

ABSTRACT
Baculoviruses encode a variety of auxiliary proteins that are not essential for viral replication but provide them with a selective advantage in nature. P10 is a 10-kDa auxiliary protein produced in the very late phase of gene transcription by Autographa californica multiple nucleopolyhedrovirus (AcMNPV). The P10 protein forms cytoskeleton-like structures in the host cell that associate with microtubules varying from filamentous forms in the cytoplasm to aggregated perinuclear tubules that form a cage-like structure around the nucleus. These P10 structures may have a role in the release of occlusion bodies (OBs) and thus mediate the horizontal transmission of the virus between insect hosts. Here, using mass spectrometric analysis, it is demonstrated that the C terminus of P10 is phosphorylated during virus infection of cells in culture. Analysis of P10 mutants encoded by recombinant baculoviruses in which putative phosphorylation residues were mutated to alanine showed that serine 93 is a site of phosphorylation. Confocal microscopy examination of the serine 93 mutant structures revealed aberrant formation of the perinuclear tubules. Thus, the phosphorylation of serine 93 may induce the aggregation of filaments to form tubules. Together, these data suggest that the phosphorylation of serine 93 affects the structural conformation of P10.
IMPORTANCE The baculovirus P10 protein has been researched intensively since it was first observed in 1969, but its role during viral infection remains unclear. It is conserved in the alphabaculoviruses and expressed at high levels during virus infection. Producing large amounts of a protein is wasteful for the virus unless it is advantageous for the survival of its progeny, and therefore, P10 presents an enigma. As P10 polymerizes to form organized cytoskeletal structures that colocalize with host cell microtubules, the structural relationship of the protein with the host cell may present a key to help understand the function and importance of this protein. This study addresses the importance of the structural changes in P10 during infection and how they may be governed by phosphorylation. The P10 structures affected by phosphorylation are closely associated with the viral progeny and thus may potentially be responsible for its dissemination and survival.
KEYWORDS: AcMNPV, P10, baculovirus, cytoskeleton, host-cell interactions, microtubules, occlusion bodies, protein phosphorylation, Trichoplusia ni
INTRODUCTION
Autographa californica multiple nucleopolyhedrovirus (AcMNPV) is a model alphabaculovirus and belongs to the family Baculoviridae. This family of viruses is characterized by a circular double-stranded DNA genome enclosed in a rod-shaped capsid and further enveloped by a membrane (1). The replication cycle of baculoviruses produces two forms of progeny virus: the budded virus (BV) and the occlusion-derived virus (ODV) (2, 3). The ODV is protected within a protein-rich matrix forming an occlusion body (OB) that is either polyhedral (nucleopolyhedroviruses) or granular (granuloviruses) in shape (4). The transcription of some baculovirus genes, notably polyhedrin (ac8) and p10 (ac137), occurs in a very late phase that initiates approximately 6 h after the onset of late gene transcription (5). While the role of polyhedrin as an OB matrix protein is well established, the P10 protein remains poorly understood. P10 is a 10-kDa protein that forms cytoskeleton-like fibrillar structures in virus-infected cells and, together with polyhedrin, accounts for the majority of the virus-encoded protein present in the host cell during the very late phase (6).
Homologues of p10 were reported in 27 alphabaculovirus and 2 betabaculovirus genomes (7); however, we found a further 25 homologues (19 alphabaculovirus and 6 betabaculovirus genomes) in the NCBI protein database (Table 1).
TABLE 1.
Recently identified P10 homologues
| Baculovirus | GenBank accession no. |
|---|---|
| Alphabaculoviruses | |
| Adoxophyes orana nucleopolyhedrovirus | YP_002300549 |
| Agrotis ipsilon multiple nucleopolyhedrovirus | YP_002268180 |
| Ampelophaga rubiginosa nucleopolyhedrovirus | ACB32175 |
| Apocheima cinerarium nucleopolyhedrovirus | YP_006607870 |
| Bombyx mandarina nucleopolyhedrovirus | ACQ57311 |
| Choristoneura murinana alphabaculovirus | AHD25508 |
| Choristoneura occidentalis alphabaculovirus | AGR56909 |
| Choristoneura rosaceana alphabaculovirus | YP_008378374 |
| Euproctis pseudoconspersa nucleopolyhedrovirus | ACO53500 |
| Hemileuca sp. nucleopolyhedrovirus | YP_008378239 |
| Lambdina fiscellaria nucleopolyhedrovirus | YP_009133322 |
| Lonomia obliqua multiple nucleopolyhedrovirus | AKN80950 |
| Mamestra brassicae multiple nucleopolyhedrovirus | YP_009011213 |
| Orgyia leucostigma nucleopolyhedrovirus | ABY65747 |
| Philosamia cynthia ricini nucleopolyhedrovirus virus | AFY62828 |
| Pseudoplusia includens single nucleopolyhedrovirus IE | AJD80709 |
| Spodoptera littoralis nucleopolyhedrovirus | AGE89872 |
| Sucra jujuba nucleopolyhedrovirus | YP_009186700 |
| Thysanoplusia orichalcea nucleopolyhedrovirus | YP_007250541 |
| Betabaculoviruses | |
| Clostera anachoreta granulovirus | AEB00304 |
| Clostera anastomosis granulovirus | AKS25391 |
| Erinnyis ello granulovirus | YP_009091893 |
| Mocis sp. granulovirus | AKR17400 |
| Pieris rapae granulovirus | ACZ63503 |
| Pseudaletia unipuncta granulovirus | YP_003422344 |
Baculovirus replication can occur in the absence of P10 (8), but studies have indicated that P10 may have a number of roles in the very late stages of the replication cycle (8–10). P10 has been implicated in nuclear lysis, as Spodoptera frugiperda cells infected with recombinant AcMNPV lacking p10 failed to release OBs, even at 2 weeks postinfection (9). In contrast, cells infected with wild-type AcMNPV released large numbers of OBs at 2 days postinfection.
Early transmission electron microscopy (TEM) studies of the P10 protein structure reported a close association between the polyhedron envelope (PE) and P10 (8–10). Virus infection of Trichoplusia ni cells with an AcMNPV p10 deletion mutant resulted in poor attachment of the PE to the surface of polyhedra (9). Studies utilizing scanning electron microscopy (EM) demonstrated that the polyhedra from Orgyia pseudotsugata larvae infected with p10-deficient recombinant Orgyia pseudotsugata multiple nucleopolyhedrovirus (OpMNPV) had pitted surfaces, from the dislodging of virions, whereas wild-type polyhedra had smooth surfaces (10).
Although P10 fibrillar structures have been described in TEM analyses dating from 1969 (11), immunofluorescence microscopy images first appeared in a study by Quant-Russell et al. (12). In OpMNPV-infected Lymantria dispar cells, P10 structures were first detected at 14 h postinfection (hpi) as “fine threads” in the cytoplasm, and by 16 hpi, these structures had “condensed into thicker rod-like” structures that formed an “interconnected network” at later stages (12). Subsequent studies by Patmanidi et al. (13) and Carpentier et al. (14) employed confocal immunofluorescence microscopy to analyze P10 structures in AcMNPV-infected S. frugiperda and T. ni cells, respectively. The P10 filamentous structures were evident at 18 hpi in AcMNPV-infected T. ni cells (TN368 cell line), and these structures had formed a network in the cytoplasm by 30 hpi, followed by distinctive perinuclear aggregates or tubules by 36 hpi (14).
