

Abstract
In vitro hepatocyte culture systems have inherent limitations in capturing known human drug toxicities that arise from complex immune responses. Therefore, we established and characterized a liver immunocompetent coculture model and evaluated diclofenac (DCF) metabolic profiles, in vitro–in vivo clearance correlations, toxicological responses, and acute phase responses using liquid chromatography–tandem mass spectrometry. DCF biotransformation was assessed after 48 hours of culture, and the major phase I and II metabolites were similar to the in vivo DCF metabolism profile in humans. Further characterization of secreted bile acids in the medium revealed that a glycine-conjugated bile acid was a sensitive marker of dose-dependent toxicity in this three-dimensional liver microphysiological system. Protein markers were significantly elevated in the culture medium at high micromolar doses of DCF, which were also observed previously for acute drug-induced toxicity in humans. In this immunocompetent model, lipopolysaccharide treatment evoked an inflammatory response that resulted in a marked increase in the overall number of acute phase proteins. Kupffer cell–mediated cytokine release recapitulated an in vivo proinflammatory response exemplified by a cohort of 11 cytokines that were differentially regulated after lipopolysaccharide induction, including interleukin (IL)-1β, IL-1Ra, IL-6, IL-8, IP-10, tumor necrosis factor-α, RANTES (regulated on activation normal T cell expressed and secreted), granulocyte colony-stimulating factor, macrophage colony-stimulating factor, macrophage inflammatory protein-1β, and IL-5. In summary, our findings indicate that three-dimensional liver microphysiological systems may serve as preclinical investigational platforms from the perspective of the discovery of a set of clinically relevant biomarkers including potential reactive metabolites, endogenous bile acids, excreted proteins, and cytokines to predict early drug-induced liver toxicity in humans.
Introduction
In the development of drug candidates, experiments with microsomal proteins, S9 fractions, or two-dimensional or suspension hepatocytes may not be physiologically sufficient to predict effects in patients. Many patients taking medication may experience chronic or acute inflammation, thus motivating an in vitro model that includes liver nonparenchymal cells, especially resident macrophages (Kupffer cells), to capture innate immune responses, including those arising from leaky gut (Roberts et al., 2007). Hepatocytes and Kupffer cells show relatively rapid functional decline in standard culture (Godoy et al., 2013; Kegel et al., 2015); thus, a variety of three-dimensional (3D) and microperfused cultures that improve in vitro physiology have been developed to address complex problems in drug biotransformation, pharmacokinetics (PK), drug-induced liver toxicity, and drug–drug interactions in the early stage of drug development (Gómez-Lechón et al., 2004; Li, 2007; Dash et al., 2009; LeCluyse et al., 2012; Ebrahimkhani et al. 2014; Kegel et al., 2015; Knöspel et al., 2016; Long et al., 2016). We developed the LiverChip, which is a microreactor that employs a 0.2-mm-thick scaffold to drive formation of 3D tissue-like structures from primary liver cells and an on-board microfluidic pump to provide controlled perfusion to the array of these 3D liver tissue-like structures (Domansky et al., 2010). The recirculation of medium directly through the 3D tissue provides both convective mass transfer of drugs and other molecules to the 3D tissue and results in an approximately physiologic drop in oxygen tension from one side of the scaffold to the other. Hepatocytes and Kupffer cells can be cocultured in a highly functional state in this microreactor for weeks using a culture medium that supports long-term maintenance of cytochrome P450 activity but is also permissive for examining inflammatory responses (Sarkar et al., 2015; Long et al., 2016; Tsamandouras et al., 2017). The reactor configuration employed here was designed to culture 0.4–0.8 million cells in 1.6–3 ml culture medium, thus providing sufficient sample volume for multiple analyses at multiple time points to gain information that could help predict the fate of drugs and their potential toxicity.
Herein, the LiverChip culture system was used to study the PK, metabolism, and dose-induced toxicity of diclofenac (DCF), a nonsteroidal anti-inflammatory drug and widely used painkiller (Brogden et al., 1980; Skoutakis et al., 1988; Tang, 2003). As noted by Knöspel et al. (2016) in their recent study of DCF metabolism in a larger (1.3 M cells) bioreactor of different configuration, quantitative analysis and comparison of DCF metabolism across different culture formats is challenging, owing to the effects of mixing (or lack of mixing in static cultures, such as the 3D static spheroids; Messner et al., 2013) and significant loss of compounds from solution due to adsorption to system components, as they observed in their reactor system. Furthermore, DCF binds to plasma proteins, further complicating quantitative interpretation of results. The LiverChip system was designed for highly quantitative analysis of exposure and fate, as it is made from components that exhibit minimal adsorption of hydrophobic compounds so that loss of drug is insignificant and exposures can be relatively well controlled (Dash et al., 2009; Long et al., 2016; Tsamandouras et al., 2017).
DCF metabolism follows two major pathways in humans: 1) phase I: major oxidative metabolite 4′-hydroxydiclofenac and minor 5′-hydroxydiclofenac, and 2) phase II: diclofenac glucuronide and hydroxydiclofenac glucuronides, which are toxic due to the formation of reactive electrophilic isoglucuronides (Bort et al., 1999).
Polarized and well differentiated hepatocytes synthesize bile acids, including amino acid–conjugated bile acids (Mörk et al., 2016). Bile acid synthesis may be limited by oxidation, further modification of hepatocytes, or bile transporters (Axelson et al., 2000). Bile acids are synthesized from cholesterol by CYP7A1 and, when conjugated, are excreted to the medium (Mörk et al., 2016). Therefore, it is reasonable to speculate that oxidative damage from DCF may alter bile acid metabolism and transport and to thus investigate bile acid concentrations in conditioned medium as a function of DCF treatment.
