

Abstract
The voltage-gated K+ channel Kv2.1 has been intimately linked with neuronal apoptosis. After ischemic, oxidative, or inflammatory insults, Kv2.1 mediates a pronounced, delayed enhancement of K+ efflux, generating an optimal intracellular environment for caspase and nuclease activity, key components of programmed cell death. This apoptosis-enabling mechanism is initiated via Zn2+-dependent dual phosphorylation of Kv2.1, increasing the interaction between the channel's intracellular C-terminus domain and the SNARE (soluble N-ethylmaleimide-sensitive factor activating protein receptor) protein syntaxin 1A. Subsequently, an upregulation of de novo channel insertion into the plasma membrane leads to the critical enhancement of K+ efflux in damaged neurons. Here, we investigated whether a strategy designed to interfere with the cell death-facilitating properties of Kv2.1, specifically its interaction with syntaxin 1A, could lead to neuroprotection following ischemic injury in vivo. The minimal syntaxin 1A-binding sequence of Kv2.1 C terminus (C1aB) was first identified via a far-Western peptide screen and used to create a protherapeutic product by conjugating C1aB to a cell-penetrating domain. The resulting peptide (TAT-C1aB) suppressed enhanced whole-cell K+ currents produced by a mutated form of Kv2.1 mimicking apoptosis in a mammalian expression system, and protected cortical neurons from slow excitotoxic injury in vitro, without influencing NMDA-induced intracellular calcium responses. Importantly, intraperitoneal administration of TAT-C1aB in mice following transient middle cerebral artery occlusion significantly reduced ischemic stroke damage and improved neurological outcome. These results provide strong evidence that targeting the proapoptotic function of Kv2.1 is an effective and highly promising neuroprotective strategy.
SIGNIFICANCE STATEMENT Kv2.1 is a critical regulator of apoptosis in central neurons. It has not been determined, however, whether the cell death-enabling function of this K+ channel can be selectively targeted to improve neuronal survival following injury in vivo. The experiments presented here demonstrate that the cell death-specific role of Kv2.1 can be uniquely modulated to provide neuroprotection in an animal model of acute ischemic stroke. We thus reveal a novel therapeutic strategy for neurological disorders that are accompanied by Kv2.1-facilitated forms of cell death.
Keywords: apoptosis, ischemia, neuroprotection, potassium channel, syntaxin, zinc
Introduction
Voltage-gated potassium channels (Kv), key regulators of cellular excitability, play an important role in cell death processes underlying several neurodegenerative conditions, including stroke (Shah and Aizenman, 2014). At physiological concentrations, intracellular K+ suppresses caspase function, inhibits active nuclease activity, and limits apoptosome formation (Hughes and Cidlowski, 1999; Yu, 2003). Thus, for cell death cascades to proceed, intracellular K+ concentrations must decrease. In the CNS, the delayed rectifier K+ channel Kv2.1 plays a prominent role in this process, particularly in cortical, hippocampal, and nigral neurons (Pal et al., 2003; Redman et al., 2006; Shen et al., 2009; Shepherd et al., 2012). This cell death-enabling mechanism is initiated by intracellular free Zn2+ released from oxidized metal-binding proteins and compromised organelles (Aizenman et al., 2000; Sensi et al., 2003; Knoch et al., 2008; Aras et al., 2009; McCord and Aizenman, 2013; Granzotto and Sensi, 2015; Medvedeva et al., 2017). Zn2+, in turn, activates a dual kinase-mediated process, resulting in the sequential phosphorylation of the intracellular Kv2.1 residues Y124 and S800 by Src and p38 MAPK, respectively (Redman et al., 2007, 2009; Shepherd et al., 2012; He et al., 2015). These channel phosphorylation events increase a Ca2+/CaMKII-dependent interaction between the proximal C terminus of Kv2.1, termed C1a (Singer-Lahat et al., 2007, 2008), and the SNARE (soluble N-ethylmaleimide-sensitive factor activating protein receptor) protein syntaxin 1A (STX1A), promoting de novo channel incorporation into the plasma membrane and the associated apoptosis-permitting K+ efflux in dying cells (Pal et al., 2006; McCord and Aizenman, 2013; McCord et al., 2014). Notably, the increased surface expression of functional Kv2.1 channels in vitro generally occurs after a delay of ∼3 h following the initiation of the injurious event (McLaughlin et al., 2001).
Experimental manipulation of this pathway at several checkpoints has strongly suggested that inhibition of the proapoptotic function of Kv2.1 has therapeutic potential. Nonphosphorylatable point mutations of either Y124 or S800 Kv2.1 residues, inhibition of upstream kinases, or interference with STX1A function have all led to a blockade of proapoptotic channel trafficking and are protective in in vitro models of neurodegeneration (McLaughlin et al., 2001; Aras and Aizenman, 2005; Redman et al., 2007, 2009; McCord and Aizenman, 2014). Not surprisingly, suppressing delayed rectifier currents directly is sufficient for improving neuronal survival both in vitro (Pal et al., 2003; Yuan et al., 2011) and in vivo (Wei et al., 2003). However, blockers of delayed rectifier currents can have significant effects on cardiomyocyte repolarization, and are associated with potentially serious side effects, such as ventricular tachycardia and respiratory failure (Graham, 1950; Iwaki et al., 1987; Nattel, 2008), making them less than ideal candidates for CNS pharmacotherapy. As such, the identification of an approach to target the cell death-specific elements of Kv2.1 is an essential step toward realizing the neuroprotective potential of manipulating this channel. With this in mind, we report the generation of a cell-permeant, highly neuroprotective peptide construct (TAT-C1aB), derived from a heretofore unidentified minimal, 9 aa, STX1A-binding Kv2.1 sequence coupled to the cell-permeable transactivator of transcription (TAT) domain from the human immunodeficiency virus.
Materials and Methods
Peptide spot array and far-Western assay.
Far-Western protein-binding affinity assays were performed as previously described (Brittain et al., 2011a). Peptide spot arrays (15 mers) spanning the proximal C-terminus residues 451–540 of rat Kv2.1 were constructed using the Spots-synthesis method. Standard 9-fluorenylmethoxy carbonyl (Fmoc) chemistry was used to synthesize the peptides and spot them onto nitrocellulose membranes prederivatized with a polyethylene glycerol spacer (Intavis). Fmoc protected and activated amino acids were spotted in 20–30 arrays on 150 by 100 mm membranes using an Intavis MultiPep robot. The nitrocellulose membrane containing the immobilized peptides was soaked in N-cyclohexyl-3-aminopropanesulfonic acid (CAPS) buffer (10 mm CAPS, pH 11.0, with 20% v/v methanol) for 30 min, washed once with Tris-buffered 0.1% Tween 20 (TBST), and then blocked for 1 h at room temperature (RT) with gentle shaking in TBST containing 5% (w/v) nonfat milk and then incubated with enriched STX1A protein for 1 h at RT with gentle shaking. Next, the membrane was incubated in primary antibody for STX1A (Millipore, catalog #AB5820-50UL, RRID:AB_2216165) for 2 h at RT with gentle shaking, followed by washing with TBST. Finally, the membrane was incubated in secondary antibody (goat anti-rabbit DyLight 800, catalog #355571, Thermo Fisher Scientific) for 45 min, washed for 30 min in TBST, and visualized by infrared fluorescence (Li-Cor). Four independent peptide spot arrays were used in this study. A second set of membranes (n = 4) was treated as above, but also in the presence of the identified peptide C1aB, which had been coupled to TAT (TAT-C1aB; 100 μm; Fig. 1). Peptides were synthesized at >95% purity (theoretical molecular weight, 2737.20 g/mol; GenScript) and were prepared in small aliquots with ultrapure water.
Figure 1.

Generation of a Kv2.1-derived, STX1A-binding peptide sequence. A, Far-Western assay of the proximal Kv2.1 C-terminus (C1a) region using 15 aa segments spanning residues Kv2.1 451–540, in overlapping 1 aa steps. Two peptides, highlighted in blue and red, flanked a region of high STX1A binding. Error bars indicate mean ± SEM of signal intensity in four independent assays in the absence and presence (black bars) of 100 μm of the derived TAT-containing STX1A-binding peptide described in C below. B, Representative peptide spot-array of the far-Western experiment. C, Final sequences of the peptides used in this study are shown. Orange sequence represents the cell-permeable HIV transactivator of transcription domain (TAT). Red sequence represents the STX1A-binding domain derived from Kv2.1. Green sequence represents a scrambled control based on the C1aB sequence.
Electrophysiology.
