion (Fig. 1A), which was confirmed by ultrasound examination.
FIGURE 1.

(A) Imaging study. Axial image from 18F-FDG PET/CT demonstrates focal, intense hypermetabolic uptake in the anterior aspect of the left kidney. (B) Pathology study. Gross pathologic examination (left panel) reveals a tumor (arrow) that is well delineated from the remaining renal parenchyma (*). Histologically, neoplastic cells with ample eosinophilic cytoplasm are arranged in nests and papillary structures. The dyscohesion between the cells results in a partially cystic appearance with tumor cells lining a fibrovascular core (arrow head) (right panel)
A partial nephrectomy was performed in order to resect the mass with a goal of preserving renal function and avoiding the need to start chronic dialysis. An intraoperative biopsy showed evidence of inflammation suggesting an infectious etiology and lymph node dissection was therefore not performed. Final pathology of the mass was consistent with RCC (Fig. 1B). Immunohistochemical staining of the biopsy was positive for nuclear TFE3, nuclear PAX8, and CD10, suggesting Xp11.2 translocation associated RCC, but fluorescence in situ hybridization analysis was negative for TFE3 gene rearrangement, resulting in a final diagnosis of RCC NOS. A repeat PET/CT scan performed 3 months after surgery did not show any focal areas of FDG uptake. The patient continues to be in remission one year after surgery.
Clinical whole exome sequencing (WES) of peripheral blood and frozen tumor samples was performed as part of the Baylor College of Medicine and Texas Children’s Hospital IRB approved BASIC3 clinical research study.10,11 Germline WES confirmed two heterozygous pathogenic variants in the methylmalonyl CoA mutase (MUT) gene, c.682C > T (p.R228X) and c.322C > T (p.R108C), which are associated with the development of MMA.12,13 The germline WES report also included a variant of uncertain significance in RHBDF2, a gene that is associated with tylosis with esophageal cancer, an autosomal dominant syndrome associated with a high lifetime risk of esophageal cancer,14 as well as three pathogenic variants in genes associated with rare auto-somal recessive Mendelian disorders unrelated to the patient’s clinical phenotype (Supplementary Table S1). Tumor WES revealed two inactivating somatic mutations in the TSC2 gene: a nonsense mutation in exon 4 (c.246G > A, p.W82X) and a 13 base pair deletion in exon 29 leading to a frameshift and a premature stop codon (c.3370_3382del, p.A1124fs). These somatic TSC2 mutations were present at variant allele fractions (VAFs) of 23% and 19%, respectively, and were confirmed by Sanger sequencing. Tumor WES also revealed novel somatic mutations in six genes that are not known to contribute to cancer pathogenesis (Supplementary Table S2).
3 DISCUSSION
Germline mutations in the TSC1 and TSC2 genes, which encode the proteins hamartin and tuberin of the TSC (Fig. 2), result in constitutive activation of the mammalian target of rapamycin (mTOR) pathway and cause the genetic disorder TSC.5 Patients with TSC are at risk for a number of renal tumors, most frequently angiomyolipoma and less commonly RCC. Somatic alterations in TSC1 and TSC2 have also been reported in adult RCC, including frequent heterozygous loss of TSC115 and mutations in TSC1 and TSC2 in 1–2% of cases.16 Similarly, the presence of biallelic TSC2 somatic mutations has been previously reported in a rat model of RCC (chemically induced non-Eker rat RCCs).17
FIGURE 2.