Additional insights into the effects of DCF, especially induced by lipopolysaccharides (LPSs), can be assessed by both targeted and shotgun proteomics to reveal alterations in known cytokines and acute phase proteins (APPs). In this study, we investigated secreted protein profiles in cultured medium from cryopreserved hepatocytes and Kupffer cells in a coculture model. To investigate whether secreted proteins correlate with toxicity, we exposed the coculture to a range of doses of DCF with or without a concurrent inflammatory cue (LPS) and profiled large molecules as putative markers of toxicity, including both targeted and shotgun proteomics approaches.
Materials and Methods
Chemicals and Reagents
13C6-DCF and d5-glycocholic acid (GCA), used as internal standards, were obtained from Thermo Fisher Scientific (Waltham, MA) and Toronto Research Chemicals (North York, ON, CA). High-performance liquid chromatography (HPLC)–grade (≥99.9%) methanol (MeOH; Chemical Abstracts Service [CAS] no. 67-56-1), acetonitrile (ACN; CAS no. 75-05-8), formic acid (FA; CAS no. 64-18-6), molecular biology–grade dimethylsulfoxide (DMSO; CAS no. 67-68-5), DCF (CAS no. 15307-86-5), urea (CAS no. 57-13-6), ammonium bicarbonate (ABC; CAS no. 1066-33-7), dl-dithiothreitol (DTT; CAS no. 3483-12-3), and iodoacetamide (IAA; CAS no. 144-48-9) were purchased from Sigma-Aldrich (St. Louis, MO). The standard tuning solution used to calibrate the quadrupole time-of-flight (QTOF) instrument was acquired from Agilent Technologies (Santa Clara, CA). Distilled water was prepared in house with double distillation. Trypsin (catalog no. V5111) and protease MAX surfactant (catalog no. V2071) were purchased from Promega (Madison, WI). The albumin depletion kit (product no. 85160) and the top 12 abundant protein depletion spin columns (product no. 85164) were purchased from Life Technologies (Carlsbad, CA). The iTRAQ Reagent–8Plex Multiplex Kit (SKU no. 4390812) and the iTRAQ Reagent–Multiplex Buffer Kit (SKU no. 4381664) were obtained from AB SCIEX (Framingham, MA). OMIX tips (product no. A57003100) and cleanup C18 pipette tips (product no. 5188-5239) were purchased from Agilent Technologies. Solid-phase extraction columns were purchased from Phenomenex (catalog no. 8B-S100-TAK; Torrance, CA).
Cell Culture
Cryopreserved primary human hepatocytes (Hu8150) and cryopreserved human Kupffer cells (HK8160) were purchased from Life Technologies (Paisley, UK). All cells were thawed according to the manufacturer’s instructions. Viability, assessed using trypan blue exclusion post-thaw, was >85%. Hepatocyte and Kupffer cell cocultures were seeded into LiverChip bioreactors (Long et al., 2016; Kostrzewski et al., 2017) simultaneously at a ratio of 10:1 hepatocytes to Kupffer cells, for a total cell number of 600,000 cells per well, in a volume of 1.6 ml Advanced Dulbecco’s Modified Eagle’s Medium containing thawing and plating supplements (Life Technologies) but without dexamethasone for the first 24 hours of culture. Flow was maintained in the downward direction at 1.0 µl/s through the scaffold for 8 hours postseeding and then switched to the upward direction for the duration of culture. From day 1 to day 3, cultures were maintained in Advanced Dulbecco’s Modified Eagle’s Medium with primary hepatocyte maintenance supplements, omitting dexamethasone. On day 3, the medium was replaced with William’s E medium (WEM) and maintenance supplements containing 100 nM hydrocortisone. At each medium change, medium samples were saved for analysis of liver-produced proteins (albumin, cytokines) and metabolites (urea). For DCF clearance studies, a complete medium change was performed and DCF (Sigma-Aldrich, Poole, UK) in DMSO [final concentration of 0.5% (v/v) for all doses] was added in a volume of 2 ml medium containing 1.25 mg/ml to yield concentrations indicted in the text. Samples (50 µl each) were removed from duplicate wells at time points of 0, 0.5, 1, 4, 6, 24, and 48 hours. No additional medium was added to replace the sample removed. For DCF toxicity, biotransformation, inflammation, and proteomic studies, cultures were dosed with DCF (Sigma-Aldrich) in DMSO [final concentration of 0.5% (v/v) for all doses] on day 5 to yield initial concentrations indicated in the text, and media were changed every 48 hours. At each medium change, fresh compound was added. To induce inflammation, LPS (Sigma-Aldrich) was dosed at 1 µg/ml.
Characterization of Perfused Cocultures in the LiverChip
Quantification of Total Protein.
Cells and scaffolds were washed once in phosphate-buffered saline and lysed using 0.5 ml of 0.1 M sodium hydroxide containing 2% sodium dodecyl sulfate. Total cellular protein was then measured using the Pierce BCA protein assay kit (Thermo Fisher, Loughborough, UK).
Hepatocyte and Kupffer Cell Phenotyping.
Albumin secretion was measured with a human albumin enzyme-linked immunosorbent assay (Assaypro, St. Charles, MO). Lactate dehydrogenase (LDH) activity was measured using the CytoTox 96 Non-Radioactive Cytotoxicity Assay (Promega, Southampton, UK).