Whole-cell patch-clamp experiments were performed on Chinese hamster ovary (CHO) cells transfected with either WT Kv2.1 or Kv2.1(S800E), together with eGFP-expressing plasmid constructs. The S800E point mutation was performed in a prior study (Redman et al., 2007) and transfection was performed as previously described (McCord et al., 2014). Briefly, CHO cells were plated on coverslips in 24-well plates at a density of 5.6 × 104 cells per well. Cells were treated for 3–4 h in serum-free medium with a total of 1.2 μl lipofectamine (Invitrogen) and 0.28 μg of DNA per well. Following transfection, cells were incubated with vehicle, 10 μm TAT-C1aB, or 10 μm scrambled control (TAT-SC) and maintained in F12 medium containing fetal bovine serum in 37°C and 5% CO2 for 24 h before recording. Current recordings were performed on eGFP-positive CHO cells using the whole-cell patch-clamp configuration as described previously (McLaughlin et al., 2001). Borosilicate glass electrodes (3–4 MΩ; 1.5 mm diameter; Sutter Instruments) were filled with internal solution composed of (in mm): 100 K-gluconate, 10 KCl, 1 MgCl2, 1 CaCl2, 10 HEPES, 11 EGTA, 2.2 ATP, 0.33 GTP. The internal solution was further adjusted to pH 7.2 and to 280 mOsm with the addition of sucrose. The pH-adjusted (7.2) external solution was composed of the following (in mm): 115 NaCl, 2.5 KCl, 2.0 MgCl2, 10 HEPES, 10 d-glucose. Partial compensation (80%) for series resistance was performed for all recordings; currents were filtered at 2 kHz and digitized at 10 kHz (Digidata 1440A, Molecular Devices). Delayed rectifier currents were evoked with a series of 200 ms voltage steps to +80 mV from the holding potential of −80 mV in +10 mV increments. Measurements were performed with an Axopatch 200B amplifier and Clampex. Steady-state current analysis was performed at 180 ms, at the +30 mV voltage step relative to baseline current. Current density (pA/pF) was calculated by normalizing the current amplitude measurement to cell capacitance, an electrical determination of cell size. Recordings were performed at RT (∼25°C).
Cortical cultures, LDH assay, and calcium measurements.
All animal protocols in this study were approved by the Institutional Animal Care and Use Committee of the University of Pittsburgh School of Medicine. Cortical neurons were prepared from embryonic day 16 rats of either sex as described previously (McCord et al., 2014). Pregnant donor rats (Charles River Laboratories) were killed by gradual CO2 inhalation, an American Veterinary Medical Association approved protocol (Leary et al., 2013). Cortices were dissociated with trypsin, and plated at 670,000 cells per well on glass coverslips in six-well plates as described previously (Hartnett et al., 1997). Non-neuronal cell proliferation was inhibited with 1–2 μm cytosine arabinoside. dl-threo-β-benzyloxaspartate (TBOA; Tocris Bioscience) excitotoxicity assays were performed on 28–32 days in vitro (DIV) cultures. Coverslips were transferred into 24-well plates containing 10 mm HEPES-supplemented MEM without phenol red. On each individual plate, coverslips were treated with vehicle or 100 μm TBOA and 0, 0.3, or 1 μm of either TAT-C1aB or TAT-SC at 37°C and incubated in 5% CO2 for 24 h. Following this, external medium was collected for LDH colorimetric measurements using a toxicity kit (Sigma-Aldrich), as previously described (Aras et al., 2001). Each experiment contained four replicates of six conditions, including three control and three TBOA treatment groups; peptide treatment groups (TAT-C1aB, TAT-SC) were evaluated in separate experiments. Cell toxicity was quantified as the LDH ratio of TBOA-treated over no-TBOA control values within each experiment. For direct visualization of individual neurons in a similar set of assays, eGFP protein transfection was performed using Lipofectamine 2000 (Invitrogen) as previously described (McCord et al., 2014). Cells were imaged at 60× using an A-1 confocal microscope (Nikon).
Intracellular Ca2+ measurements were performed essentially as previously described (Aizenman et al., 1990; Reynolds et al., 1990) on the same cortical culture preparations as above, but with 20 DIV cells plated on MatTek glass-bottom 35 mm culture dishes. At this developmental stage, neurons robustly express both GluN2A and GluN2B subunits of the NMDA receptor (Sinor et al., 2000). Before imaging, cultures were incubated overnight with 1 μm of either TAT-C1aB or TAT-SC. These treatments were removed just before the Ca2+ measurements. Neurons were incubated with the fluorescent Ca2+ indicator Fura-2 AM ester (5 μm; Invitrogen) with 0.02% Pluronic F-127 (Invitrogen) for 1 h at 37°C. Culture dishes were then mounted on an inverted microscope stage (Olympus) and continuously perfused with a 10 mm HEPES-buffered normal salt solution. Perfusion rate (5 ml/min) was controlled with a gravity flow and a rapid-switching local perfusion system (Warner Instruments). Firmly attached refractile cells were identified as regions of interest (ROIs; 50 cells/coverslip; three coverslips per condition). A ratio of fluorescence emission (F) at 510 nm in response to excitations at 340 and 380 nm was acquired at 1 Hz (Lambda DG-4 and 10-B SmartShutter, Sutter Instruments) via camera (ORCA-ER, Hamamatsu) and saved to a computer using HCImage (Hamamatsu). Baseline Ca2+ signals were recorded for 2 min before a 20 min application of NMDA (30 μm plus 10 μm glycine) followed by a 5 min washout period. Peak increases in intracellular calcium concentration were measured by calculating ΔF/Fo (ΔF, peak fluorescence; Fo, average signal across 2 min baseline period). The area under the response for the first 15 min of NMDA application was also calculated.
Fluorescently labeled peptide brain imaging.
For two-photon imaging of TAT-C1aB-FitC in cortical vasculature, a cranial window was opened over the temporal lobe area in head-fixed young-adult mice under isoflurane anesthesia (induction: 3% in oxygen; maintenance: 1.5% in oxygen; Butler Schein). Mode-locked infrared laser light (940 nm, 100–200 mW intensity at the back focal plane of the objective; MaiTai HP, Newport) was delivered through a galvanometer-based scanning two-photon microscope (Scientifica) controlled with ScanImage (Pologruto et al., 2003), using a 40×, 0.8 numerical aperture objective (Olympus). After obtaining a stable imaging location (field of view, 145 × 145 μm; 100–200 μm below the cortical surface), a single intraperitoneal injection of TAT-C1aB-FitC (6 nmol/g) was administered to the animal. We imaged FitC fluorescence emission with a photomultiplier tube (PMT) and a green emission filter (FF03-525/50, Semrock) at 5–10 min intervals for 15 min before and 2 h after intraperitoneal injection. The laser power and PMT voltage remained constant throughout the imaging session. Quantification of ROI signal intensity was evaluated by ImageJ analysis software (National Institutes of Health).
To confirm blood–brain barrier permeability of the TAT-linked peptide, mice were thoroughly perfused with ice-cold saline 1 h after intraperitoneal injection of either TAT-C1aB or TAT-C1aB-FitC (6 nmol/g). Brains were quickly removed, sectioned into 2 mm slices, and imaged using an Olympus MVX10 microscope with a SPOT RT3 camera. TAT-C1aB-labeled and TAT-C1aB-FitC-labeled brains were imaged sequentially with identical camera settings. Generation of the 8-bit fluorescence signal heat map was performed using the ImageJ plugin HeatMap Histogram (http://www.samuelpean.com/heatmap-from-stack/).
In vivo cerebral blood flow measurements.
Cerebral blood flow was monitored using a two-dimensional laser speckle contrast analysis system (PeriCam PSI High Resolution with PIMSoft, Perimed). Anesthesia was induced by 3% isoflurane, and maintained at 1.5% isoflurane (Butler Schein) in 3:1 NO/O2gas mixture using a vaporizer (General Anesthetic Service). Throughout the experimental procedure, rectal temperature was maintained between 36.5 and 37.0°C using a feedback-controlled heating system (PhysioSuite). The skull of the animal was secured in a stereotactic frame (David Kopf Instruments). A midline incision was made in the scalp and the skull surface was cleaned with sterile normal saline. At 40 min before middle-cerebral artery occlusion (MCAO), 15 min into MCAO, and 15 min after reperfusion, blood perfusion images were taken with a charge-coupled device camera placed 10 cm above the skull. Raw speckle images were taken in a 1.6 × 1.4 cm field at 19 frames/s, 57 frames averaging, with the resolution of 0.02 mm. Five consecutive images at each time point per animal were averaged for analysis using oval-shaped ROIs covering the frontal and parietal bone plates of the ipsilateral and contralateral sides. Percentage signal intensity was calculated by comparing the ipsilateral side mean signal intensity to that of the contralateral side at each time point.
MCAO procedure.