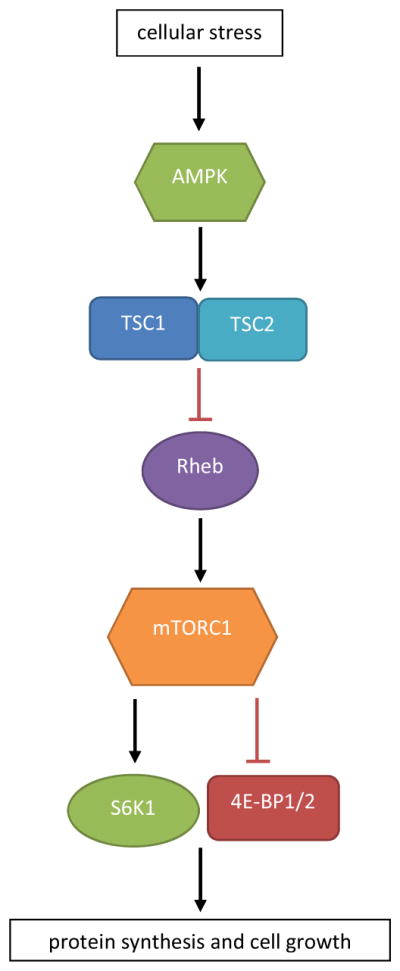
Overview of the tuberous sclerosis complex and mTOR pathway. Cellular stress, including ischemia, hypoxia, and low glucose, is sensed by AMP kinase (AMPK), which stimulates the TSC.23 This complex has an inhibitory effect on the protein Rheb, which in turn activates the mTorc1 complex. MTorc1 stimulates two proteins, S6K1 and 4E-BP1/2, which both, through different mechanisms, result in an increase in protein synthesis
Inhibitors of mTOR signaling (temsirolimus and everolimus) are FDA-approved for the treatment of relapsed or metastatic RCC in adults.18,19 There is emerging evidence that tumor mutations in mTOR pathway genes (e.g., TSC1, TSC2, MTOR) predict response to mTOR inhibition in adult RCC patients, including one series in which the tumors of three of five exceptional responders harbored somatic mutations in TSC1 or MTOR.19 Similarly, a larger study found mutations in TSC1, TSC2, or MTOR to be significantly more frequent in responders than nonresponders.18 The two inactivating mutations in TSC2 discovered in our patient are, to our knowledge, the first reported in a pediatric RCC patient, with VAFs consistent with those reported for TSC1/2 mutations in adult RCC.18 Although this child currently has no evidence of tumor, these data suggest that the use of an mTOR inhibitor could be considered in the event of tumor recurrence. This could be particularly important in our patient given that further resection of kidney tissue could result in end stage renal disease.
Mut-type MMA is an autosomal recessive disorder of propionate metabolism12 in which defects in methyolmalonyl-CoA mutase, a critical Krebs cycle enzyme, lead to accumulation of methylmalonic acid and fluids within the tissues. Clinically, this causes episodes of ketoacidosis and hyperammonemia, characterized by vomiting, lethargy, hypertonia, and abnormal movements. Long-term effects of MMA include chronic kidney disease and renal failure.12,20 Liver transplantation is potentially curative and is often performed in conjunction with kidney transplant. Although there is no clear increase in cancer risk for children with MMA, rare cases of hepatoblastoma have been described.21 Our patient represents the first report of RCC in an MMA patient. One could speculate that our patient’s somatic TSC2 mutations could be the result of chemically induced damage stemming from her underlying metabolic disorder and longstanding kidney disease. Of note, other metabolic disorders, such as Fabry disease that result in chronic kidney disease, have also rarely been associated with RCC.22 Defects in succinate dehydrogenase, another Krebs cycle enzyme, are also associated with a predisposition to development of RCC.7
This case provides an example of the potential clinical utility of paired tumor and germline genomic sequencing for pediatric cancer patients. Sequencing a blood sample from this patient enabled us to confirm the pathogenic variants underlying her diagnosis of MMA, while analysis of her tumor identified potentially actionable somatic variants (in the event of tumor recurrence) in the mTOR signaling pathway. In the absence of paired sequencing, we would not have been able to confirm that the TSC2 mutations identified were somatic rather than germline, potentially necessitating additional clinical follow-up and testing even in this young child without signs or symptoms of TSC.