A study by Cheley et al. (15) revealed that S. frugiperda cells infected with recombinant AcMNPV encoding the catalytic subunit of Aplysia protein kinase A (PKA) developed cellular projections. Analysis of these cells using TEM showed that these projections were a result of extended microtubules (MTs). Moreover, 32Pi labeling of paclitaxel (originally named taxol)-stabilized MTs from cells infected with the PKA recombinant baculovirus showed high levels of phosphorylated P10. However, no phosphorylated P10 was observed in MTs prepared from cells infected with the wild-type virus. These data allowed those authors to conclude that the cellular projections were a result of MT elongation induced by phosphorylated P10. Additionally, it was shown that P10 was phosphorylated by Aplysia PKA at the C terminus. Further analysis of virus-infected cells revealed that phosphorylated P10 was associated with MTs, but it could not bundle them. The interaction of P10 with MTs during wild-type virus infection was later confirmed in S. frugiperda and T. ni cells (13, 14). These studies demonstrated that the initial P10 filamentous structures in the cytoplasm coalign with MTs. Furthermore, the formation of P10 filamentous structures was inhibited upon treatment with colchicine, which inhibits microtubule polymerization (14). Together, these data suggest that P10's interaction with MTs is important for its formation and stabilization.
Previous work by Cheley et al. (15) showed that phosphorylation of P10 by Aplysia PKA affected the P10 structure. It is not known whether this phenomenon occurs in wild-type AcMNPV infection and in a natural AcMNPV host. In this study, we provide evidence for P10 phosphorylation in AcMNPV infection of T. ni cells via mass spectrometric analysis and also identify the phosphoacceptor site within P10. The structural consequences of P10 phosphorylation were investigated by alanine mutagenesis and confocal microscopy analyses.
RESULTS
Temporal analysis of P10 structures by confocal microscopy.
Wild-type AcMNPV P10 structures in virus-infected cells were analyzed by laser scanning confocal microscopy to visualize the major changes that occur from their peak expression time at 48 hpi (16) until they are semidisintegrated, typically at 96 hpi (Fig. 1), extending data from previous studies which examined P10 structures until 72 hpi (14). TN368 cells were infected in triplicate culture dishes with AcMNPV at a multiplicity of infection (MOI) of 10 PFU cell−1; fixed at 48, 72, and 96 hpi; and then immunostained to detect P10 and host MTs.
FIG 1.

Temporal changes in P10 structures during AcMNPV infection of TN368 cells. (A) The amino acid sequence of AcMNPV P10 reveals three distinct regions, a coiled-coil domain at the N terminus (blue residues), a proline-rich region in the variable region (green residues), and a positively charged basic region at the C terminus (red residues) (R is arginine, and K is lysine). Amino acid residues of the heptad repeat in the coiled-coil region are denoted abcdefg, in which a and d are hydrophobic, whereas e and g are charged residues. (B) Wild-type virus-infected TN368 cells were analyzed at 48, 72, and 96 hpi by using confocal laser scanning microscopy. Cells were stained with anti-P10 and Alexa Fluor 488 antibodies to visualize P10 (green) and with anti-α-tubulin and Alexa Fluor 568 antibodies to visualize MTs (red). P10 and α-tubulin channels were merged to show coalignment. The position of the OB-filled nucleus is shown in the bright-field images. At 48 and 72 hpi, P10 filaments were coaligned with MTs and spanned the host cytoplasm; bundling of these filaments was evident at 72 hpi. P10 also formed perinuclear tubular structures that were present from 48 hpi and most developed at 96 hpi. The P10 cytoplasmic filaments appeared detached from the perinuclear tubule and partially disintegrated at 96 hpi. Bars, 30 μm.
At 48 hpi (Fig. 1, left), P10 formed filamentous structures in the cytoplasm and around the nucleus of the host cell. Aggregated filaments surrounding the nucleus were also observed. The orientation of P10 filaments in the cytoplasm was similar to that of the host MTs, and thus, both structures were coaligned, most prominently in regions of stable MTs. At 72 hpi (Fig. 1, middle), the P10 cytoplasmic filaments showed further bundling and were still coaligned with MTs. At this time point, thicker tubule-like structures, possibly resulting from the aggregation of finer P10 filaments, were also observed to surround the OB-filled nucleus. At 96 hpi (Fig. 1, right), the P10 perinuclear tubular structures had fully matured, and the cytoplasmic filaments were mostly detached and/or disintegrated. Some detached filaments had a loop-like terminal structure. Additionally, a layer of P10 was also observed to envelope the OBs inside the host nucleus at 72 and 96 hpi.
Phosphorylation of P10 in wild-type AcMNPV infection.
This study utilized matrix-assisted laser desorption ionization–time of flight (MALDI-TOF) mass spectrometry to analyze changes in the mass of the P10 C terminus that Cheley et al. (15) had reported to be the domain phosphorylated by Aplysia PKA. Analysis of the amino acid sequence of P10 showed that the C terminus contained three potential phosphorylation sites, at serines 70, 92, and 93. For MALDI-TOF analysis, T. ni cells were infected with wild-type AcMNPV at an MOI of 10 and harvested at 72 hpi. This time point was selected as it showed both forms of P10 structures (Fig. 1). Lysates were separated by using SDS-PAGE and stained with a Coomassie solution. Digestion of the P10 protein was carried out by using endoproteinase GluC (Staphylococcus aureus protease V8) to cleave peptide bonds C terminally to glutamic acid residues. The MALDI-TOF spectrum was analyzed for peaks corresponding to the C-terminal peptide containing serines 92 and 93.
Figure 2 shows a MALDI-TOF spectrum containing the mass-to-charge ratio (m/z) peaks (labeled) corresponding to the C-terminal peptide 82LDSDARRGKRSSK94, a product of endoproteinase GluC digestion of P10. The MALDI-TOF spectrum shows a peak ([M + H]+ 1,475.81) corresponding to the peptide 82LDSDARRGKRSSK94 (calculated mass, 1,475.80). The phosphorylation of the P10 C terminus was confirmed by the presence of the peak ([M + H]+ 1,555.75) corresponding to 82LDSDARRGKRSSK94 + 1P (PO32−) (calculated mass, 1,555.77). This finding suggests that either serine 92 or serine 93 in the peptide 82LDSDARRGKRSSK94 is a phosphoacceptor site of the P10 protein. Another peptide, 55IQSILTGDIVPDLPDSLKPKLKSQAFE81, was also examined by using MALDI-TOF analysis but did not reveal a form consistent with the phosphorylation of serine 70 (data not shown).
FIG 2.

MALDI-TOF mass spectrometric analysis of the P10 C terminus. The AcMNPV P10 protein was harvested at 72 hpi and digested with endoproteinase GluC to cleave peptide bonds C terminally to glutamic acid residues. The peptide products were analyzed by MALDI-TOF MS (Ultraflex; Bruker Daltonics) in the linear mode. The image shows a portion of the spectrum containing the peptides of interest from the P10 C terminus. The x axis represents m/z values, and the y axis represents the absolute intensity. Peaks with m/z values of 1,475.81 and 1,555.75 corresponded to the non- and monophosphorylated states of the P10 C-terminal peptide 82LDSDARRGKRSSK94. a.u., arbitrary units.
Temporal analysis of P10 mutant structures by confocal microscopy.
Recombinant viruses containing serine-alanine mutations at positions 92 and 93 or at both residues within P10 were generated (Fig. 3). These viruses, AcP10S9293A, AcP10S92A, AcP10S93A, and a control, AcP10wt, were used to infect TN368 cells at an MOI of 10, which were examined subsequently by using confocal microscopy. The images were obtained from a number of different cells at each time point and are representative; the heterogeneous nature of the TN368 cell line was taken into account during the analysis.