Each cohort of young-adult mice (ages, 8–10 weeks; male; 24–29 g; Jackson Laboratory) were randomized to each group to account for possible confounding factors in the experimental order and body weight on the day of the surgery. However, as body weight did not vary that much, we were able to use the same model/thickness of single-use silicon-coated sutures for all animals (#602212PK10, Doccol Corp.). The suture was advanced from the junction of the external and the common carotid artery into the internal carotid artery for ∼9 mm or until resistance was felt. The suture was secured in this position for the duration of the ischemia time (40/50 min). Mice were anesthetized with isoflurane and maintained at physiological body temperature as described above. Animals were only anesthetized during the surgery. Researchers performing MCAO (C-Y.Y.), drug injections (C-Y.Y.), neurological assessment (A.M.B.), and quantitative analysis of the infarct size (E.A.) were all blinded to the treatment groups. Treatments (saline, TAT-C1aB, or TAT-SC) had been previously randomized by an additional experimenter (K.A.H., A.M.B.).
Infarct ratio measurements.
For quantification of the infarct area, whole brains were extracted from each animal and dissected into 2 mm sections before being stained with 2,3,5-triphenyltetrazolium (TTC; Sigma-Aldrich) with the optimal staining protocol specifications described previously (Joshi et al., 2004): 0.05% TTC in PBS, at 37°C, for 30 min. Brain slices were scanned after TTC staining and the infarct ratio was measured as the total or section infarct area/total area. Infarct size was measured through ImageJ analysis software. Percentage swelling was calculated as follows: (ipsilateral volume/contralateral volume) × 100%.
Neurological testing.
Neurological deficits were assessed on days 1, 2, 3, 5, 7, 10, and 14 following MCAO surgery. Mice were evaluated by a blinded experimenter on an eight-point scale as described previously (Xia et al., 2006), adapted for left-side MCAO. Briefly, animals were scored as follows: 0, no neurological deficit; 1, right forelimb flexion when suspended by tail or failure to extend left forepaw fully; 2, right shoulder adduction when suspended by tail; 3, reduced resistance to lateral push toward the right; 4, spontaneous movement in all directions with circling to the right exhibited only if pulled by tail; 5, circle or walk spontaneously only to the right; 6, walk only when stimulated; 7, no response to stimulation; 8, stroke-related death.
Results
Establishing the sequence of TAT-C1aB
To identify the minimal Kv2.1 C-terminal sequence that can bind syntaxin 1A (STX1A), a far-Western assay (Brittain et al., 2011a) was performed on a peptide spot array of 76 15 aa sequences derived from Kv2.1, spanning residues 451–540 (Rattus norvegicus; accession #NP_037318.1; McCord and Aizenman, 2014) in 1 aa overlapping steps (Fig. 1A). STX1A-enriched lysates derived from CHO cells overexpressing the SNARE protein were used to probe against the bait Kv2.1 peptides. Subsequent immunofluorescence revealed two strongly interacting fragments flanking a region of high STX1A binding (Fig. 1A,B, red, blue). These two fragments contain an overlapping 9 aa sequence, HLSPNKWKW from N terminus to C terminus, corresponding to rat Kv2.1 amino acid residues 478–486. Of note, this exact sequence is present in mouse Kv2.1, corresponding to amino acid residues 482–490 (Mus musculus; accession #NP_032446.2). This sequence likely represents the minimal Kv2.1 C-terminus (C1a) STX1A-binding sequence (C1aB). Addition of the HIV TAT cell-permeable domain to the N terminus of C1aB yielded TAT-C1aB: YGRKKRRQRRRHLSPNKWKW (Fig. 1C). A second set of membranes were used to confirm a displacement of the spotted peptides to STX1A by TAT-C1aB (100 μm, n = 4; Fig. 1A,B). A BLAST search of C1aB revealed no identical sequence in any other mammalian protein, with only an analogously similar (77%), but not identical, sequence in the Kv2.1 cognate Kv2.2 (HLSPSRWKW, Rattus norvegicus and Mus musculus; accession #NP_446452.2 and #NP_001091998.1, respectively), a channel that has not yet been implicated in apoptotic processes, possibly as it lacks a p38 target site analogous to Kv2.1 S800 and flanking sequences. Randomizing the C1aB domain of the Kv2.1-derived, STX1A-binding peptide yielded a scrambled control (TAT-SC; YGRKKRRQRRRNLKWSHPKW). BLAST search of the scrambled C1aB sequence resulted in no identifiable mammalian proteins. A diagram summarizing our overall experimental approach is illustrated in Figure 2, where we hypothesize that the isolated STX1A-binding sequence in TAT-C1aB competes for STX1A and prevents the increase of functional Kv2.1 channels on the plasma membrane during apoptosis and is thus neuroprotective.
Figure 2.

An illustration of the enhancement of Kv2.1 surface expression during neuronal apoptosis model and the protective mechanism of TAT-C1aB. A, In an untreated neuron facing lethal injury, the increased interaction between Kv2.1 and the SNARE protein STX1A through the Kv2.1 C1aB domain promotes channel incorporation into the plasma membrane (Pal et al., 2006). This enhances K+ efflux and enables apoptosis. B, A cell-permeable peptide (TAT-C1aB) is created to contain the Kv2.1-derived STX1A-binding domain C1aB. By competitively binding the Kv2.1-binding site on STX1A, TAT-C1aB provides neuroprotection by attenuating the enhancement of proapoptotic K+ efflux.
TAT-C1aB suppresses apoptotic Kv2.1-mediated K+ currents and provides neuroprotection from “slow” excitotoxicity in vitro without influencing NMDA-evoked Ca2+ responses
Previously, we demonstrated that p38 MAPK phosphorylates Kv2.1 at serine residue S800 to induce the proapoptotic increase in K+ currents (Redman et al., 2007). A point mutation of the serine to a negatively charged amino acid (E or D) at this position results in apoptotic-like enhanced currents in the CHO cell expression system, provided that tyrosine residue Y124 remains intact (Redman et al., 2007, 2009; He et al., 2015). It is noteworthy that CHO cells do not express any endogenous voltage-gated K+ channels (Yu and Kerchner, 1998), but contain all relevant signaling components that lead to proapoptotic trafficking of Kv2.1 (Pal et al., 2003; Aras and Aizenman, 2005). This offers an advantageous preparation to evaluate the effects of the peptide, as it focuses on the STX1A binding-mediated insertion process itself, in the absence of potentially confounding signaling events and other apoptosis-related processes. We observed that overnight exposure to 10 μm TAT-C1aB beginning immediately following the transfection protocol significantly prevented Kv2.1(S800E)-mediated enhanced currents. In fact, current densities in TAT-C1aB-treated, Kv2.1(S800E)-expressing CHO cells were not different from those observed in CHO cells expressing WT Kv2.1 (Fig. 3A). Importantly, the identical TAT-C1aB treatment did not reduce the current density of WT Kv2.1 channels, indicating that normal channel trafficking was not affected by the peptide. Moreover, the control, scrambled peptide TAT-SC (10 μm) had no measurable effects on the current density of either Kv2.1 construct (Fig. 3A). Although these results strongly suggest that TAT-C1aB prevents apoptotic trafficking of Kv2.1, a nonequivocal demonstration of this process will require single-particle tracking of fluorescently labeled channels with techniques such as total internal reflection microscopy.
Figure 3.
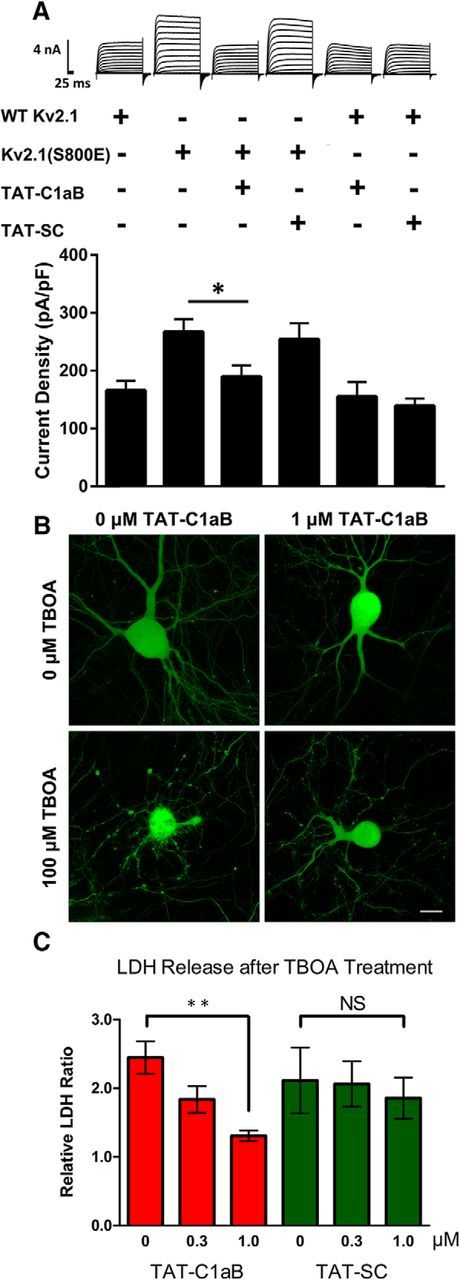
TAT-C1aB prevents enhanced currents mediated by Kv2.1(S800E) and ameliorates TBOA-induced neuronal damage in vitro. A, Representative whole-cell K+ current traces and pooled means ± SEM of current densities recorded from CHO cells expressing WT Kv2.1 treated with vehicle (n = 11), 10 μm TAT-C1aB (n = 12), or 10 μm TAT-SC (n = 11) and CHO cells expressing Kv2.1(S800E) treated with vehicle (n = 11), 10 μm TAT-C1aB (n = 11), or 10 μm TAT-SC (n = 11). Overnight TAT-C1aB incubation significantly blocked the enhanced currents present in Kv2.1(S800E)-expressing cells (current density at +30 mV; vehicle vs TAT-C1aB: 267.2 ± 22.0 vs 189.5 ± 19.6 pA/pF mean ± SEM; ANOVA/Dunnett, *p < 0.05). Scale bar, 4 nA/25 ms. B, 100 μm TBOA treatment induces toxicity in GFP-expressing rat cortical neurons in vitro (28–32 DIV), but not in the presence of 1 μm TAT-C1aB. Scale bar,10 μm. C, LDH release, as an index of cell toxicity, was measured 24 h following TBOA treatment in 28–32 DIV rat cortical neuronal cultures. Coincubation of 1 μm TAT-C1aB mitigated cellular damage of TBOA-treated neurons as indicated by decreased LDH release (1.31 ± 0.077 vs 2.45 ± 0.24 mean ± SEM normalized colorimetric ratio; ANOVA/Dunnett, **p < 0.01; n = 4 independent experiments, each performed in quadruplicate). Coincubation with TAT-SC had no protective effects (n = 6 independent experiments, each performed in quadruplicate).