Supplementary Material
Acknowledgments
Grant sponsor: NHGRI/NCI; Grant number: 1U01HG006485.
The authors are grateful to the patient and her family as well as the clinical and research staff at Texas Children’s Cancer Center for their participation in the study. The BASIC3 study is a Clinical Sequencing Exploratory Research (CSER) program project supported by NHGRI/NCI 1U01HG006485. Dr. Potter is a Houston Junior Woman’s Club Fellow.
Abbreviations
- MMA
methylmalonic acidemia
- MTOR
mammalian target of rapamycin
- PET/CT
positron emission tomography-computed tomography
- PTHrP
parathyroid hormone related peptide
- RCC
renal cell carcinoma
- TSC
tuberous sclerosis complex
- WES
whole exome sequencing
Footnotes
CONFLICT OF INTEREST
Baylor College of Medicine (BCM) and Miraca Holdings, Inc. have formed a joint venture with shared ownership and governance of the Baylor Miraca Genetics Laboratories (BMGL), which perform exome sequencing. SEP is an employee of BCM and serves on the Scientific Advisory board of BMGL. CE is the Chief Quality Officer and Chief Medical Officer of the BMGL.
Additional Supporting Information may be found online in the supporting information tab for this article.
References
- 1.Young EE, Brown CT, Merguerian PA, Akhavan A. Pediatric and adolescent renal cell carcinoma. Urol Oncol. 2016;34(1):42–49. doi: 10.1016/j.urolonc.2015.06.009. [DOI] [PubMed] [Google Scholar]
- 2.Akhavan A, Richards M, Shnorhavorian M, Goldin A, Gow K, Merguerian PA. Renal cell carcinoma in children, adolescents and young adults: a National Cancer Database study. J Urol. 2015;193(4):1336–1341. doi: 10.1016/j.juro.2014.10.108. [DOI] [PubMed] [Google Scholar]
- 3.Geller JI, Argani P, Adeniran A, et al. Translocation renal cell carcinoma: lack of negative impact due to lymph node spread. Cancer. 2008;112(7):1607–1616. doi: 10.1002/cncr.23331. [DOI] [PubMed] [Google Scholar]
- 4.Cajaiba MM, Jennings LJ, Rohan SM, et al. ALK-rearranged renal cell carcinomas in children. Genes Chromosomes Cancer. 2016;55(5):442–451. doi: 10.1002/gcc.22346. [DOI] [PubMed] [Google Scholar]
- 5.Linehan WM. Genetic basis of kidney cancer: role of genomics for the development of disease-based therapeutics. Genome Res. 2012;22(11):2089–2100. doi: 10.1101/gr.131110.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Henske EP. The genetic basis of kidney cancer: why is tuberous sclerosis complex often overlooked? Curr Mol Med. 2004;4(8):825–831. doi: 10.2174/1566524043359610. [DOI] [PubMed] [Google Scholar]
- 7.Tuthill M, Barod R, Pyle L, et al. A report of succinate dehydrogenase B deficiency associated with metastatic papillary renal cell carcinoma: successful treatment with the multi-targeted tyrosine kinase inhibitor sunitinib. BMJ Case Rep. 2009;2009 doi: 10.1136/bcr.08.2008.0732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Medeiros LJ, Palmedo G, Krigman HR, Kovacs G, Beckwith JB. Oncocytoid renal cell carcinoma after neuroblastoma: a report of four cases of a distinct clinicopathologic entity. Am J Surg Pathol. 1999;23(7):772–780. doi: 10.1097/00000478-199907000-00004. [DOI] [PubMed] [Google Scholar]
- 9.Dhall D, Al-Ahmadie HA, Dhall G, Shen-Schwarz S, Tickoo SK. Pediatric renal cell carcinoma with oncocytoid features occurring in a child after chemotherapy for cardiac leiomyosarcoma. Urology. 2007;70(1):178, e113–e175. doi: 10.1016/j.urology.2007.03.055. [DOI] [PubMed] [Google Scholar]