FIG 3.

Construction of recombinant viruses. (A) Wild-type or mutant p10 flanked by XbaI and XmaI restriction sites was inserted downstream of the polyhedrin promoter in the transfer vector pBacPAK8. Recombinant baculoviruses were made by allowing homologous recombination of the transfer vector and flashBACULTRA. Four viruses were constructed; in the single mutants AcP10S92A and AcP10S93A, serines 92 and 93 were mutated to alanine, respectively. In the double mutant AcP10S9293A, both serines 92 and 93 were mutated to alanine. AcP10wt contained wild-type p10. (B) pAcUW2B was used to construct the His-tagged wild-type and mutant p10-carrying viruses. This vector included a complete polh gene. The P10 fragment was inserted downstream of the P10 promoter in pAcUW2B by using the PstI and SpeI restriction sites. Six histidine residues followed by the TEV cleavage site residues were added at the N terminus. Two recombinant viruses were constructed by cotransfecting pAcUW2B-modified vectors with flashBACULTRA: AcUW2B-His-P10wt, containing the wild-type p10 gene, and AcUW2B-His-P10S93A. The displayed genes are not to scale.
Figure 4 shows P10 mutant and control structures at 72 hpi (Fig. 4, top) and 96 hpi (Fig. 4, bottom) when under the control of the polh promoter. The P10 structures of AcP10wt exhibited the same profile as that of the wild-type AcMNPV structures at 72 and 96 hpi (compare Fig. 4, left, and Fig. 1); both cytoplasmic filaments and perinuclear tubules were present. Similar to wild-type AcMNPV infection (Fig. 1), a few cytoplasmic filaments were detached at 72 hpi; however, by 96 hpi, all filaments appeared detached, and a distinctive P10 tubular structure surrounding the nucleus in a ring-like form was visible at both 72 and 96 hpi. This structure remained intact at 96 hpi and disjointed from the cytoplasmic filaments.
FIG 4.

Analysis of wild-type and mutant P10 structures. TN368 cells were infected with AcP10wt, AcP10S92A, AcP10S93A, or AcP10S9293A and then fixed at 72 and 96 hpi. P10 structures were visualized by using anti-P10- and Alexa Fluor 488 antibodies; microtubules (red) were visualized by using anti-α-tubulin and Alexa Fluor 568 antibodies. P10 and α-tubulin channels were merged to show coalignment. At 72 hpi, cells infected with AcP10wt or AcP10S92A showed both P10 perinuclear tubules (NT) and cytoplasmic filaments (CF). By 96 hpi, the perinuclear tubules had matured, and most cytoplasmic filaments were detached from the central tubule. Cells infected with AcP10S93A or AcP10S9293A lacked perinuclear tubules and displayed rigid and angular cytoplasmic filaments that were not fully detached from the nucleus. Images are representative. Bars, 30 μm.
The P10 mutant structures of AcP10S92A were similar to those of AcP10wt and developed at the same time (Fig. 4). However, the P10 mutant structures of AcP10S93A and AcP10S9293A revealed conformational differences compared to the AcP10wt control (Fig. 4). The ring-like form of the P10 perinuclear tubule is absent and is replaced by thin filaments surrounding the nucleus at 96 hpi in these mutants. The mutant cytoplasmic filaments were disorganized and displayed a thinner and rigid conformation at 72 and 96 hpi, indicating a structural aberration. In addition, these filaments displayed a delay in detachment from the cell nucleus compared to those from AcP10wt (Fig. 4) or wild-type AcMNPV (Fig. 1) infection. These results suggest that a single mutation of P10 serine residue 93 affects the organization of P10 filaments and consequently disrupts their detachment from the nucleus. It does not, however, completely abolish detachment, as some filaments were detached from the nucleus at both 72 and 96 hpi. Detachment was further analyzed in relation to microtubules. The colocalization of P10 mutant structures with microtubules was compared to that of the wild type (Fig. 4).
Mass spectrometric analysis of the C termini of P10 mutants.
MALDI-TOF mass spectrometry was employed to identify the site of phosphorylation in AcMNPV P10. This was done by analyzing the phosphorylation-associated mass shifts in mutant and wild-type P10 proteins. The consensus sequence for PKA includes the motif XRXS(T)X (where R is arginine, K is lysine, X is any amino acid, S is serine, and T is threonine) (17), and serine 93 of P10 fulfils this requirement (the last 5 residues of P10 are KRSS*K, where * is serine 93). Therefore, the serine 93 mutant of P10 was selected for mass spectrometric analysis. Furthermore, the P10 C terminus has been shown to be efficiently phosphorylated by PKA (15).
MALDI-TOF analysis was performed following infection of T. ni cells with the recombinant viruses AcP10S93A and AcP10wt at an MOI of 10. The MALDI-TOF spectrum was analyzed for peaks corresponding to the C-terminal peptides containing the serine 93 (unmodified) or alanine 93 (mutant) residues from AcP10wt and AcP10S93A, respectively.
Figure 5 shows the MALDI-TOF spectra containing the m/z peaks corresponding to the C-terminal peptide 82LDSDARRGKRS(S/A)K94, a product of endoproteinase GluC digestion of P10. In these spectra, the labeled m/z peaks correspond to the P10 C-terminal peptide 82LDSDARRGKRSSK94 ([M + H]+ 1,475.81; calculated mass, 1,475.80) or 82LDSDARRGKRSAK94 ([M + H]+ 1,459.85; calculated mass, 1,459.81). Phosphorylation of the P10 C-terminal peptide from AcP10wt is evident by the presence of the peak ([M + H]+ 1,555.75) corresponding to the peptide 82LDSDARRGKRSSK94 + 1P (PO32−) (calculated mass, 1,555.77). However, in the spectra of the P10 mutant peptide from AcP10S93A, no peak is detected at a mass of 1,539.81, the mass of the phosphorylated peptide 82LDSDARRGKRSAK94 + 1P (PO32−).
FIG 5.

MALDI-TOF mass spectrometric analysis of the P10 peptides from AcP10wt and AcP10S93A. In-gel digestion of P10 proteins (separated by SDS-PAGE) from AcP10wt and AcP10S93A was carried out with endoproteinase GluC; this cleaved peptide bonds C terminally to glutamic acid residues in ammonium carbonate buffer. The peptide fragments were analyzed by MALDI-TOF MS (Ultraflex; Bruker Daltonics) in the linear mode. The image shows a portion of the spectrum containing the P10 C-terminal peptides of interest. The x axis represents m/z values, and the y axis represents absolute intensity as measured by the detector. The top panel shows the MALDI-TOF spectrum of the P10 C-terminal peptide from AcP10wt, in which wild-type P10 expression was driven by the polyhedrin gene promoter. The MALDI-TOF spectrum shows peaks with m/z values of 1,475.81 and 1,555.75 that corresponded to the non- and monophosphorylated states of the P10 peptide 82LDSDARRGKRSSK94. The bottom panel shows the MALDI-TOF spectrum of the P10 peptide from AcP10S93A. In this recombinant virus, the P10 serine 93 residue was mutated to alanine, and mutant expression was driven by the polh promoter. The MALDI-TOF spectrum shows a signal at [M + H]+ 1,459.85, corresponding to the peptide 82LDSDARRGKRSAK94; however, no phosphorylated form of this peptide was observed (no signal at m/z 1,539).
These data confirm, by exclusion, that the P10 serine 93 residue, and not serine 92, is the substrate residue for a kinase. The presence of the phosphorylated P10 peptide from AcP10wt provides further evidence of phosphorylation in native P10.