As TAT-C1aB could effectively prevent enhanced, proapoptotic Kv2.1 currents, we next evaluated whether the peptide would be neuroprotective in an in vitro neuronal system. Embryonically derived rat cortical cultures were treated with 100 μm TBOA at 28–32 DIV, in the absence or the presence of 0.3 or 1 μm of either TAT-C1aB or TAT-SC. TBOA, like other glutamate transporter blockers, elicits slow NMDA receptor-mediated excitotoxicity in neuronal cultures (Blitzblau et al., 1996; Wang et al., 1998). We opted for this cell toxicity model as it has long been established that relatively mild exposure to NMDA receptor agonists, over long periods of time, can induce apoptotic injury (Bonfoco et al., 1995; Leist and Nicotera, 1998). Moreover, apoptotic excitotoxic stimuli can elicit K+ current increases (Yao et al., 2009), and NMDA receptor activation has been closely associated with ischemic stroke injury (Meldrum et al., 1987; Aarts et al., 2002). Within 24 h, TBOA incubation led to the appearance of dendritic and membrane blebs. Critically, the presence of 1 μm TAT-C1aB seemed to be sufficient to ameliorate the TBOA-mediated toxicity, as visualized via prior transfection of the neurons with eGFP (Fig. 3B). To quantify the degree of cellular damage induced by TBOA and neuroprotection via TAT-C1aB, a LDH viability assay was performed 24 h after TBOA treatment. The release of long-lived cytosolic proteins, such as LDH, is indicative of compromised cellular integrity (Koh and Choi, 1987; Aras et al., 2001). In corroboration with our qualitative assessment, we found that TAT-C1aB treatment significantly decreased TBOA-induced injury in cortical cultures. In contrast, TAT-SC afforded no neuroprotection (Fig. 3C).
Finally, we evaluated whether TAT-C1aB could directly influence NMDA-evoked Ca2+ responses. Intracellular Ca2+ recordings were performed as described previously (Aizenman et al., 1990; Reynolds et al., 1990) in neurons that had been exposed to 1 μm of either TAT-C1aB or TAT-SC overnight (∼18 h), until just immediately before recordings. Fura-2 measurements revealed that initial Ca2+ responses to 30 μm NMDA plus 10 μm glycine, as well as delayed calcium dysregulation profiles (Brittain et al., 2012) were not significantly different between both groups of cells (50 cells per coverslip; n = 3 coverslips per group; total 150 cells per condition; Fig. 4). These data strongly suggest that the neuroprotective actions of the peptide occur well downstream from NMDA receptor activation, as predicted by our model (Shah and Aizenman, 2014) and by the observed delayed enhancement of apoptotic potassium currents following injury (∼3 h) described in prior work (McLaughlin et al., 2001).
Figure 4.

TAT-C1aB does not influence NMDA-evoked Ca2+ responses in cortical neurons in vitro. A, Representative Ca2+ transient traces illustrating the average response of 50 rat cortical neurons from a single coverslip previously exposed to either (1 μm; 18 h) TAT-SC (black) or TAT-C1aB (gray). NMDA (30 μm plus 10 μm glycine) was applied for 20 min following a 2 min baseline recording, and later washed for 5 min. B, Pooled means ± SEM (n = 50 cells/coverslip; 3 coverslips per condition) show there is no significant difference in ΔF/Fo (0.69 ± 0.14 vs 0.58 ± 0.06; t test, p > 0.05) or area under the curve for the first 15 min of NMDA application (350.2 ± 59.6 vs 292.1 ± 41.1; t test, p > 0.05) between both peptide treatments.
TAT-C1aB provides neuroprotection in vivo
Once we established that TAT-C1aB was effective in both inhibiting proapoptotic Kv2.1-mediated currents and providing neuroprotection in vitro, we evaluated the in vivo efficacy of TAT-C1aB using a transient ischemic stroke model. First, however, we investigated whether intraperitoneal administration of TAT-C1aB in mice reached the CNS vasculature within a therapeutically relevant timeframe. For this purpose, a fluorescein (FitC) fluorophore was conjugated to the C terminus of TAT-C1aB for live in vivo two-photon imaging in young-adult C57BL/6J male mice (24–29 g; n = 3). A single intraperitoneal injection of TAT-C1aB-FitC (6 nmol/g) was administered after a stable imaging position had been reached at 100–200 μm depth from the cortical surface through a craniotomy window over the temporal cortex. A rapid rise in FitC fluorescence throughout cerebral vessel structures was observed within 10 min of the intraperitoneal injection (Fig. 5A). Vessel fluorescence intensity continued to increase and peaked at 30 min after the injection (Fig. 5B).
Figure 5.

Intraperitoneal TAT-C1aB administration reaches the brain vasculature. A, A representative montage of the in vivo two-photon imaging of FitC-tagged TAT-C1aB (TAT-C1aB-FitC) fluorescent signals through a cranial window. TAT-C1aB-FitC was injected intraperitoneally at 0 min (6 nmol/g). An example ROI (blood vessel) evaluated for fluorescence intensity over time is denoted by white square. Scale bar, 50 μm. B, Quantification of the two-photon imaging data. FitC fluorescence intensity was normalized to the preinjection baseline. Error bars indicate SEM of signal intensity at the ROIs such as that shown in prior panels (n = 3 animals; 2–4 ROIs per animal). C, Injection of TAT-C1aB-FitC (6 nmol/g, i.p.), but not TAT-C1aB, increased fluorescence intensity throughout the brain nervous tissue at 1 h after injection. Animals were transcardially perfused thoroughly before brain sections (2 mm) were obtained. Shown are, from left to right, bright field images, fluorescence images, and heat maps generated from the fluorescent images. Note the higher signals present in section obtained from TAT-C1aB-FitC-labeled brains (n = 3). Scale bar, 2 mm.
Further, we confirmed CNS penetration by the TAT peptide using low-power fluorescence microscopy. After complete saline transcardial perfusion, animals previously injected with TAT-C1aB-FitC (6 nmol/g) were found to present increased fluorescent signal throughout the brain, when compared with animals injected with the nonfluorescent TAT-C1aB (Fig. 5C). These findings are in line with previous characterization of the CNS penetrance of other TAT-linked peptides (Schwarze et al., 1999; Stalmans et al., 2015). Because other TAT-linked peptides have been shown to positively influence CNS neurons in various animal models of ischemic stroke (Kilic et al., 2006; Brittain et al., 2011b; Cook et al., 2012; Zou et al., 2013), and based on our own observations here, we concluded that TAT-C1aB can reach its intended target in a therapeutically realistic fashion following an intraperitoneal injection. Our observations, in fact, directly confirm that a TAT-linked peptide can rapidly be detected within the brain vasculature and brain parenchyma following a peripherally intraperitoneally administered injection.