- 10.Parsons DW, Roy A, Yang Y, et al. Diagnostic yield of clinical tumor and germline whole-exome sequencing for children with solid tumors. JAMA Oncol. 2016 doi: 10.1001/jamaoncol.2015.5699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Scollon S, Bergstrom K, Kerstein RA, et al. Obtaining informed consent for clinical tumor and germline exome sequencing of newly diagnosed childhood cancer patients. Genome Med. 2014;6(9):69. doi: 10.1186/s13073-014-0069-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Acquaviva C, Benoist JF, Pereira S, et al. Molecular basis of methylmalonyl-CoA mutase apoenzyme defect in 40 European patients affected by mut(o) and mut- forms of methylmalonic acidemia: identification of 29 novel mutations in the MUT gene. Hum Mut. 2005;25(2):167–176. doi: 10.1002/humu.20128. [DOI] [PubMed] [Google Scholar]
- 13.Worgan LC, Niles K, Tirone JC, et al. Spectrum of mutations in mut methylmalonic acidemia and identification of a common Hispanic mutation and haplotype. Hum Mut. 2006;27(1):31–43. doi: 10.1002/humu.20258. [DOI] [PubMed] [Google Scholar]
- 14.Blaydon DC, Etheridge SL, Risk JM, et al. RHBDF2 mutations are associated with tylosis, a familial esophageal cancer syndrome. Am J Hum Genet. 2012;90(2):340–346. doi: 10.1016/j.ajhg.2011.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cancer Genome Atlas Research N. Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature. 2013;499(7456):43–49. doi: 10.1038/nature12222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. [Accessed April 1, 2015];COSMIC: catalogue of somatic mutations in cancer. http://cancer.sanger.ac.uk/cosmic.
- 17.Urakami S, Tokuzen R, Tsuda H, Igawa M, Hino O. Somatic mutation of the tuberous sclerosis (TSC2) tumor suppressor gene in chemically induced rat renal carcinoma cell. J Urol. 1997;158(1):275–278. doi: 10.1097/00005392-199707000-00085. [DOI] [PubMed] [Google Scholar]
- 18.Kwiatkowski DJ, Choueiri TK, Fay AP, et al. Mutations in TSC1, TSC2, and MTOR are associated with response to rapalogs in patients with metastatic renal cell carcinoma. Clin Cancer Res. 2016;22(10):2445–2452. doi: 10.1158/1078-0432.CCR-15-2631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Voss MH, Hakimi AA, Pham CG, et al. Tumor genetic analyses of patients with metastatic renal cell carcinoma and extended benefit from mTOR inhibitor therapy. Clin Cancer Res. 2014;20(7):1955–1964. doi: 10.1158/1078-0432.CCR-13-2345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zsengeller ZK, Aljinovic N, Teot LA, et al. Methylmalonic acidemia: a megamitochondrial disorder affecting the kidney. Pediatr Nephrol. 2014;29(11):2139–2146. doi: 10.1007/s00467-014-2847-y. [DOI] [PubMed] [Google Scholar]
- 21.Chan R, Mascarenhas L, Boles RG, Kerkar N, Genyk Y, Venkatramani R. Hepatoblastoma in a patient with methylmalonic aciduria. Am J Med Gen. 2015;167A(3):635–638. doi: 10.1002/ajmg.a.36925. [DOI] [PubMed] [Google Scholar]
- 22.Erez A, Shchelochkov OA, Plon SE, Scaglia F, Lee B. Insights into the pathogenesis and treatment of cancer from inborn errors of metabolism. Am J Hum Genet. 2011;88(4):402–421. doi: 10.1016/j.ajhg.2011.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kahn BB, Alquier T, Carling D, Hardie DG. AMP-activated protein kinase: ancient energy gauge provides clues to modern understanding of metabolism. Cell Metab. 2005;1(1):15–25. doi: 10.1016/j.cmet.2004.12.003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.