Circular dichroism.
The circular dichroism (CD) profile of a protein varies with the different secondary structure elements or folds. Circular dichroism was therefore used to analyze the secondary structure of wild-type P10 and its serine 93 mutant in order to determine whether phosphorylation may affect secondary structure characteristics.
Purified protein samples were prepared for CD spectroscopy using the recombinant viruses AcUW2B-His-P10wt and AcUW2B-His-P10S93A, encoding His-tagged wild-type and mutant (serine 93) P10 proteins, respectively (Fig. 3). The CD profile of wild-type P10 showed minima at 221 nm and at 208 nm; the serine 93 mutant showed minima at 228.5 nm and at 205 to 215 nm (Fig. 6). To determine the secondary structure of the serine 93 mutant and wild-type P10 from the CD spectra, deconvolution analysis of the spectra was performed by using the LINCOMB (linear combination of the CD spectrum) method. This method uses an algorithm based on a least-squares fit and a set of reference spectra (18). For the P10 CD spectrum analysis, the set comprised typical CD curves of α-helix, β-pleated sheet (antiparallel), β-turns, disordered protein, and aromatic/disulfide (or nonpeptide).
FIG 6.
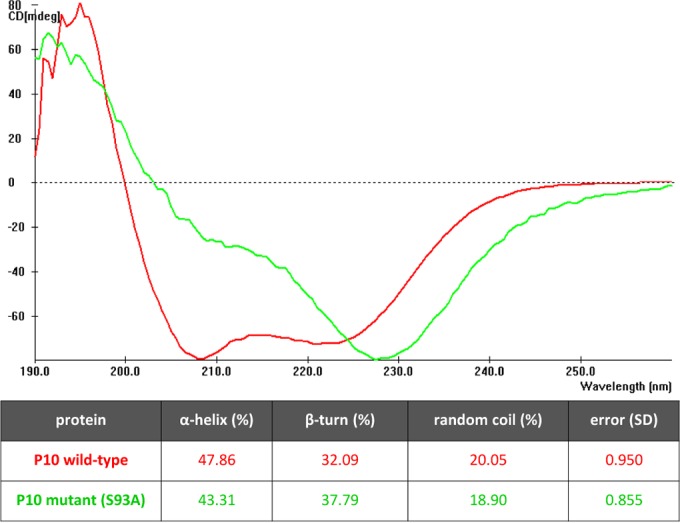
Secondary structure of wild-type P10 and its serine 93 mutant. Spectra were averaged from 4 to 16 scans in the wavelength range of 260 to 190 nm. CD was measured in ellipticity units of millidegrees (mdeg). The CD spectra of the serine 93 mutant and wild-type P10 revealed differences in the minima. The table shows the percentages of different secondary structures in the two proteins following LINCOMB analysis of spectra.
Deconvolution of the P10 wild-type spectrum showed that the protein comprised an α-helix (47.86%) followed by β-turns (32.09%), while the remaining structure was random coils (20.05%). In comparison, the P10 serine 93 mutant revealed a slightly reduced content of α-helix (43.31%) and a higher content of β-turns (37.79%). The percent content of random coils in the two samples did not vary.
DISCUSSION
The formation of P10 cytoskeleton-like structures during AcMNPV infection has been established in a number of previous studies (12–14). In this study, examination of P10 structures at 48, 72, and 96 hpi using confocal microscopy revealed that they undergo a transition during this period of the AcMNPV infection cycle. The P10 tubular structure, which surrounded the host nucleus, was present from 48 hpi and developed into a discrete ring-like form disjointed from the P10 cytoplasmic filaments by 96 hpi. In contrast, the P10 cytoplasmic filaments became detached and disintegrated by 96 hpi. Previously (14), these structures were described at 48 hpi. To better understand the significance of P10 structures during infection, we analyzed how these structures transform until a very late time point, such as 96 hpi, when cells undergo lysis. These results are also consistent with findings from studies using pulse-labeling experiments (16) that reported the high-level synthesis of P10 from 33 to 99 hpi. The fact that P10 continues to form structures in the host cell following the viral replication cycle is one of the key findings of this study. This phenomenon is indeed indicative of the requirement of this protein at this postreplication stage.
Phosphorylation of P10 has been postulated in a number of previous studies (12, 14, 15); however, there was no evidence to suggest that the phenomenon occurred in wild-type virus infection. Here we report the phosphorylation of P10 in wild-type AcMNPV infection at 72 hpi using mass spectrometric analysis of P10 (Fig. 2). Due to the different anticipated ionization efficiencies between the phosphorylated and nonphosphorylated peptides observed, the amount of phosphorylated peptide observed in this assay will not be truly reflective of the total amount of phosphorylated P10 present during infection (19). MALDI-TOF analysis of P10 mutants found that the C-terminal serine 93 residue is the site of phosphorylation (Fig. 5). This phosphorylation site is conserved in P10 sequences from six members of the alphabaculoviruses, and there are potential phosphorylation sites in the C-terminal basic domain of P10 whose distribution is highly conserved in alphabaculovirus P10 homologues (7). Baculoviruses are known to encode several kinases, including the serine/threonine kinases PK-1 and PK-2, which are expressed very late and late, respectively (20, 21). Thus, it is likely that P10 is phosphorylated by PK-1 or -2 encoded by the virus.
Phosphorylated P10 was also found in cells infected with the recombinant viruses and AcP10wt. This suggested that the dynamics of P10 phosphorylation in the recombinant viruses were comparable to those of the wild type and thus unaffected by the use of the polyhedrin promoter. Phosphorylation was inhibited in the mutant P10 protein from cells infected with the recombinant virus AcP10S93A, in which the phosphorylation site, serine 93, was replaced with alanine.
P10 structures of AcP10S93A and AcP10S9293A revealed significant differences compared to the wild-type control virus AcP10wt (Fig. 4) and the wild-type virus (Fig. 1). Mutation of the phosphorylation site at serine 93 resulted in the aberrant formation of P10 perinuclear tubules; it also affected the conformation of the cytoplasmic filaments. Therefore, it is very likely that phosphorylation of the P10 C terminus facilitates the aggregation of P10 in order to form the very distinctive tubular structures in the final stages of infection. The timing of P10 phosphorylation (72 hpi) observed in the mass spectrometry data also correlates with the formation of these structures. The serine 93 mutants also showed a delay in the detachment of filaments from the nucleus, suggesting that the aggregated filaments facilitated this process. Indeed, phosphorylation modulates the aggregation propensity of several proteins and peptides (22–24); these include tau, synuclein, and peptide model systems. The aggregation of self-assembling proteins is particularly regulated by phosphorylation (25), and this may also be true for P10, which also self-assembles (26).
The changes observed in the P10 mutant structures are unlikely to be caused by the replacement of serine with alanine at position 93, as both alanine and serine have neutral pHs. Additionally, the observation that the P10 structures of the serine 92 mutant are similar to the wild-type P10 structures further confirms that a single replacement of serine with alanine, in close proximity of the penultimate residue, does not have any observable influence on the protein conformation.
This study analyzed the secondary structures of wild-type and serine 93 mutant P10 proteins using CD spectroscopy. No previous work has been done to reveal the secondary structure of P10. A reduction in the α-helical content of P10 was observed upon mutation of serine 93. Although not analyzed, the serine 92 mutant most likely retained the wild-type conformation, as no differences in the P10 structures were observed by confocal microscopy. Moreover, results from the secondary structure prediction software PSIPRED (27) showed no differences in the secondary structure of P10 upon the replacement of serine 92 or serine 93 with alanine. Therefore, it is unlikely that the replacement of serine 92 or 93 with alanine produced a significant change in the secondary structure of P10 unless there is a posttranslational modification of the protein. Thus, the change in secondary structure could be a result of the addition of a phosphate moiety to a protein that is known to affect the electrostatic forces in a protein determining its folding. The type and extent of change in folding vary with the location of phosphorylation and are not entirely predictable. It is likely that the phosphorylation of the P10 penultimate residue plays a role in the stabilization of the entire protein.