The Longa method of transient MCAO (Longa et al., 1989) was used in young-adult C57BL/6J mice (Jackson Laboratory; age, 8–10 weeks; male; 24–29 g; n = 67 for entire study) to evaluate TAT-C1aB's neuroprotective efficacy. Successful induction of the MCAO procedure was first validated in a small cohort of animals (n = 3) by monitoring changes of cerebral blood perfusion using a laser Doppler camera before, during, and after 50 min of MCAO. We found that compared with the pre-MCAO baseline (Fig. 6A), the procedure reliably reduced ipsilateral Doppler signal intensity by 50%, compared with the contralateral, noninfarcted side (Fig. 6B). Removal of the suture for blood reperfusion achieved partial recovery of the cerebral blood flow after 15 min (Fig. 6C,D).
Figure 6.

In vivo validation of the MCAO model. A–C, Laser sparkle Doppler images through the skull of an anesthetized mouse undergoing MCAO treatment. Images were taken at ∼40 min before MCAO (A), 15 min into MCAO (B), and 15 min after MCAO (C). Scale bar indicates relative signal intensity. D, Quantification of the mean Doppler signal intensity at each time point. A 50% decrease in Doppler signal intensity was observed during MCAO. Perfusion was partially recovered after suture was withdrawn. Bar graph indicates mean ± SEM of percentage perfusion versus contralateral of each time point (n = 3; 2-way ANOVA, Sidak's multiple comparison, ***p < 0.001, *p < 0.05).
The half-life for TAT-conjugated peptides is cargo-dependent and has been found to range from 1 h to as high as 18 h (Krosl et al., 2003; Bach et al., 2012; Wang et al., 2016). Our two-photon data indicated that the FitC-tagged TAT-C1aB remained detectable above baseline in the CNS vasculature for ≥2 h after intraperitoneal injection (Fig. 5B), suggesting a turnover rate within the range of comparable compounds. In vitro, the proapoptotic channel insertion process is known to take place by 3 h following an acute injury (Pal et al., 2006), while neuronal delayed rectifier currents have been observed to remain elevated 24 h after MCAO (Wu et al., 2015). Based on all of this information, we designed our experimental protocol to consist of two separate intraperitoneal peptide injections, 1 and 6 h following the initiation of reperfusion (6 nmol/g per injection; Fig. 7A). Remarkably, in TAT-C1aB-treated animals (n = 7), TTC staining at 24 h after reperfusion revealed a 40% decrease in total brain infarct ratio compared with that of the TAT-SC-treated (n = 8) or saline-treated (n = 10) animals (Fig. 7B,C). Analysis of individual 2 mm coronal sections revealed that the reduction in infarct ratio provided by TAT-C1aB treatment was most prominent in the central infarct area (Fig. 7D). Of note, a ∼13% increase in MCAO-induced ipsilateral swelling was observed in all animal groups, regardless of the presence or absence of the peptide treatments. That is, ipsilateral swelling was nearly identical in all three treatment groups (saline: 112.9 ± 1.7%, n = 10; TAT-SC: 114.0 ± 3.5%, n = 8; TAT-C1aB: 115.2 ± 2.2%, n = 7; one-way ANOVA p = 0.7811).
Figure 7.

TAT-C1aB ameliorates ischemic stroke damage and improves neurological deficits in mice after MCAO. A, Timeline of the experiment. Note that TAT-C1aB was injected twice intraperitoneally at 1 and 6 h following the initiation of reperfusion. B, Representative images of brain sections after TTC staining at 24 h after 50 min of MCAO injury. Injections of TAT-C1aB (6 nmol/g, i.p., at 1 and 6 h reperfusion) seemed to drastically reduce infarct size. Scale bar, 5 mm. C, Quantification of total infarct ratio is shown (infarct ratio is defined as infarcted area/total area). The total infarct ratio of TAT-C1aB-treated animals (n = 7) was significantly decreased compared with that of either saline-treated (n = 10) or TAT-SC-treated (n = 8) animals (0.12 ± 0.02 vs 0.20 ± 0.02 and 0.20 ± 0.02 mean ± SEM; ANOVA/Dunnett, **p < 0.01) at 24 h reperfusion. D, The majority of the neuroprotective actions of TAT-C1aB were located within the core infarct area. The infarct ratio at 4 (0.16 ± 0.02) and 6 mm (0.16 ± 0.02) coronal section of TAT-C1aB-treated animals was significantly reduced compared with that of either vehicle-treated (0.28 ± 0.02; 0.27 ± 0.03) or TAT-SC-treated animals (0.29 ± 0.03; 0.30 ± 0.02; ANOVA/Dunnett, **p < 0.01). E, Evaluation of neurological scores of mice for 14 d following a 40 min MCAO using an eight-point neurological scoring system (see Materials and Methods). TAT-C1aB-treated animals (n = 13) exhibited a significant improvement in overall neurological outcome when compared with that of the TAT-SC-treated animals (n = 13; 2-way ANOVA, *p = 0.0174). Error bars indicate mean ± SEM of neurological score. Please note that treatments were randomized for all in vivo studies. Investigators performing surgeries, injections, and image/behavioral analyses were all blinded to the treatment group assignments.
We next evaluated the extent of neurological deficit amelioration provided by TAT-C1aB's in vivo neuroprotection. In preliminary studies, animals treated with 50 min MCAO had an overall survival rate of 38.5% by 2 weeks (TAT-C1aB: 42.9%, n = 3 of 7; TAT-SC: 33.3%, n = 2 of 6; Fisher's exact test, not significant), making it difficult to adequately assess behavioral deficits over time. By reducing the ischemia time to 40 min, the survival rate at 2 weeks was improved (TAT-C1aB: 92.3%, n = 12 of 13; TAT-SC: 76.9%, n = 10 of 13; Fisher's exact test, not significant). We thus evaluated the neurological score of animals exposed to 40 min MCAO and treated with either TAT-C1aB or TAT-SC, as described above (6 nmol/g, at 1 and 6 h following reperfusion), on an eight-point neurological-deficit scale over 14 d (see Materials and Methods; Xia et al., 2006). Consistent with the neuroprotection profile afforded by TAT-C1aB, a significant treatment group effect was observed in TAT-C1aB-treated animals, which had an overall improved (lower) neurological deficits score when compared with TAT-SC-treated mice (Fig. 7E). Notably, relatively similar numbers of animals (4 of 13 for TAT-C1aB; 3 of 12 for TAT-SC) exhibited seizure-like behavior following MCAO. These in vivo results suggest that the degree of neuroprotection provided by TAT-C1aB is functionally significant.
Discussion
Ischemic stroke is a leading cause of death worldwide (Thrift et al., 2017). Stroke survivors are often afflicted with permanent physical disabilities due to the loss of central neurons. While acute stroke causes almost immediate necrotic cell death in the ischemic core, programmed neuronal apoptosis in the penumbral region occurs for hours and can continue for several days (Bretón and Rodríguez, 2012). Unfortunately, over the past three decades, essentially all successful stroke treatments observed in preclinical studies have failed to demonstrate any therapeutic benefits in subsequent human trials (Kikuchi et al., 2014; Tymianski, 2014). Only recently, the excitotoxicity-ameliorating NA-1 (TAT-NR2B9c) has shown efficacy in a phase II clinical trial (Evaluating Neuroprotection in Aneurysm Coiling Therapy). This represents a potential novel approach to ischemic stroke treatment that does not involve a thrombolytic agent but does involve a TAT-linked peptide directed at interfering with an ion channel-associated lethal cascade (Hill et al., 2012), which is relevant to the present study. Nonetheless, no stroke treatment yet exists that is both approved by the U.S. Food & Drug Administration (FDA) and is intrinsically neuroprotective against delayed cell death (Tymianski, 2013). The results presented here suggest that the inhibition of the apoptotic-specific functions of Kv2.1 is a potential novel approach to address this medicinal shortcoming.
Recent findings have indirectly implicated the possible involvement of Kv2.1 antagonism as the underlying mechanism in other modes of neuroprotection. For instance, the FDA-approved anticonvulsant and KCNQ (Kv7) channel opener retigabine was shown to provide neuroprotection in an MCAO rodent model, an effect that was initially proposed to be mediated via regulation of neuronal excitability (Bierbower et al., 2015). Interestingly, however, retigabine was also recently shown to inhibit Kv2.1 currents via a poorly reversible, open channel block mechanism, which could account for its neuroprotective actions (Stas et al., 2016). A similar mechanistic convergence may also be present in an alternative model of Kv2.1-mediated neuronal apoptosis. Mutation of Kv2.1 N-terminal cysteine residue C73A to an alanine prevents cysteine-targeted oxidation of the channel, a step suggested to also be intimately linked to neuronal injury (Sesti, 2016; Yu et al., 2016). Rodents carrying this mutation were found to show some protection in a model of traumatic brain injury (Yu et al., 2016), an injurious process with many molecular cell death cascades parallel to those observed in ischemic stroke (Quillinan et al., 2016). Interestingly, we have found that the C73A mutation also effectively prevents p38 phosphorylation of serine residue S800 and the associated K+ current elevation (He et al., 2015), thereby linking the oxidative and the membrane insertion processes of the channel during cell death cascades. Regardless, these observations offer support to the notion of suppressing enhanced Kv2.1 currents after lethal injury as a potentially important and novel neuroprotective approach.