DCF Biotransformation
Protein Precipitation and Extraction of Metabolites.
Internal standard (13C6-DCF) was added to 25 µl coculture medium to give a final concentration of 20 μM DCF when at 50–100× Cmax (4.4 µM was Cmax for our study), and 2 μM with DCF at 1× Cmax of d5-GCA (0.5–1 μM) was added as an internal standard prior to sample extraction for bile acid measurements. The choice was arbitrary. 4.4 is a low value in the range of the physiologic dose, and higher values were chosen to represent overdosing. MeOH was then added at a 1:4 ratio [(v/v); 25 µl sample/100 µl MeOH). Resulting suspensions were maintained at −20°C for 5 minutes, vortexed for 20 seconds, and subjected to gentle shaking for 5 minutes on a Fisher Vortex Genie 2 with a vortex adapter. The samples were then maintained at −20°C for 5 minutes and centrifuged at 15,000 rpm for 10 minutes. The supernatants were then collected carefully (without disturbing the protein pellet) and dried in a SpeedVac (Savant Instruments, Holbrook, NY). Samples were prepared immediately for liquid chromatography (LC)–mass spectrometry (MS) analysis by resuspension in 2% ACN containing 0.1% FA. Injections of 1–5 µl were analyzed on an Agilent QTOF 6530 using parameters described previously (Sarkar et al., 2015).
Metabolite Profiling.
LC-MS analyses were performed on an Agilent 6530 Accurate-Mass LC-QTOF mass spectrometer with an Agilent Jet Stream electrospray ionization (ESI) source and a MassHunter workstation (version B.06). The mass spectrometer was interfaced with an Agilent 1290 ultra-HPLC system. The column was an Agilent Extend-C18 (2.1 × 50 mm, 1.8 μm; Agilent Technologies). The column compartment temperature was set at 40°C. The QTOF instrument was calibrated daily before runs using the standard tuning solution from Agilent Technologies. ESI mass spectra were acquired in positive ion mode for total and free DCF measurements. Mass data were collected between a mass-to-charge ratio (m/z) of 70 and 1000 at either two scans per second or four scans per second. The settings were as follows: ion spray voltage, 3800 V; heated capillary temperature, 350°C; drying gas, 8 l/min; nebulizer, 30 psi; sheath gas temperature, 380°C; and sheath gas flow, 12 l/min. Two reference masses (m/z 121.0509, C5H4N4; and m/z 922.0098, C18H18O6N3P3F24) were infused continuously to allow constant mass correction during the run. Variation of retention times and m/z values was ≤0.2 minutes and <5 ppm mass error, respectively, and the relative standard deviations of peak areas were <20%. Mobile phases consisted of double-distilled water containing 0.1% FA (A) and ACN containing 0.1% FA (B). Linear gradients were from 2% to 95% B over 12 minutes at a flow rate of 0.4 ml/min.
Data Processing, Analysis, and Metabolite Identification.
DCF clearance and GCA were measured by targeted mass spectrometry on the Agilent QTOF instrument. Data were processed using Agilent MassHunter qualitative analysis software (version B.06). Peak areas of DCF (m/z 296.0245), 13C6-DCF, GCA (m/z 466.3169), and d5-GCA as internal standard were obtained using the extracted ion chromatogram (EIC) function. MS/MS spectra of DCF metabolites were analyzed manually with the fragmentor tool in ChemDraw (PerkinElmer, Waltham, MA) and with the molecular structure correlator function in MassHunter (i.e., all signals associated with a given analyte, with intensities >2000–5000, were used to profile metabolites at a 5-ppm mass accuracy threshold).
Tandem mass spectra were generated with an Agilent QTOF 6530 mass spectrometer to further confirm the identity of metabolites. For this analysis, the matched exact masses of parent and fragmented ions (<5 ppm mass error), and associated retention times (<20 seconds) were used to create a target list. Isotope patterns were also used to identify Cl-containing DCF metabolites.
DCF PK Using a One-Compartment Model
The PK properties of DCF in the LiverChip bioreactor were investigated after administration of 4.4 μM DCF to a coculture of hepatocytes and Kupffer cells (10:1). High mass accuracy LC-MS/MS was used to follow DCF concentrations over time. PK parameters were obtained using MATLAB software (version R2014; MathWorks Inc., Natick, MA). Based on the initial observation of the DCF concentration profile over time, a one-compartmental PK model was used to estimate the DCF elimination rate (kel) and the volume of distribution (Vd). Due to sampling from the bioreactor, Vd was used as the average of the volumes at t = 0 and the end point. The following equations were used (eqs. 1 and 2):
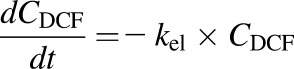 |
(1) |
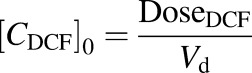 |
(2) |
where CDCF is the DCF concentration, [CDCF]0 is the initial DCF concentration in the liver bioreactor, and DoseDCF is the DCF dose, respectively. The elimination half-life (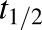 ) of DCF and the clearance (CL) were calculated using eqs. 3 and 4:
) of DCF and the clearance (CL) were calculated using eqs. 3 and 4:
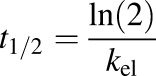 |
(3) |
| (4) |
Modeling of DCF and Bovine Serum Albumin Equilibrium Binding
The binding equilibrium between DCF and bovine serum albumin (BSA) (1.25 mg/ml and 25 mg/ml) was modeled based on the following equation (eq. 5):
 |
(5) |
where [BSA] is the total BSA concentration, [DCFBound] and [DCFFree] are bound and free DCF concentrations, and 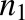 and
and 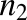 are the number of binding sites on BSA for DCF, which are 2.15 and 12.45, respectively (Dutta et al., 2006). Similarly, 0.88 × 105 M−1 and 0.727 × 103 M−1 are the association constants
are the number of binding sites on BSA for DCF, which are 2.15 and 12.45, respectively (Dutta et al., 2006). Similarly, 0.88 × 105 M−1 and 0.727 × 103 M−1 are the association constants 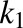 and
and 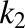 , respectively. These values were estimated from the experimental data in Dutta et al. (2006).