Taken together, the results of this study support the hypothesis that the phosphorylation of P10 at the C terminus regulates its structural organization. This phenomenon could be involved in multiple roles of P10 during virus infection. The P10 perinuclear tubules surround the polyhedra inside the cell nucleus, which indicates that they may have a protective role in the terminal stages of infection. These tubules may also stabilize the host nucleus to allow the complete maturation of polyhedra to take place. Without the tubules, the polyhedra may be susceptible to digestion by viral cathepsin that is activated upon cell death (28). This could also explain the results from an early study by Gross et al. (10) in which the peripheries of the polyhedra were affected upon the deletion of the p10 gene. The phosphorylation-driven aggregation of P10 cytoplasmic filaments may also be involved in the timely destruction of the host cell to release viral enzymes. This is consistent with data from a study in which the deletion of P10 delayed the release of chitinase by 24 h (28).
Microtubule-associated proteins (MAPs) such as tau have a basic C terminus that interacts with the negatively charged residues in tubulin (29). The phosphorylation of the MAP tau allows it to dissociate from the MTs because of the negative charge introduced by phosphorylation (30). Furthermore, the aggregation of tau is also facilitated by phosphorylation (31). The C terminus of P10, which is also the site of phosphorylation, is basic in nature since it is rich in lysine and arginine residues. Similarly to the tau protein, the interaction of P10 with MTs may be facilitated through these basic residues, and the phosphorylation of P10 may influence its affinity for MTs and self-aggregation properties. Although no differences were observed in the coalignment of mutant filaments and MTs in comparison to the wild type, the mutants showed a rigid conformation and delayed detachment from the nucleus (results not shown). These observations could be the result of the altered affinity of P10 structures for MTs upon the inhibition of phosphorylation. Testing this model could explain one of the mechanisms through which baculoviruses are able to take control of the host cytoskeleton.
MATERIALS AND METHODS
Cells and viruses.
This study utilized cell lines derived from the ovary of T. ni (High Five; TN368). TN368 (32) cells were grown in TC-100 insect medium (Gibco) with 10% (vol/vol) fetal bovine serum (Sigma-Aldrich) as an adherent culture. High Five (33) cells were grown in Ex-Cell 405 (Sigma-Aldrich) as a suspension culture. Sf9 (34) and Sf21 (35) cells, derived from the ovary of S. frugiperda, were used for the process of recombinant baculovirus generation and plaque assays. Sf9 cells were grown in InsectExpress Sf9-S2 (PAA), and Sf21 cells were grown in TC-100 insect medium (Gibco) with 10% (vol/vol) fetal bovine serum (Sigma-Aldrich). Both Sf21 and Sf9 cells were maintained as suspension cultures. The wild-type virus used in this work was AcMNPV C6 (36).
Site-directed mutagenesis of p10 and generation of recombinant viruses.
Site-directed mutagenesis was performed by using the QuikChange kit (Stratagene) according to methods described previously by Vandeyar et al. (37). In essence, DNA was synthesized by PCR using a high-fidelity Pfu DNA polymerase (Agilent Technologies) and custom primers. This DNA was treated with the DpnI endonuclease to digest the parental DNA template. The DpnI-treated DNA was used to transform competent bacterial cells. All mutations were confirmed by DNA sequencing.
Plasmid pCRII-TOPO-P10wt was constructed by subcloning of the p10 gene (amplified from the AcMNPV strain C6) into pCRII-TOPO (Invitrogen) using the XbaI and EcoRI restriction sites. The p10 codons for serines 92 and 93 were then each mutated to specify alanine. In the first step, serine 92 was mutated to alanine by PCR using primers P10_S92AF and P10_S92AR (Table 2). In a second PCR, serine 93 was mutated to alanine by using primers P10_S93AF and P10_S93AR. The resulting plasmid was named pCRII-TOPO-P10S9293A.
TABLE 2.
PCR primers
| Primer | Sequence |
|---|---|
| P10_S92AF | 5′-CGTGGTAAACGCGCGTCCAAGTAAGAATTC-3′ |
| P10_S92AR | 5′-TTCTTACTTGGACGCGCGTTTACCACGAC-3′ |
| P10_S93AF | 5′-GGTAAACGCGCGGCGAAGTAAGAATTCG-3′ |
| P10_S93AR | 5′-GAATTCTTACTTCGCCGCGCGTTTACCAC-3′ |
| P10_S9293A_pBP8F | 5′-CATGTAGCTCTAGAATGTCAAAGCCTAACG-3′ |
| P10_S9293A_pBP8R | 5′-CATGTAGCCCCGGGTTACTTCGCCGCGC-3′ |
| P10_S92A_pBP8F | 5′-CGTCGTGGTAAACGCGCTTCCAAGTAACCCGGG-3′ |
| P10_S92A_pBP8R | 5′-CCCGGGTTACTTGGAAGCGCGTTTACCACGACG-3′ |
| P10_S93A_pBP8F | 5′-CTCGTCGTGGTAAACGCAGTGCCAAGTAACCC-3′ |
| P10_S93A_pBP8R | 5′-GGGTTACTTGGCACTGCGTTTACCACGACGAG-3′ |
| P10_wtF | 5′-CATGTAGCTCTAGAATGTCAAAGCCTAACG-3′ |
| P10_wtR | 5′-CATGTAGCCCCGGGTTACTTGGAACTGCG-3′ |
| P10_wt_pW2BF | 5′-GTAGCCTGCAGTTACTTGGAACTGCGTTTACC-3′ |
| P10_wt_pW2BR | 5′-GTAGCAAGCCTATGTCAAAGCCTAACGTTTTGACG-3′ |
| P10_S93A_pW2BF | 5′-GCTCTGCAGTTACTTGGCACTGCGTTTACCACGAC-3′ |
| P10_S93A_pW2BR | 5′-GTCGTGGTAAACGCAGTGCCAAGTAACTGCAGAGC-3′ |
| HISP10_wt_pW2BF | 5′-GTAGCCTGCAGTTACTTGGAACTGCGTTTACCACGACGAGCGTC-3′ |
| HISP10_wt_pW2BR | 5′-GCACTAGTATGCATCACCATCACCATCACGAAAACCTGTATTTTCAGGGCTCAAAGCCTAACG-3′ |
| HISP10_S93A_pW2BF | 5′-GTAGCCTGCAGTTACTTGGAACTGCGTTTACCACGACGAGCGTC-3′ |
| HISP10_S93A_pW2BR | 5′-GTAGCCTGCAGTTACTTGGCACTGCGTTTACCACGACGAGC-3′ |
The modified p10 fragment was amplified from plasmid pCRII-TOPO-P10S9293A by PCR using primers P10_S9293A_pBP8F and P10_S9293A_pBP8R. The product was digested with XbaI and XmaI and then subcloned into pBacPAK8, downstream of the polyhedrin promoter, to generate pBacPAK8-P10S9293A.
Plasmid pBacPAK8-P10S9293A was used to generate single P10 mutants in which either serine 92 or 93 codons were mutated to specify alanine instead. Single mutations were created through site-specific mutagenesis using primers P10_S92A_pBP8F and P10_S92A_pBP8R to derive pBacPAK8-P10S92A and primers P10_S93A_pBP8F and P10_S93A_pBP8R to derive pBacPAK8-P10S93A.