The minimal STX1A-binding domain within the Kv2.1 C terminus is seemingly unique to this channel as we were unable to identify via a BLAST search any other proteins containing this specific sequence, except, as noted earlier, for a sequence within Kv2.2 containing 7 of the 9 aa found in Kv2.1 C1aB. In an overexpression system, Kv2.2 has indeed been shown to interact with STX1A, albeit in a manner different from that observed for Kv2.1 (Wolf-Goldberg et al., 2006). Specifically, Kv2.1/STX1A interactions are dramatically influenced by the presence of the additional SNARE protein SNAP-25, while this is not the case for Kv2.2 (Michaelevski et al., 2003; Wolf-Goldberg et al., 2006). We have previously found that enzymatic cleavage of either STX1A or SNAP-25 alone is sufficient to eliminate proapoptotic trafficking of Kv2.1 (Pal et al., 2006), strongly suggesting that a formation of the STX1A/SNAP-25 SNARE complex is necessary for the observed enhanced currents. It is entirely possible that STX1A can indeed bind and influence Kv2.2 function in neurons, but given the factors noted here, as well as a lack of a p38 phosphorylation site in Kv2.2 that is analogous to S800 in Kv2.1, we believe that the effects of our peptide reported in our study are mostly, if not perhaps exclusively, mediated through a Kv2.1-directed mechanism.
As Kv2.1 is only expressed in the CNS in neurons (Murakoshi and Trimmer, 1999; Speca et al., 2014), TAT-C1aB may represent a unique and direct approach to suppressing cell death programs associated with neuronal enhanced K+ currents. Although traditional Kv blockers, such as tetraethylammonium and clofilium, have been shown to ameliorate ischemic damage (Wei et al., 2003), these drugs are also associated with increased vulnerability to epileptic seizures and ventricular tachycardia (Graham, 1950; Fueta and Avoli, 1993; Batey and Coker, 2002). These off-target effects make these molecules less than optimal candidates for stroke treatment. In contrast, we have observed no effects of TAT-C1aB on baseline currents, a finding similar to those related to our previously reported actions of C1a overexpression and neuroprotection, which also specifically disrupts Kv2.1/STX1A interactions during apoptotic injury (McCord et al., 2014). In agreement with these findings, no increase in seizure-like behavior was observed in TAT-C1aB-treated animals after ischemic stroke. The lack of reduction in poststroke cerebral swelling also ruled out indirect protective mechanisms that could accompany ion flow manipulation in endothelial cells. Altogether, these data strongly suggest that TAT-C1aB targets a unique property of Kv2.1 that is intimately linked to a cell death process and, importantly, does not influence upstream processes, such as NMDA receptor-mediated Ca2+ responses. Along with the growing body of evidence indicating the significant involvement of proapoptotic Kv2.1 functions in many other neurodegenerative conditions (McCord and Aizenman, 2014; Shah and Aizenman, 2014; Yu et al., 2016), our observations may foreshadow the development of a new generation of highly robust neuroprotectants for human neurological conditions.
Footnotes
This work was supported by National Institutes of Health Grants NS043277 (E.A.) and DC007905 (T.T.). C-Y.Y. was supported by the National Institutes of Health Training Grant 5T32NS007433-18 and the American Heart Association individual predoctoral Fellowship 16PRE29170009. We thank J. Trimmer (University of California Davis) for providing the WT Kv2.1 plasmid and R. Di Maio (University of Pittsburgh) and J. Justice (University of Pittsburgh) for expert advice. We also thank E. Levitan (University of Pittsburgh) and I. Lotan (Tel Aviv University, Israel) for encouragement and advice throughout this project, as well as B. Davis (University of Pittsburgh) for allowing us access to his equipment to complete this work.
E.A. has filed a patent application for the neuroprotective strategy presented in this study. The remaining authors declare no competing financial interest.
References
- Aarts M, Liu Y, Liu L, Besshoh S, Arundine M, Gurd JW, Wang YT, Salter MW, Tymianski M (2002) Treatment of ischemic brain damage by perturbing NMDA receptor-PSD-95 protein interactions. Science 298:846–850. 10.1126/science.1072873 [DOI] [PubMed] [Google Scholar]
- Aizenman E, Hartnett KA, Reynolds IJ (1990) Oxygen free radicals regulate NMDA receptor function via a redox modulatory site. Neuron 5:841–846. 10.1016/0896-6273(90)90343-E [DOI] [PubMed] [Google Scholar]
- Aizenman E, Stout AK, Hartnett KA, Dineley KE, McLaughlin B, Reynolds IJ (2000) Induction of neuronal apoptosis by thiol oxidation. J Neurochem 75:1878–1888. [DOI] [PubMed] [Google Scholar]
- Aras MA, Aizenman E (2005) Obligatory role of ASK1 in the apoptotic surge of K+ currents. Neurosci Lett 387:136–140. 10.1016/j.neulet.2005.06.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aras MA, Hara H, Hartnett KA, Kandler K, Aizenman E (2009) Protein kinase C regulation of neuronal zinc signaling mediates survival during preconditioning. J Neurochem 110:106–117. 10.1111/j.1471-4159.2009.06106.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aras MA, Hartnett KA, Aizenman E (2008) Assessment of cell viability in primary neuronal cultures. Curr Protoc Neurosci Chapter 7: Unit 7.18. 10.1002/0471142301.ns0718s44 [DOI] [PubMed] [Google Scholar]
- Bach A, Clausen BH, Møller M, Vestergaard B, Chi CN, Round A, Sørensen PL, Nissen KB, Kastrup JS, Gajhede M, Jemth P, Kristensen AS, Lundström P, Lambertsen KL, Strømgaard K (2012) A high-affinity, dimeric inhibitor of PSD-95 bivalently interacts with PDZ1–2 and protects against ischemic brain damage. Proc Natl Acad Sci U S A 109:3317–3322. 10.1073/pnas.1113761109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batey AJ, Coker SJ (2002) Proarrhythmic potential of halofantrine, terfenadine and clofilium in a modified in vivo model of torsade de pointes. Br J Pharmacol 135:1003–1012. 10.1038/sj.bjp.0704550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bierbower SM, Choveau FS, Lechleiter JD, Shapiro MS (2015) Augmentation of M-type (KCNQ) potassium channels as a novel strategy to reduce stroke-induced brain injury. J Neurosci 35:2101–2111. 10.1523/JNEUROSCI.3805-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blitzblau R, Gupta S, Djali S, Robinson MB, Rosenberg PA (1996) The glutamate transport inhibitor L-trans-pyrrolidine-2,4-dicarboxylate indirectly evokes NMDA receptor mediated neurotoxicity in rat cortical cultures. Eur J Neurosci 8:1840–1852. 10.1111/j.1460-9568.1996.tb01328.x [DOI] [PubMed] [Google Scholar]
- Bonfoco E, Krainc D, Ankarcrona M, Nicotera P, Lipton SA (1995) Apoptosis and necrosis: two distinct events induced, respectively, by mild and intense insults with N-methyl-D-aspartate or nitric oxide/superoxide in cortical cell cultures. Proc Natl Acad Sci U S A 92:7162–7166. 10.1073/pnas.92.16.7162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bretón RR, Rodríguez JC (2012) Excitotoxicity and oxidative stress in acute ischemic stroke. In: Acute ischemic stroke (Rodríguez JC, ed), pp 29–58. Rijeka, Croatia: InTech; 10.5772/1438 [DOI] [Google Scholar]