, respectively. These values were estimated from the experimental data in Dutta et al. (2006).
Protein Digestion and Peptide Fractionation.
Prior to digestion, BSA was depleted from the coculture medium using albumin depletion columns according to the manufacturer’s instructions. Proteins were then resuspended in 15 µl of 8 M urea (dissolved in 50 mM ABC) followed by the addition of 20 µl 0.2% ProteaseMAX (Promega) surfactant, 50 µl of 50 mM ABC, and 2.12 µl of 400 mM DTT. Disulfide bonds were reduced by incubating the samples at 56°C for 30 minutes and were alkylated by the addition of 6 µl of 550 mM IAA, followed by incubation for 30 minutes at room temperature in the dark. To prevent alkylation of trypsin, excess IAA was inactivated by the addition of 2.12 µl DTT and incubated for an additional 30 minutes in the dark. Proteins were digested by adding 3.7 µl of 0.5 µg/µL trypsin (1:27 trypsin/protein) and 1 µl 1% ProteaseMAX, followed by a 3-hour incubation at 37°C. After digestion, trypsin was inactivated by addition of 20% trifluoroacetic acid to a final concentration of 0.5%. The tryptic peptides were concentrated and desalted with OMIX tips from Agilent Technologies according to the manufacturer’s instructions and were dehydrated to dryness in a SpeedVac.
To fractionate the peptides by isoelectric focusing, samples were resuspended in 3.6 ml 1× off-gel buffer and then loaded onto an Agilent off-gel fractionator with Immobilized pH Gradient (IPG) strips (pH 3–11) according to the manufacturer’s instructions. For the first experiment, the 24 fractions were pooled into 20 fractions (i.e., combining 1 and 24, 2 and 23, 3 and 22, 4 and 21, and 5 and 20, without combining fractions 6–19). All fractions were dried in a SpeedVac prior to resuspension in 20 µl 98% water, 2% ACN, and 0.1% FA for LC-MS analysis as described below.
Protein Profiling by LC-MS/MS.
These experiments were carried out on the Agilent 6530 QTOF mass spectrometer, interfaced with an Agilent 1290 series ultra-HPLC containing a binary pump, a degasser, a well-plate autosampler with a thermostat, and a temperature-controlled column compartment. Mass spectra were acquired in the 3200-Da extended dynamic range mode (2 GHz) using the following settings: ESI capillary voltage, 3800 V; fragmentor, 150 V; nebulizer gas, 30 psi; drying gas, 8 l/min; and drying temperature, 380°C. Data were acquired at 6 mass spectra per second and 3 tandem mass spectra per second in the mass ranges of m/z 100–2000 for MS and 50–2500 for MS/MS, with a maximum of five precursors per cycle, and were stored in profile mode. Fragmentation energy was applied at a slope of 3.0 V/100 Da with a 2.8 offset. Mass accuracy was maintained by continually spraying internal reference ions in positive ion mode, with m/z 121.0509 and 922.0098.
An Agilent ZORBAX 300SB-C18 RRHD column (2.1 × 100 mm, 1.8 μm) was used for all analyses. The LC parameters were as follows: autosampler temperature, 4°C; injection volume, 20 µl; and column temperature, 40°C. Mobile phases were 0.1% FA in water (A) and 0.1% FA in ACN (B). The gradient started at 2% B at 400 µl/min for 1 minute, increased to 50% B from 1 to 19 minutes with a flow rate of 250 µl/min, then increased to 95% B from 19 to 23 minutes with an increased flow rate of 400 µl/min and was held up to 27 minutes at 95% B before decreasing to 2% B at 27.2 minutes and ending at 30 minutes, and was followed by a 2-minute postrun at 2% B.
Proteomics Data Processing.
Raw data were extracted and searched with the Spectrum Mill search engine (B.04.00.127; Agilent Technologies) using published parameters (Ravindra et al., 2015). Protein expression values (spectrum counts) were determined with Scaffold software using the imported peptide hits from Spectrum Mill. The threshold for considering a protein identification included a minimum of two distinct peptides with 95% confidence.
iTRAQ 8plex: Protein Digestion, Labeling, and Chromatography.
The albumin was depleted prior to processing the samples for iTRAQ labeling. For these experiments, 100 μg total protein from individual samples was reduced in 2 mM tris(2-carboxyethyl)phosphine at 37°C for 30 minutes, and the cysteine residues were blocked in 10 mM methyl methanethiosulfonate at room temperature for 1 hour, followed by trypsin digestion (modified trypsin from Promega) at a protease/protein ratio of 1:40 (w/w) at 37°C overnight. iTRAQ-8plex labeling reagents were added to the peptide samples, which were incubated at room temperature for 3 hours with the addition of isopropyl alcohol. The reaction was stopped by the addition of 10 mM monopotassium phosphate and 25% ACN, pH 2.6 (solvent A), followed by centrifugation at 14,000g for 10 minutes to remove aggregated proteins. All individual samples were pooled and purified using solid-phase extraction columns prior to fractionation. Then, digested protein samples were separated by using the Agilent 3100 OFFGEL fractionator along the 3–11 pH range into 24 fractions based on peptide isoelectric points. The individual fractions were collected and concentrated in a SpeedVac before MS analysis. Individual fractions were further cleaned with C18 pipette tips. All identified proteins, as well as their spectral counts and quantitative values, are shown in Table 1 and Supplemental Table 1.