To generate a control, the p10 gene was amplified from pCRIITOPO-P10wt by using primers P10_wtF and P10_wtR. The PCR product was digested with XbaI and XmaI and then subcloned into pBacPAK8 downstream of the polyhedrin gene promoter to derive pBacPAK8-P10wt.
Plasmids pBacPAK8-P10S9293, pBacPAK8-P10S92A, pBacPAK8-P10S93A, and pBacPAK8-P10wt were used for cotransfection with flashBACULTRA genomic DNA (Oxford Expression Technologies Ltd.) to generate recombinant viruses. Deletion of the p10 gene in the flashBACULTRA genomic DNA meant that parental p10 could not interfere with the analysis. In addition, the flashBACULTRA genomic DNA also lacked the polh, chiA, v-cath, p26, and p74 genes. The resulting viruses were designated AcP10S9293A, AcP10S92A, AcP10S93A, and AcP10wt, respectively.
To construct polyhedrin-positive viruses, p10 was amplified from plasmid pBacPAK8-P10wt by PCR using primers P10_wt_pW2BF and P10_wt_pW2BR. The DNA product was digested with PstI (this restriction site was introduced into plasmid pAcUW2B through site-specific mutagenesis) and inserted into pAcUW2B, downstream of the p10 promoter, to derive pAcUW2B-P10wt. This plasmid was then used to generate the p10 mutants. The P10 serine 93 residue was mutated to alanine through site-specific mutagenesis using primers P10_S93A_pW2BF and P10_S93A_pW2BR to derive pAcUW2B-P10S93A.
To derive a pAcUW2B transfer vector encoding polyhistidine-tagged P10 (wild type and serine 93 mutant), plasmids pAcUW2B-P10wt and pAcUW2B-P10S93A were used. The wild-type p10 gene was PCR amplified from plasmid pAcUW2B-P10wt by using primers HISP10_wt_pW2BF and HISP10_wt_pW2BR, introducing a 6× histidine tag and a tobacco etch virus (TEV) cleavage site at the N terminus. The PCR fragment and pAcUW2B were digested with PstI and SpeI and ligated together to produce pAcUW2B-His-P10wt. The serine 93 mutant p10 gene was PCR amplified from plasmid pAcUW2B-P10S93A by using primers HISP10_S93A_pW2BF and HISP10_S93A_pW2BR, introducing a 6× histidine tag and a TEV cleavage site at the N terminus. The PCR fragment and pAcUW2B were digested with PstI and SpeI; the fragment was then ligated into pAcUW2B, downstream of the p10 promoter, to derive pAcUW2B-His-P10S93A. Recombinant viruses (AcUW2B-His-P10wt and AcUW2B-His-P10S93A) were generated as described above.
Generation of recombinant viruses.
Cell cultures dishes (35 mm) were seeded with Sf9 cells at a density of 0.5 × 106 cells ml−1. Cotransfection mixtures were prepared by using 1 ml of the appropriate cell culture medium, 5 μl of Lipofectin reagent (Invitrogen), 100 ng of flashBACULTRA (Oxford Expression Technologies Ltd.), and 500 ng of the transfer vector according to methods described previously by King and Possee (38). The medium containing the recombinant virus was collected on the fifth day. Viruses were amplified in Sf9 cell cultures, and titers were determined as PFU per milliliter by a plaque assay using Sf21 cells.
Immunofluorescence.
For confocal immunofluorescence microscopy, TN368 cells were employed. These cells were seeded onto glass coverslips (22-mm diameter) in 35-mm cell culture dishes at a density of 1 × 105 cells ml−1 and were left to settle overnight at 28°C. To infect cells, the medium was removed from the dishes, and 100 μl of the appropriate dilution of the virus inoculum was added dropwise to the cells. For mock infection, 100 μl of cell culture medium was used. Cells were infected with each type of virus in triplicate. Cells were incubated at room temperature for 1 h to allow virus adsorption. The inoculum was then removed, and 2 ml of fresh media were added to the cells (this time point was defined as 0 hpi). Cells were incubated at 28°C until the desired time point, medium was removed from the dishes, and cells were washed twice with 1 ml of phosphate-buffered saline (PBS). For chemical fixation, cells were treated with 1 ml of 4% (vol/vol) paraformaldehyde for 1 h, washed once with 1 ml of PBS, and stored at 4°C until required for immunostaining.
Tubulin was stained by using a mouse monoclonal anti-α-tubulin antibody (Sigma-Aldrich) and anti-mouse Alexa Fluor 568 (Invitrogen). P10 was stained by using a guinea pig polyclonal antibody (13) and anti-guinea pig Alexa Fluor 488 (Invitrogen). For immunofluorescence staining, fixed cells were treated with permeabilization buffer (1% [wt/vol] bovine serum albumin [BSA] and 0.1% [vol/vol] Triton X-100 in PBS) for 10 min. Cells were washed with 1 ml of PBS followed by 1 ml of 1% (wt/vol) BSA in PBS (PBS-BSA). Cells were probed with primary antibody, diluted in PBS-BSA, for 50 min. Unbound antibody was removed by washing cells with PBS-BSA three times. Cells were probed with secondary antibody diluted in PBS-BSA for 50 min and then washed three times with PBS. Following immunofluorescence staining, coverslips were mounted onto glass slides by using Vectashield mounting medium (Vector Laboratories). Coverslips were sealed by using clear nail varnish, and slides were stored at 4°C, protected from light.
Confocal microscopy.
Confocal laser scanning microscopy of immunostained cells was performed by using the Zeiss LSM 510 Meta system with an Axio Imager-Z1 upright microscope. Images were acquired by using an EC Plan-Neofluar 40× (1.3-numerical-aperture) or a Plan-Apochromat 63× (1.4-numerical-aperture) oil immersion objective. A multitrack setup was employed to prevent signal crossover. Fluorescence from Alexa Fluor 488 and Alexa Fluor 568 was recorded through the laser lines at 488 and 543 nm, respectively. Projection three-dimensional (3D) images were generated by using the Zeiss LSM Image Browser (v4.2). Images shown were selected to be representative of a large number of individual cells examined (n > 100).
Mass spectrometry.
Coomassie-stained protein gel bands of P10 were excised, cut into small pieces (1 to 2 mm3), and transferred to a 1.5-ml tube. Gel pieces were shaken vigorously for 18 h in destaining solution (1 ml containing 50% [vol/vol] methanol and 5% [vol/vol] acetic acid). Further destaining was carried out for 2 to 3 h with fresh destaining solution. The destaining solution was removed, and gel pieces were dehydrated in 200 μl of acetonitrile for 5 min. Acetonitrile was removed, and the dehydration step was repeated. Reduction was carried out with 30 μl of 10 mM dithiothreitol buffer for 30 min. The reduction buffer was removed and replaced with alkylation buffer; alkylation was carried out with 30 μl of 50 mM iodoacetamide buffer for 30 min. The alkylation buffer was removed, and gel pieces were dehydrated in 200 μl acetonitrile for 5 min. Acetonitrile was removed, and gel pieces were rehydrated in 200 μl of a 100 mM ammonium bicarbonate solution for 10 min. The dehydration step was repeated with 200 μl acetonitrile for 5 min, and the solution was removed.