- Brittain JM, Duarte DB, Wilson SM, Zhu W, Ballard C, Johnson PL, Liu N, Xiong W, Ripsch MS, Wang Y, Fehrenbacher JC, Fitz SD, Khanna M, Park CK, Schmutzler BS, Cheon BM, Due MR, Brustovetsky T, Ashpole NM, Hudmon A et al. (2011a) Suppression of inflammatory and neuropathic pain by uncoupling CRMP-2 from the presynaptic Ca2+ channel complex. Nat Med 17:822–829. 10.1038/nm.2345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brittain JM, Chen L, Wilson SM, Brustovetsky T, Gao X, Ashpole NM, Molosh AI, You H, Hudmon A, Shekhar A, White FA, Zamponi GW, Brustovetsky N, Chen J, Khanna R (2011b) Neuroprotection against traumatic brain injury by a peptide derived from the collapsin response mediator protein 2 (CRMP2). J Biol Chem 286:37778–37792. 10.1074/jbc.M111.255455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brittain MK, Brustovetsky T, Sheets PL, Brittain JM, Khanna R, Cummins TR, Brustovetsky N (2012) Delayed calcium dysregulation in neurons requires both the NMDA receptor and the reverse Na+/Ca2+ exchanger. Neurobiol Dis 46:109–117. 10.1016/j.nbd.2011.12.051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook DJ, Teves L, Tymianski M (2012) Treatment of stroke with a PSD-95 inhibitor in the gyrencephalic primate brain. Nature 483:213–217. 10.1038/nature10841 [DOI] [PubMed] [Google Scholar]
- Fueta Y, Avoli M (1993) Tetraethylammonium-induced epileptiform activity in young and adult rat hippocampus. Brain Res Dev Brain Res 72:51–58. 10.1016/0165-3806(93)90158-7 [DOI] [PubMed] [Google Scholar]
- Graham AJ. (1950) Toxic effects of tetraethylammonium bromide. Br Med J 2:321. 10.1136/bmj.2.4674.321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granzotto A, Sensi SL (2015) Intracellular zinc is a critical intermediate in the excitotoxic cascade. Neurobiol Dis 81:25–37. 10.1016/j.nbd.2015.04.010 [DOI] [PubMed] [Google Scholar]
- Hartnett KA, Stout AK, Rajdev S, Rosenberg PA, Reynolds IJ, Aizenman E (1997) NMDA receptor-mediated neurotoxicity: a paradoxical requirement for extracellular Mg2+ in Na+/Ca2+-free solutions in rat cortical neurons in vitro. J Neurochem 68:1836–1845. [DOI] [PubMed] [Google Scholar]
- He K, McCord MC, Hartnett KA, Aizenman E (2015) Regulation of pro-apoptotic phosphorylation of Kv2. 1 K+ channels. PloS One 10:e0129498. 10.1371/journal.pone.0129498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill MD, Martin RH, Mikulis D, Wong JH, Silver FL, Terbrugge KG, Milot G, Clark WM, Macdonald RL, Kelly ME, Boulton M, Fleetwood I, McDougall C, Gunnarsson T, Chow M, Lum C, Dodd R, Poublanc J, Krings T, Demchuk AM, et al. (2012) Safety and efficacy of NA-1 in patients with iatrogenic stroke after endovascular aneurysm repair (ENACT): a phase 2, randomised, double-blind, placebo-controlled trial. Lancet Neurol 11:942–950. 10.1016/S1474-4422(12)70225-9 [DOI] [PubMed] [Google Scholar]
- Hughes FM Jr, Cidlowski JA (1999) Potassium is a critical regulator of apoptotic enzymes in vitro and in vivo. Adv Enzyme Regul 39:157–171. 10.1016/S0065-2571(98)00010-7 [DOI] [PubMed] [Google Scholar]
- Iwaki M, Mizobuchi S, Nakaya Y, Kawano K, Niki T, Mori H (1987) Tetraethylammonium induced coronary spasm in isolated perfused rabbit heart: a hypothesis for the mechanism of coronary spasm. Cardiovasc Res 21:130–139. 10.1093/cvr/21.2.130 [DOI] [PubMed] [Google Scholar]
- Joshi CN, Jain SK, Murthy PS (2004) An optimized triphenyltetrazolium chloride method for identification of cerebral infarcts. Brain Res protoc 13:11–17. 10.1016/j.brainresprot.2003.12.001 [DOI] [PubMed] [Google Scholar]
- Kikuchi K, Tanaka E, Murai Y, Tancharoen S (2014) Clinical trials in acute ischemic stroke. CNS Drugs 28:929–938. 10.1007/s40263-014-0199-6 [DOI] [PubMed] [Google Scholar]
- Kilic E, Kilic U, Hermann DM (2006) TAT fusion proteins against ischemic stroke: current status and future perspectives. Front Biosci 11:1716–1721. 10.2741/1917 [DOI] [PubMed] [Google Scholar]
- Knoch ME, Hartnett KA, Hara H, Kandler K, Aizenman E (2008) Microglia induce neurotoxicity via intraneuronal Zn(2+) release and a K(+) current surge. Glia 56:89–96. 10.1002/glia.20592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh JY, Choi DW (1987) Quantitative determination of glutamate mediated cortical neuronal injury in cell culture by lactate dehydrogenase efflux assay. J Neurosci Methods 20:83–90. 10.1016/0165-0270(87)90041-0 [DOI] [PubMed] [Google Scholar]
- Krosl J, Austin P, Beslu N, Kroon E, Humphries RK, Sauvageau G (2003) In vitro expansion of hematopoietic stem cells by recombinant TAT-HOXB4 protein. Nat Med 9:1428–1432. 10.1038/nm951 [DOI] [PubMed] [Google Scholar]
- Leary S, Underwood W, Anthony R, Cartner S, Corey D, Grandin T, Greenacre CB, Gwaltney-Bran S, McCrackin MA, Meyer R (2013) AVMA guidelines for the euthanasia of animals: 2013 edition. Schaumberg, IL: American Veterinary Medical Association. [Google Scholar]
- Leist M, Nicotera P (1998) Apoptosis, excitotoxicity, and neuropathology. Exp Cell Res 239:183–201. 10.1006/excr.1997.4026 [DOI] [PubMed] [Google Scholar]
- Longa EZ, Weinstein PR, Carlson S, Cummins R (1989) Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke 20:84–91. 10.1161/01.STR.20.1.84 [DOI] [PubMed] [Google Scholar]
- McCord MC, Aizenman E (2013) Convergent Ca2+ and Zn2+ signaling regulates apoptotic Kv2.1 K+ currents. Proc Natl Acad Sci U S A 110:13988–13993. 10.1073/pnas.1306238110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCord MC, Aizenman E (2014) The role of intracellular zinc release in aging, oxidative stress, and Alzheimer's disease. Front Aging Neurosci 6:77. 10.3389/fnagi.2014.00077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCord MC, Kullmann PH, He K, Hartnett KA, Horn JP, Lotan I, Aizenman E (2014) Syntaxin-binding domain of Kv2.1 is essential for the expression of apoptotic K+ currents. J Physiol 592:3511–3521. 10.1113/jphysiol.2014.276964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin B, Pal S, Tran MP, Parsons AA, Barone FC, Erhardt JA, Aizenman E (2001) p38 activation is required upstream of potassium current enhancement and caspase cleavage in thiol oxidant-induced neuronal apoptosis. J Neurosci 21:3303–3311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medvedeva YV, Ji SG, Yin HZ, Weiss JH (2017) Differential Vulnerability of CA1 versus CA3 pyramidal neurons after ischemia: possible relationship to sources of Zn2+ accumulation and its entry into and prolonged effects on mitochondria. J Neurosci 37:726–737. 10.1523/JNEUROSCI.3270-16.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meldrum BS, Evans MC, Swan JH, Simon RP (1987) Protection against hypoxic/ischaemic brain damage with excitatory amino acid antagonists. Med Biol 65:153–157. [PubMed] [Google Scholar]
- Michaelevski I, Chikvashvili D, Tsuk S, Singer-Lahat D, Kang Y, Linial M, Gaisano HY, Fili O, Lotan I (2003) Direct interaction of target SNAREs with the Kv2.1 channel. Modal regulation of channel activation and inactivation gating. J Biol Chem 278:34320–34330. 10.1074/jbc.M304943200 [DOI] [PubMed] [Google Scholar]
- Murakoshi H, Trimmer JS (1999) Identification of the Kv2.1 K+ channel as a major component of the delayed rectifier K+ current in rat hippocampal neurons. J Neurosci 19:1728–1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nattel S. (2008) Delayed-rectifier potassium currents and the control of cardiac repolarization: Noble and Tsien 40 years after. J Physiol 586:5849–5852. 10.1113/jphysiol.2008.163089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pal S, Hartnett KA, Nerbonne JM, Levitan ES, Aizenman E (2003) Mediation of neuronal apoptosis by Kv2.1-encoded potassium channels. J Neurosci 23:4798–4802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pal SK, Takimoto K, Aizenman E, Levitan ES (2006) Apoptotic surface delivery of K+ channels. Cell Death Differ 13:661–667. 10.1038/sj.cdd.4401792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pologruto TA, Sabatini BL, Svoboda K (2003) ScanImage: flexible software for operating laser scanning microscopes. Biomed Eng Online 2:13. 10.1186/1475-925X-2-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quillinan N, Herson PS, Traystman RJ (2016) Neuropathophysiology of brain injury. Anesthesiol Clin 34:453–464. 10.1016/j.anclin.2016.04.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redman PT, Jefferson BS, Ziegler CB, Mortensen OV, Torres GE, Levitan ES, Aizenman E (2006) A vital role for voltage-dependent potassium channels in dopamine transporter-mediated 6-hydroxydopamine neurotoxicity. Neuroscience 143:1–6. 10.1016/j.neuroscience.2006.08.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redman PT, He K, Hartnett KA, Jefferson BS, Hu L, Rosenberg PA, Levitan ES, Aizenman E (2007) Apoptotic surge of potassium currents is mediated by p38 phosphorylation of Kv2.1. Proc Natl Acad Sci U S A 104:3568–3573. 10.1073/pnas.0610159104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redman PT, Hartnett KA, Aras MA, Levitan ES, Aizenman E (2009) Regulation of apoptotic potassium currents by coordinated zinc-dependent signalling. J Physiol 587:4393–4404. 10.1113/jphysiol.2009.176321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds IJ, Rush EA, Aizenman E (1990) Reduction of NMDA receptors with dithiothreitol increases [3H]-MK-801 binding and NMDA-induced Ca2+ fluxes. Br J Pharmacol 101:178–182. 10.1111/j.1476-5381.1990.tb12109.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarze SR, Ho A, Vocero-Akbani A, Dowdy SF (1999) In vivo protein transduction: delivery of a biologically active protein into the mouse. Science 285:1569–1572. 10.1126/science.285.5433.1569 [DOI] [PubMed] [Google Scholar]