TABLE 1.
The ln2 quantitative iTRAQ values of APPs from day 7
| Drug/Control | LPS/Control | [LPS Plus Drug]/LPS | Protein |
|---|---|---|---|
| 1.4 | 3.0 | −1.2 | Transferrin |
| 1.1 | 4.2 | −1.9 | α1-antitrypsin |
| 1.0 | 4.2 | −2.0 | α1 antiproteinase |
| 0.5 | 3.8 | −1.3 | Haptoglobin |
| 3.4 | 3.9 | 0.1 | Serum albumin |
| 3.4 | 3.9 | 0.1 | Hypothetical protein |
| −0.8 | 2.1 | −1.0 | Vitamin D binding protein |
| 1.2 | 3.8 | −0.9 | Ceruloplasmin |
| 1.7 | 2.4 | 0.0 | Liver carboxylesterase 1 isoform a |
| 2.3 | 3.7 | −1.2 | Angiotensinogen |
| 3.6 | 7.0 | −1.3 | α1-acid glycoprotein 1 |
| −1.6 | 5.0 | −1.5 | α1-acid glycoprotein 2 |
| 0.3 | 1.5 | −1.0 | α2-HS glycoprotein |
| −1.0 | 3.3 | −2.0 | α2 macroglobulin |
| 2.7 | 5.0 | −1.9 | Retinol binding protein 4 |
| 1.8 | 5.1 | −1.7 | Complement component C3 |
| 1.2 | 1.3 | −0.9 | Transthyretin |
| 1.5 | 2.8 | −0.5 | Leucine-rich α2-glycoprotein |
| 2.3 | −1.7 | 8.5 | Amyloid β A4 protein isoform b |
The drug- and LPS-treated samples were normalized to untreated samples (control). The LPS and LPS and drug treatment samples were normalized to LPS samples. As the values indicate, the treatment of drug (440 μM DCF) suppressed the synthesis of APPs.
Cytokine Analysis
Multiplex Immunoassays.
Analysis of 62 unique cytokines, chemokines, growth factors, and matrix metalloproteinases was performed using the human group 1 27-plex and 40-plex chemokine panels, as well as select singleplexes of group II 21-plex and inflammation panel 1 37-plex (Supplemental Fig. 1). Assays were completed according to the manufacturer’s instructions (Bio-Rad Laboratories, Hercules, CA), with the exception that coupled beads, biotinylated detection antibodies, and streptavidin-phycoerythrin fluorescent reporters were diluted 2-fold. All analytes were evaluated in neat, undiluted samples; a total volume of 50 μl sample from coculture medium was analyzed per multiplex panel. To attain a measurement for interleukin (IL)-8 within the working range of the assay, samples were diluted 8-fold. Standard and sample diluents consisted of WEM (Life Technologies) in the presence of 0.75% BSA (Sigma-Aldrich) as a final concentration.
Assays were performed in parallel (unmixed) to avoid antibody crossreactivity between groups. Prepared arrays were assessed by the 3D suspension array system (Bio-Rad Laboratories) utilizing xMAP technology licensed by Luminex Corporation (Austin, TX). Data were collected with xPONENT for FLEXMAP 3D software (version 4.2; Luminex Corporation) and results were evaluated initially in BioPlex Manager Software (version 6.1; Bio-Rad Laboratories). Median fluorescence intensity values were converted to absolute concentrations via calibration to 15-point standard series that implemented a 2-fold serial dilution. Assay performance metrics for each analyte are summarized in Supplemental Table 2.
Data Processing and Statistical Analyses.
To quantify the concentration of each analyte, the five-parameter logistic model was used for the best curve fit of standards. Regression analysis minimized the weighted sum of squared errors. In general, the weights are set equal to the inverse variance; however, for immunoassays, the high-response end of a curve approaches saturation of the detector; thus, variance is approximated more appropriately by a power function as shown in eq. 6:
| (6) |
where A is a function of the magnitudes of the responses and 1.0 ≤ B ≤ 2.0 for immunoassays (Finney, 1987). Curve-fitting techniques were completed in BioPlex Manager Software (version 6.1; Bio-Rad Laboratories). Parameters including the weighted sum of squared errors, residual variance, and fit probability are provided in Supplemental Table 2.
Multivariate statistical techniques, such as unsupervised hierarchical clustering, were evaluated in MATLAB (version 2012b).
Bioinformatics.
Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes pathway enrichment analyses (P > 0.05) were performed by using the functional annotation tool DAVID (Huang et al., 2009). ClueGO, a Cytoscape plug-in (Bindea et al., 2009), was used to facilitate identification of the functional and pathway analyses for DCF- and LPS-treated hepatocyte culture medium.