Digestion was carried out by using Staphylococcus aureus protease V8 endoproteinase GluC (NEB), which was prepared by adding 1 ml of ice-cold 50 mM ammonium bicarbonate to 20 μg of GluC (final concentration, 20 ng/μl). Gel pieces were rehydrated with 30 μl of the GluC solution on ice for 10 min and then briefly centrifuged to allow the removal of the excess enzyme solution. After the addition of 5 μl of 50 mM ammonium bicarbonate buffer solution to the gel pieces, digestion was performed at 37°C for 18 h.
Peptides were extracted from the gel pieces during each of three successive 10-min incubations with (i) 50 μl of 50 mM ammonium bicarbonate buffer, (ii) 50 μl of extraction buffer 1 (50% [vol/vol] acetonitrile, 5% [vol/vol] formic acid), and (iii) 50 μl of extraction buffer 2 (85% [vol/vol] acetonitrile, 5% [vol/vol] formic acid). The peptide solution was dried completely in a vacuum centrifuge and resuspended in 20 μl of a buffer solution (2% [vol/vol] acetonitrile, 0.1% [vol/vol] formic acid).
For MALDI-TOF mass spectrometry analysis, 1 μl of the peptide solution was mixed with 1 μl of matrix (α-cyano-4-hydroxycinnamic acid) and spotted onto a MALDI target. Samples were measured by using a MALDI-TOF instrument (Ultraflex; Bruker Daltonics) in the linear mode. The MALDI-TOF spectra were analyzed by using flexAnalysis software (Bruker Daltonics) as described previously (39).
Protein purification.
A shaking suspension culture of T. ni High Five cells was set up at a density of 0.5 × 106 cells ml−1 in a total volume of 500 ml. Cells were infected with the virus expressing the His-tagged protein at an MOI of 5 and incubated at 28°C. At the required time point, cells were harvested by centrifugation at 10,000 × g for 15 min. The supernatant was removed, and cells were washed with 50 ml of ice-cold PBS. Cells were lysed with the CytoBuster protein extraction reagent (Novagen) and spun at 14,000 × g for 30 min to remove all insoluble material. After centrifugation, the supernatant was filtered through a 0.45-μm membrane to prevent clogging of purification resin in subsequent steps. His-tagged protein purification was carried out by using the His-Bind purification kit (Novagen) according to the manufacturer's instructions. In brief, iminodiacetic acid (IDA) agarose resin was used in a spin column to purify His-tagged proteins. IDA agarose resin was activated with charge buffer (50 mM NiSO4) and equilibrated with binding buffer (0.5 M NaCl, 20 mM Tris-HCl, 5 mM imidazole [pH 7.9]). Prepared soluble lysates were passed through the spin column. The resin was treated with binding buffer and then wash buffer (0.5 M NaCl, 60 mM imidazole, 20 mM Tris-HCl [pH 7.9]) to remove any nonspecific binding of proteins with the resin. Elution was performed with buffer containing 400 mM imidazole, 0.5 M NaCl, and 20 mM Tris-HCl at pH 7.9. The purified protein was assessed for purity by Coomassie staining.
Circular dichroism.
CD spectra were recorded on a Jasco J-720 spectropolarimeter (Jasco GmbH) using a 0.05-cm-path-length quartz cell. Spectra of 100 μg ml−1 of the protein solution in 10 mM phosphate buffer were averaged from 4 to 16 scans (260 to 190 nm) and corrected by using a buffer blank. The CD spectra were analyzed with CD analyzer system (V2.02) software using the LINCOMB method (18).
ACKNOWLEDGMENTS
We are grateful to John Runions (Oxford Brookes University) and David Staunton (University of Oxford) for their technical expertise with confocal microscopy and circular dichroism, respectively.
The latter work was performed at the University of Oxford with a research grant from the Santander Group.
REFERENCES
- 1.Federici BA. 1986. Ultrastructure of baculoviruses, p 61–88. In Granados, RR, Federici, BA (ed), The biology of baculoviruses. CRC Press, Boca Raton, FL. [Google Scholar]
- 2.Vaughn JL, Faulkner P. 1963. Susceptibility of an insect tissue culture to infection by virus preparations of the nuclear polyhedrosis of the silkworm (Bombyx mori L.). Virology 20:484–489. doi: 10.1016/0042-6822(63)90098-9. [DOI] [PubMed] [Google Scholar]
- 3.Summers MD, Volkman LE. 1976. Comparison of biophysical and morphological properties of occluded and extracellular nonoccluded baculovirus from in vivo and in vitro host systems. J Virol 17:962–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bilimoria S. 1986. Taxonomy and identification of baculoviruses, p 37–59. In Granados R, Federeci B (ed), The biology of baculoviruses. CRC Press, Boca Raton, FL. [Google Scholar]
- 5.Thiem SM, Miller LK. 1990. Differential gene expression mediated by late, very late and hybrid baculovirus promoters. Gene 91:87–94. doi: 10.1016/0378-1119(90)90166-O. [DOI] [PubMed] [Google Scholar]
- 6.Rohel DZ, Cochran MA, Faulkner P. 1983. Characterization of two abundant mRNAs of Autographa californica nuclear polyhedrosis virus present late in infection. Virology 124:357–365. doi: 10.1016/0042-6822(83)90352-5. [DOI] [PubMed] [Google Scholar]
- 7.Carpentier DCJ, King LA. 2009. The long road to understanding the baculovirus P10 protein. Virol Sin 24:227–242. doi: 10.1007/s12250-009-3045-0. [DOI] [Google Scholar]
- 8.Vlak JM, Klinkenberg FA, Zaal KJ, Usmany M, Klinge-Roode EC, Geervliet JB, Roosien J, van Lent JW. 1988. Functional studies on the p10 gene of Autographa californica nuclear polyhedrosis virus using a recombinant expressing a p10-beta-galactosidase fusion gene. J Gen Virol 69(Part 4):765–776. doi: 10.1099/0022-1317-69-4-765. [DOI] [PubMed] [Google Scholar]
- 9.Williams GV, Rohel DZ, Kuzio J, Faulkner P. 1989. A cytopathological investigation of Autographa californica nuclear polyhedrosis virus p10 gene function using insertion/deletion mutants. J Gen Virol 70(Part 1):187–202. doi: 10.1099/0022-1317-70-1-187. [DOI] [PubMed] [Google Scholar]
- 10.Gross CH, Russell RL, Rohrmann GF. 1994. Orgyia pseudotsugata baculovirus p10 and polyhedron envelope protein genes: analysis of their relative expression levels and role in polyhedron structure. J Gen Virol 75(Part 5):1115–1123. doi: 10.1099/0022-1317-75-5-1115. [DOI] [PubMed] [Google Scholar]
- 11.Summers MD, Arnott HJ. 1969. Ultrastructural studies on inclusion formation and virus occlusion in nuclear polyhedrosis and granulosis virus-infected cells of Trichoplusia ni (Hübner). J Ultrastruct Res 28:462–480. doi: 10.1016/S0022-5320(69)80034-1. [DOI] [PubMed] [Google Scholar]
- 12.Quant-Russell RL, Pearson MN, Rohrmann GF, Beaudreau GS. 1987. Characterization of baculovirus p10 synthesis using monoclonal antibodies. Virology 160:9–19. doi: 10.1016/0042-6822(87)90038-9. [DOI] [PubMed] [Google Scholar]