- Sensi SL, Ton-That D, Sullivan PG, Jonas EA, Gee KR, Kaczmarek LK, Weiss JH (2003) Modulation of mitochondrial function by endogenous Zn2+ pools. Proc Natl Acad Sci U S A 100:6157–6162. 10.1073/pnas.1031598100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sesti F. (2016) Oxidation of K(+) channels in aging and neurodegeneration. Aging Dis 7:130–135. 10.14336/AD.2015.0901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah NH, Aizenman E (2014) Voltage-gated potassium channels at the crossroads of neuronal function, ischemic tolerance, and neurodegeneration. Transl Stroke Res 5:38–58. 10.1007/s12975-013-0297-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen QJ, Zhao YM, Cao DX, Wang XL (2009) Contribution of Kv channel subunits to glutamate-induced apoptosis in cultured rat hippocampal neurons. J Neurosci Res 87:3153–3160. 10.1002/jnr.22136 [DOI] [PubMed] [Google Scholar]
- Shepherd AJ, Loo L, Gupte RP, Mickle AD, Mohapatra DP (2012) Distinct modifications in Kv2.1 channel via chemokine receptor CXCR4 regulate neuronal survival-death dynamics. J Neurosci 32:17725–17739. 10.1523/JNEUROSCI.3029-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singer-Lahat D, Sheinin A, Chikvashvili D, Tsuk S, Greitzer D, Friedrich R, Feinshreiber L, Ashery U, Benveniste M, Levitan ES, Lotan I (2007) K+ channel facilitation of exocytosis by dynamic interaction with syntaxin. J Neurosci 27:1651–1658. 10.1523/JNEUROSCI.4006-06.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singer-Lahat D, Chikvashvili D, Lotan I (2008) Direct interaction of endogenous Kv channels with syntaxin enhances exocytosis by neuroendocrine cells. PLoS One 3:e1381. 10.1371/journal.pone.0001381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinor JD, Du S, Venneti S, Blitzblau RC, Leszkiewicz DN, Rosenberg PA, Aizenman E (2000) NMDA and glutamate evoke excitotoxicity at distinct cellular locations in rat cortical neurons in vitro. J Neurosci 20:8831–8837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speca DJ, Ogata G, Mandikian D, Bishop HI, Wiler SW, Eum K, Wenzel HJ, Doisy ET, Matt L, Campi KL, Golub MS, Nerbonne JM, Hell JW, Trainor BC, Sack JT, Schwartzkroin PA, Trimmer JS (2014) Deletion of the Kv2.1 delayed rectifier potassium channel leads to neuronal and behavioral hyperexcitability. Genes Brain Behav 13:394–408. 10.1111/gbb.12120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stalmans S, Bracke N, Wynendaele E, Gevaert B, Peremans K, Burvenich C, Polis I, De Spiegeleer B (2015) Cell-penetrating peptides selectively cross the blood–brain barrier in vivo. PloS One 10:e0139652. 10.1371/journal.pone.0139652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stas JI, Bocksteins E, Jensen CS, Schmitt N, Snyders DJ (2016) The anticonvulsant retigabine suppresses neuronal KV2-mediated currents. Sci Rep 6:35080. 10.1038/srep35080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thrift AG, Thayabaranathan T, Howard G, Howard VJ, Rothwell PM, Feigin VL, Norrving B, Donnan GA, Cadilhac DA (2017) Global stroke statistics. Int J Stroke 12:13–32. 10.1177/1747493016676285 [DOI] [PubMed] [Google Scholar]
- Tymianski M. (2013) Novel approaches to neuroprotection trials in acute ischemic stroke. Stroke 44:2942–2950. 10.1161/STROKEAHA.113.000731 [DOI] [PubMed] [Google Scholar]
- Tymianski M. (2014) Stroke in 2013: disappointments and advances in acute stroke intervention. Nat Rev Neurol 10:66–68. 10.1038/nrneurol.2013.271 [DOI] [PubMed] [Google Scholar]
- Wang GJ, Chung HJ, Schnuer J, Lea E, Robinson MB, Potthoff WK, Aizenman E, Rosenberg PA (1998) Dihydrokainate-sensitive neuronal glutamate transport is required for protection of rat cortical neurons in culture against synaptically released glutamate. Eur J Neurosci 10:2523–2531. 10.1046/j.1460-9568.1998.00256.x [DOI] [PubMed] [Google Scholar]
- Wang M, Zhi D, Wang H, Ru Y, Ren H, Wang N, Liu Y, Li Y, Li H (2016) TAT-HSA-α-MSH fusion protein with extended half-life inhibits tumor necrosis factor-α in brain inflammation of mice. Appl Microbiol Biotechnol 100:5353–5361. 10.1007/s00253-015-7251-4 [DOI] [PubMed] [Google Scholar]
- Wei L, Yu SP, Gottron F, Snider BJ, Zipfel GJ, Choi DW (2003) Potassium channel blockers attenuate hypoxia- and ischemia-induced neuronal death in vitro and in vivo. Stroke 34:1281–1286. 10.1161/01.STR.0000065828.18661.FE [DOI] [PubMed] [Google Scholar]
- Wolf-Goldberg T, Michaelevski I, Sheu L, Gaisano HY, Chikvashvili D, Lotan I (2006) Target soluble N-ethylmaleimide-sensitive factor attachment protein receptors (t-SNAREs) differently regulate activation and inactivation gating of Kv2.2 and Kv2.1: implications on pancreatic islet cell Kv channels. Mol Pharmacol 70:818–828. 10.1124/mol.105.021717 [DOI] [PubMed] [Google Scholar]
- Wu KW, Yang P, Li SS, Liu CW, Sun FY (2015) VEGF attenuated increase of outward delayed-rectifier potassium currents in hippocampal neurons induced by focal ischemia via PI3-K pathway. Neuroscience 298:94–101. 10.1016/j.neuroscience.2015.04.015 [DOI] [PubMed] [Google Scholar]
- Xia CF, Smith RS Jr, Shen B, Yang ZR, Borlongan CV, Chao L, Chao J (2006) Postischemic brain injury is exacerbated in mice lacking the kinin B2 receptor. Hypertension 47:752–761. 10.1161/01.HYP.0000214867.35632.0e [DOI] [PubMed] [Google Scholar]
- Yao H, Zhou K, Yan D, Li M, Wang Y (2009) The Kv2.1 channels mediate neuronal apoptosis induced by excitotoxicity. J Neurochem 108:909-919. 10.1111/j.1471-4159.2008.05834.x [DOI] [PubMed] [Google Scholar]
- Yuan H, Wang WP, Feng N, Wang L, Wang XL (2011) Donepezil attenuated oxygen–glucose deprivation insult by blocking Kv2.1 potassium channels. Eur J Pharmacol 657:76–83. 10.1016/j.ejphar.2011.01.054 [DOI] [PubMed] [Google Scholar]
- Yu SP. (2003) Regulation and critical role of potassium homeostasis in apoptosis. Prog Neurobiol 70:363–386. 10.1016/S0301-0082(03)00090-X [DOI] [PubMed] [Google Scholar]
- Yu SP, Kerchner GA (1998) Endogenous voltage-gated potassium channels in human embryonic kidney (HEK293) cells. J Neurosci Res 52:612–617. 10.1002/(SICI)1097-4547(19980601)52:5%3C612::AID-JNR13%3E3.0.CO%3B2-3 [DOI] [PubMed] [Google Scholar]
- Yu W, Parakramaweera R, Teng S, Gowda M, Sharad Y, Thakker-Varia S, Alder J, Sesti F (2016) Oxidation of KCNB1 potassium channels causes neurotoxicity and cognitive impairment in a mouse model of traumatic brain injury. J Neurosci 36:11084–11096. 10.1523/JNEUROSCI.2273-16.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou LL, Ma JL, Wang T, Yang TB, Liu CB (2013) Cell-penetrating peptide-mediated therapeutic molecule delivery into the central nervous system. Curr Neuropharmacol 11:197–208. 10.2174/1570159X11311020006 [DOI] [PMC free article] [PubMed] [Google Scholar]