Results
Concentration-Dependent Binding Equilibrium of DCF and BSA
DCF in plasma exists in equilibrium between free and albumin-bound forms, with the free form available for metabolism (Dutta et al., 2006; Zhang et al., 2015). Albumin contains multiple different binding sites for lipophilic compounds; hence, equilibrium binding can be a complex function of concentration with multiple molecules of drug bound to the same albumin molecule in sites of different affinities, competing with other lipophilic molecules such as steroid hormones (Dutta et al., 2006). DCF equilibrium with albumin has been fit to a model comprising two high-affinity and 12 low-affinity sites (Dutta et al., 2006; see the Materials and Methods). Chemically defined liver cell culture media often contain BSA or other albumin sources as carriers of lipophilic nutrients. Although the normal human plasma concentration of albumin is 35–55 g/l (530 µM), lower albumin concentrations in the range of 1–10 g/l (15–150 µM) are typically used in cell culture. Even at these lower concentrations, the presence of albumin can influence the free concentration of drugs present, affecting the PK properties. We thus assessed these equilibria experimentally across our dose range to build appropriate PK models of our experimental clearance and metabolism data using our culture media, which contain other albumin-binding lipophilic molecules including cortisol. We first built a binding landscape from published data (Zhang et al., 2015) (Fig. 1A), illustrating the nonlinear nature of binding to two sites on albumin. We then experimentally determined the DCF-albumin binding equilibria in the culture medium used for the clearance and metabolism studies in this work, and we found that measured outcomes were in agreement with the literature for the 1.25 g/l BSA concentration used in these studies and total DCF concentrations up to 100 µM (Fig. 1B). Hence, the binding curve described in the Materials and Methods was used for further PK analysis.
Fig. 1.

Binding equilibrium between DCF and BSA. (A) Literature values of DCF-albumin equilibrium constants were used to predict a landscape of bound/free values for the interaction between DCF and BSA. The free DCF fraction was calculated with respect to BCA and DCF concentrations using the Scatchard equation (Dutta et al., 2006). (B) Free DCF concentrations were quantified in the cell culture medium with 1.25 mg/ml BSA (20 µM), the concentration used for metabolism studies in this work. The experimental data agreed well with the literature-based predictions. Data correspond to mean values of two technical replicates.
Baseline DCF Clearance and Metabolism in the LiverChip
Duplicate wells of cocultured hepatocytes and Kupffer cells in standard media containing 1.25 g/l BSA were examined for two doses of DCF. Medium samples were collected and analyzed as described in the Materials and Methods. We first confirmed that DCF exhibited no detectable binding to the LiverChip components by dosing LiverChip wells in the absence of cells and monitoring the concentration over 24 hours (data not shown). This finding is in concordance with previous reports that the LiverChip exhibits low drug binding (Tsamandouras et al., 2017).
Drug clearance was quantified after dosing with a pharmacological 4.4-µM dose and a suprapharmacological 440-µM dose of DCF in medium containing 1.25 g/l BSA (Fig. 2; Supplemental Table 3). Clearance parameters were calculated from the concentration profiles of total DCF as a function of time, taking into account the reactor mixing properties and albumin binding, using a PK model as described in the Materials and Methods. The t1/2 of DCF at a pharmacological dose was estimated to be 14.6 hours in the LiverChip bioreactor. Clearance was also investigated at a higher dose (440 µM) (Fig. 2B), as this condition was used to assess metabolite production in addition to clearance.
Fig. 2.

DCF pharmacokinetics in the LiverChip. (A and B) A 4.4-µM pharmacological dose of DCF (A) and a 440-µM suprapharmacological dose of DCF (B) were administered to separate wells of the LiverChip in duplicate wells for each dose. Data for total DCF concentrations as a function of time are plotted for each dose (circles) and were used to generate a PK model taking into account both mixing and equilibrium binding to BSA (solid line). Each well was sampled twice to evaluate technical variation; individual samples were assessed two times consecutively in positive ion mode using reversed-phase ultra-HPLC-TOF-MS.
Extrapolation of In Vitro DCF Clearance Data to Predict Intrinsic Clearance
In vivo–in vitro correlation provides valuable information for the first in human dosing. Here, the retrospective in vivo–in vitro correlation analysis of the DCF clearance using t1/2 and Vd to calculate intrinsic clearance (CLint) by accounting for the scaling factors detailed in eq. 7 (Davies and Morris 1993; Obach et al., 1997; Obach, 1999; Sarkar et al., 2015):
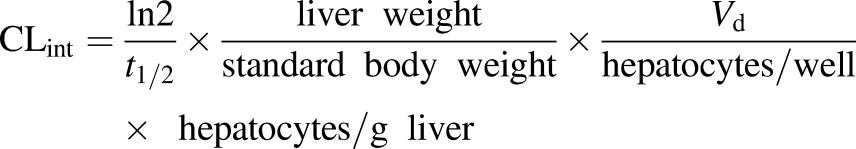 |
(7) |
Values of scaling parameters and intrinsic clearance are found in Supplemental Table 3. The predicted CLh was 0.55 ml/min per kilogram, which is <7-fold underpredicted than in vivo plasma clearance (3.8 ml/min per kilogram) (data also available at http://www.capkr.man.ac.uk). Although predicted CLh is generally underpredicted for DCF, the contribution of the gut and other organs must be considered to obtain better predicted plasma clearance in humans (Brown et al., 2007; Hallifax et al., 2010).