- 13.Patmanidi AL, Possee RD, King LA. 2003. Formation of P10 tubular structures during AcMNPV infection depends on the integrity of host-cell microtubules. Virology 317:308–320. doi: 10.1016/j.virol.2003.08.035. [DOI] [PubMed] [Google Scholar]
- 14.Carpentier DCJ, Griffiths CM, King LA. 2008. The baculovirus P10 protein of Autographa californica nucleopolyhedrovirus forms two distinct cytoskeletal-like structures and associates with polyhedral occlusion bodies during infection. Virology 371:278–291. doi: 10.1016/j.virol.2007.09.043. [DOI] [PubMed] [Google Scholar]
- 15.Cheley S, Kosik KS, Paskevich P, Bakalis S, Bayley H. 1992. Phosphorylated baculovirus p10 is a heat-stable microtubule-associated protein associated with process formation in Sf9 cells. J Cell Sci 102(Part 4):739–752. [DOI] [PubMed] [Google Scholar]
- 16.Smith GE, Vlak JM, Summers MD. 1983. Physical analysis of Autographa californica nuclear polyhedrosis virus transcripts for polyhedrin and 10,000-molecular-weight protein. J Virol 45:215–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hennrich ML, Marino F, Groenewold V, Kops GJPL, Mohammed S, Heck AJR. 2013. Universal quantitative kinase assay based on diagonal SCX chromatography and stable isotope dimethyl labeling provides high-definition kinase consensus motifs for PKA and human Mps1. J Proteome Res 12:2214–2224. doi: 10.1021/pr400074f. [DOI] [PubMed] [Google Scholar]
- 18.Perczel A, Park K, Fasman GD. 1992. Analysis of the circular dichroism spectrum of proteins using the convex constraint algorithm: a practical guide. Anal Biochem 203:83–93. doi: 10.1016/0003-2697(92)90046-A. [DOI] [PubMed] [Google Scholar]
- 19.Janek K, Wenschuh H, Bienert M, Krause E. 2001. Phosphopeptide analysis by positive and negative ion matrix-assisted laser desorption/ionization mass spectrometry. Rapid Commun Mass Spectrom 15:1593–1599. doi: 10.1002/rcm.417. [DOI] [PubMed] [Google Scholar]
- 20.Reilly LM, Guarino LA. 1994. The pk-1 gene of Autographa californica multinucleocapsid nuclear polyhedrosis virus encodes a protein kinase. J Gen Virol 75(Part 11):2999–3006. doi: 10.1099/0022-1317-75-11-2999. [DOI] [PubMed] [Google Scholar]
- 21.Li Y, Miller LK. 1995. Expression and functional analysis of a baculovirus gene encoding a truncated protein kinase homolog. Virology 206:314–323. doi: 10.1016/S0042-6822(95)80047-6. [DOI] [PubMed] [Google Scholar]
- 22.Schneider A, Biernat J, von Bergen M, Mandelkow E, Mandelkow EM. 1999. Phosphorylation that detaches tau protein from microtubules (Ser262, Ser214) also protects it against aggregation into Alzheimer paired helical filaments. Biochemistry 38:3549–3558. doi: 10.1021/bi981874p. [DOI] [PubMed] [Google Scholar]
- 23.Paleologou KE, Schmid AW, Rospigliosi CC, Kim H-Y, Lamberto GR, Fredenburg RA, Lansbury PT, Fernandez CO, Eliezer D, Zweckstetter M, Lashuel HA. 2008. Phosphorylation at Ser-129 but not the phosphomimics S129E/D inhibits the fibrillation of alpha-synuclein. J Biol Chem 283:16895–16905. doi: 10.1074/jbc.M800747200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kühnle H, Börner HG. 2009. Biotransformation on polymer-peptide conjugates: a versatile tool to trigger microstructure formation. Angew Chem Int Ed Engl 48:6431–6434. doi: 10.1002/anie.200805768. [DOI] [PubMed] [Google Scholar]
- 25.Valette NM, Radford SE, Harris SA, Warriner SL. 2012. Phosphorylation as a tool to modulate aggregation propensity and to predict fibril architecture. Chembiochem 13:271–281. doi: 10.1002/cbic.201100607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Alaoui-Ismaili MH, Richardson CD. 1998. Insect virus proteins (FALPE and p10) self-associate to form filaments in infected cells. J Virol 72:2213–2223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Buchan DWA, Minneci F, Nugent TCO, Bryson K, Jones DT. 2013. Scalable Web services for the PSIPRED protein analysis workbench. Nucleic Acids Res 41:W349–W357. doi: 10.1093/nar/gkt381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Thomas CJ, Brown HL, Hawes CR, Lee BY, Min MK, King LA, Possee RD. 1998. Localization of a baculovirus-induced chitinase in the insect cell endoplasmic reticulum. J Virol 72:10207–10212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Morris M, Maeda S, Vossel K, Mucke L. 2011. The many faces of tau. Neuron 70:410–426. doi: 10.1016/j.neuron.2011.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bramblett GT, Goedert M, Jakes R, Merrick SE, Trojanowski JQ, Lee VM. 1993. Abnormal tau phosphorylation at Ser396 in Alzheimer's disease recapitulates development and contributes to reduced microtubule binding. Neuron 10:1089–1099. doi: 10.1016/0896-6273(93)90057-X. [DOI] [PubMed] [Google Scholar]
- 31.Alonso A, Zaidi T, Novak M, Grundke-Iqbal I, Iqbal K. 2001. Hyperphosphorylation induces self-assembly of tau into tangles of paired helical filaments/straight filaments. Proc Natl Acad Sci U S A 98:6923–6928. doi: 10.1073/pnas.121119298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hink WF. 1970. Established insect cell line from the cabbage looper, Trichoplusia ni. Nature 226:466–467. doi: 10.1038/226466b0. [DOI] [PubMed] [Google Scholar]
- 33.Granados RR, Guoxun L, Derksen ACG, McKenna KA. 1994. A new insect cell line from Trichoplusia ni (BTI-Tn-5B1-4) susceptible to Trichoplusia ni single enveloped nuclear polyhedrosis virus. J Invertebr Pathol 64:260–266. doi: 10.1016/S0022-2011(94)90400-6. [DOI] [Google Scholar]
- 34.Luckow VA, Summers MD. 1988. Trends in the development of baculovirus expression vectors. Nat Biotechnol 6:47–55. doi: 10.1038/nbt0188-47. [DOI] [Google Scholar]
- 35.Vaughn JL, Goodwin RH, Tompkins GJ, McCawley P. 1977. The establishment of two cell lines from the insect Spodoptera frugiperda (Lepidoptera; Noctuidae). In Vitro 13:213–217. doi: 10.1007/BF02615077. [DOI] [PubMed] [Google Scholar]
- 36.Possee RD. 1986. Cell-surface expression of influenza virus haemagglutinin in insect cells using a baculovirus vector. Virus Res 5:43–59. doi: 10.1016/0168-1702(86)90064-X. [DOI] [PubMed] [Google Scholar]
- 37.Vandeyar MA, Weiner MP, Hutton CJ, Batt CA. 1988. A simple and rapid method for the selection of oligodeoxynucleotide-directed mutants. Gene 65:129–133. doi: 10.1016/0378-1119(88)90425-8. [DOI] [PubMed] [Google Scholar]
- 38.King LA, Possee RD. 1992. The baculovirus expression system: a laboratory guide. Chapman & Hall, New York, NY. [Google Scholar]
- 39.Kramer HB, Lavender KJ, Qin L, Stacey AR, Liu MKP, di Gleria K, Simmons A, Gasper-Smith N, Haynes BF, McMichael AJ, Borrow P, Kessler BM. 2010. Elevation of intact and proteolytic fragments of acute phase proteins constitutes the earliest systemic antiviral response in HIV-1 infection. PLoS Pathog 6:e1000893. doi: 10.1371/journal.ppat.1000893. [DOI] [PMC free article] [PubMed] [Google Scholar]