Assessment of Toxicity of DCF in the LiverChip
The toxicity of DCF in the LiverChip was assessed using a repeat dosing protocol with fresh drug containing medium added on day 5, 7 and 9. Cell viability was assessed using a tetrazolium salt reagent 2-(2-methoxy-4-nitrophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H-tetrazolium (WST-1) on day 11 (Fig. 3A), resulting in an IC50 of 227 µM for the primary hepatocyte–Kupffer cell cocultures in the LiverChip. This is comparable to the IC50 for DCF generated from spheroid culture of primary hepatocytes and nonparenchymal cells (Messner et al., 2013). To evaluate the temporal reduction in function and cell death over the multiple DCF doses, LDH (Fig. 3B) and albumin (Fig. 3C) were measured 48 hours after each dose. Throughout the culture period, vehicle controls [0.1% (v/v) DMSO] showed sustained levels of albumin secretion, indicating that hepatocytes in the cocultures remained functional. At high DCF concentrations (>50×), albumin secretion declined after a single dose, whereas LDH release became more pronounced after the second dose. Codosing of LPS resulted in the production of the proinflammatory cytokines tumor necrosis factor (TNF)-α and IL-6 (data not shown) but that result does not significantly change the toxicity profile of DCF as assessed by gross markers of functionality and cell death.
Fig. 3.

Toxicity of DCF in the LiverChip under basal and inflamed conditions. (A–C) DCF was administered to primary human hepatocyte and Kupffer cell cocultures in the LiverChip and the response of the assayed using WST-1 (A), LDH release (B), and albumin secretion (C). Data correspond to mean values of two replicates for (A), and S.D. was based on n = 3 for (B) and (C).
DCF Biotransformation by Phase I and II Metabolism under Basal and Inflamed Conditions
Accurate masses, tandem mass spectra, and available standards corresponding to major DCF metabolites from the liver coculture system were used to elucidate structures. EIC% values of metabolites were only used to understand the relative amounts formed in this coculture system.
We observed 4-hydroxy-DCF and 5-hydroxy DCF as major and minor phase I metabolites, respectively, and acylglucuronides of DCF, hydroxy-DCF, and DCF-sulfate as major phase II metabolites (Fig. 4). UDP-glucuronosyltransferase (UGT) and sulfotransferase activities were confirmed to be stable in this human liver model, as measured by DCF phase II metabolites. Under the culture conditions, three glucuronides of DCF and hydroxylated DCF were observed, which might be a result of isomeric acylglucuronides.
Fig. 4.

DCF biotrasformation. DCF metabolites from the liver coculture system were observed and included 4-hydroxy-DCF and 5-hydroxy DCF as major and minor phase I metabolites, respectively, and acylglucuronides of DCF and hydroxy-DCF as the major phase II metabolites. These metabolites were accounted for by CYP2C9, CYP2C8, and UGT2B7 metabolism in humans as well. SULT, sulfotransferase.
As assessed by the production of 4-hydroxy-DCF, CYP2C9 activity on day 5 was 1.5-fold greater than that on day 7. A minor methoxylated hydroxy DCF metabolite was detected and was approximately 2.8-fold higher when the LiverChip was treated with the drug and LPS in combination. Using our LC-MS method, we did not observe glutathione-DCF–related adducts in the culture medium. Minor metabolites were putatively predicted by ≤5 ppm mass accuracy and chlorine isotopic signature.
DCF and LPS treatment downregulated the CYP450-dependent formation of 4-hydroxy-DCF 2-fold, upregulated UGT-dependent formation of total DCF-acylglucuronides 1.5-fold, and hydroxylated-DCF acyl glucuronides 2.4 fold; no significant changes in sulphonated product were observed (data not shown). The assignment of human P450 enzymes to the formation of phase I and II DCF metabolites in this liver microphysiological system is based on previous work by Boelsterli (2003) and Tang (2003).
Endogenous GCA as a Model Bile Acid Marker of DCF-Induced Toxicity
The LC/MS methods developed for the analyses of hydrocortisone and DCF capture data for all compounds in the solutions that are present in detectable amounts. Analysis of these data with software allows untargeted searches and extracts molecular weights for detectable compounds in the media and highlights those that change with various experimental conditions. The molecular weights for compounds in these subsets can then be searched against databases that suggest possible structures, and some of these can be identified by interpreting the mass spectra from these compounds. In this instance, a prominent compound that decreased with drug treatment was identified as the bile acid GCA. This in turn suggested a targeted search (of the same data sets) for other bile acids, several of which were detected and putatively identified based on mass accuracy (<5–10 ppm), and some available standards (Fig. 5). These also declined in concentrations after treatment with DCF at various levels; due to the unavailable reference standards, the formation of other detected bile acids was determined by accurate MS area units. GCA was considered as a model bile acid in this 3D liver culture. Peak intensity (EIC%) relative values were for guidance only. They do not represent absolute amounts present in these experiments. GCA peak area was considered as 100, and the relative EICs without DCF and with DCF are listed for other detected bile acids (Table 2). The extracellular concentration of GCA in the absence of DCF was 3 to 4 μM, and there was a constant level in the system for up to 2 weeks. No GCA was detected in the starting medium at the zero time point, indicating that GCA was synthesized by the 3D LiverChip system. The Cmax for the recommended dose of DCF in humans is 4.4 µM and experiments were carried out up to 100 Cmax. Over a wide range of concentrations, there was a continuous dose-response relationship for DCF-induced toxicity as assessed by suppression of GCA production (Fig. 6A). Figure 6B demonstrates the effect of LPS alone and together with DCF, showing that there is a synergistic effect of inflammation on drug toxicity. This effect provides subtle early indications of impaired liver function that precedes cell death.
Fig. 5.

Bile acid synthesis. Bile acid synthesis and biotransformation in this LiverChip model may primarily stem from cholesterol metabolism by CYP7A1. Cholic, glycocholic, taurocholic, and glycochinodeoxycholic acids were detected by ultra-HPLC-MS in the medium. A glycine-conjugated bile acid (e.g., GCA) was the most abundant bile acid identified and was characterized as a biomarker in this coculture system. Each well was sampled twice to evaluate technical variation. BA, bile acid.
TABLE 2.
Levels of GCA detected after 48 h
| Bile Acid | Ratio of Bile Acid Provided Compared with GCA Signal | Levels of Detected Bile Acids after 48 h |
||
|---|---|---|---|---|
| 0 μM DCF | 110 μM DCF | 440 μM DCF | ||
| % | ||||
| Cholic acid | 1 | 100 | 66 | 12 |
| Taurocholic acid | 2 | 100 | 77 | 10 |
| GCA | 100 | 100 | 57 | 2 |
| Glycochenodeoxycholic acid | 10 | 100 | 50 | 15 |
| Glycochenodeoxycholic acid 3-glucuronide | 0.2 | 100 | 69 | 14 |
DCF concentrations of 440 µM and 110 µM reduced the relative amounts of bile acids at the earliest time point. At day 5, cholic acid, taurocholic acid, glycochenodeoxycholic acid, and GCA were detected as the major bile acids. GCA was the most abundant bile acid identified in this 3D liver culture. Peak intensity (EIC%) relative values were for guidance only. They do not represent absolute amounts present in these experiments. Bile acid peak areas were compared with GCA peak areas and were found to be lower. GCA production was relatively stable over several days (data not shown).
Fig. 6.

DCF-induced toxicity and the effect of LPS. (A) The microphysiological system was treated with different doses of DCF (0, 44, 110, 220, and 440 µM) and analyzed by the LC-MS/MS method with d5-GCA as the internal standard. GCA was found to be the most prevalent and sensitive small molecule marker and may be a candidate early-indicator biomarker of liver toxicity. (B) Untargeted metabolomics revealed changes in bile acid production when the liver microphysiological system was treated with LPS alone, DCF, and LPS plus DCF only. Data correspond to mean values of two technical replicates.
Secreted Proteins as Toxicity Markers
In most of our study, we focused on high-dose (440 μM) DCF-treated culture medium. Prior to processing the samples, albumin was depleted from the culture medium and then analyzed by shotgun proteomics and quantified by spectral counting. Here, we refer to albumin as BSA provided within commercial medium. All detectable proteins from culture medium were searched against the NCBInr human proteome database (National Center for Biotechnology Information, Bethesda, MD) using Agilent Spectrum Mill. Proteins identified are listed in Supplemental Table 1.
Spectral counting of experimental data indicated a significant difference between samples treated with 440 μM DCF and assessed on day 5 compared with day 7 (Fig. 7, A and B). Fifty-four proteins were common between the control and the DCF culture medium on day 5, whereas only 13 proteins were different between the control and DCF-treated samples (Fig. 7A). In contrast, the Venn diagram in Fig. 7B illustrates that 52 proteins were common between the control and DCF-treated samples on day 7. Notably, the high dose of DCF treatment elevated an additional 68 proteins, which were not identified in the control. However, between day 5 and day 7 of dosed culture medium, nearly 53 proteins were common and nearly 67 proteins were highly elevated on day 7 (Fig. 7C).
Fig. 7.

Venn diagrams summarize the proteins identified in the liver bioreactor. (A) The overlapping proteins identified in Day 5 co-culture medium between Control and DCF treatment. Nearly 54 proteins are common between the two sets. (B) On Day 7, the treatment of DCF elevates the secretion of proteins in the hepatocytes-Kupffer cells co-culture. (C) The comparison of Day 5 and Day 7 drug treated sample indicates the higher number of proteins identified in the Day 7 co-culture conditioned medium. (D) Overview of the function of secreted proteins identified in hepatocytes cultured medium by DCF treatment based on relevant functional process (GO terms). (E) The cellular location of secreted proteins.
The pie chart in Fig. 7D shows that nearly 66% of the proteins were released into the medium, most of which are regulated by the intrinsic apoptotic-signaling pathway in response to oxidative stress. Another 24% are from the cellular aldehyde metabolic process. Of the 120 hepatic proteins identified in the culture medium, 45% were intracellular or membrane proteins, 21% were plasma proteins, 12% were extracellular membrane proteins, and 22% were identified as miscellaneous (Fig. 7E).
Functional Analysis by Assessing Secreted Proteins under Inflammatory Conditions
Secreted proteins under LPS-induced inflammation were identified and quantified using iTRAQ-based LC-MS/MS (Supplemental Table 4). The selected positive and negative APPs are listed in Table 1. Figure 8 shows the Venn diagrams of the major APPs (19 selected proteins) compared among three different conditions at day 7. Sixteen APPs were common between the control and LPS samples, with only three additional proteins evoked during LPS treatment (Fig. 8A). Between the control and drug treatment samples, only seven APPs were common between the two sets (Fig. 8B), but the high concentration of drug completely suppressed the secretion of APPs. In comparison, between LPS and LPS with DCF, only eight APPs were common to the two treatments, and another 12 were present in only LPS-treated conditioned medium.
Fig. 8.

Venn diagrams summarize selected APPs (only 19, shown in Table 1) identified in the liver bioreactor on Day 7. (A) The comparison of Control and LPS treatment indicates 16 proteins are common between two sets. (B) Between the Control and Drug treatment, only 7 APPs are common, and the remaining APPs are present only in Control. (C) Overlapping of LPS and LPS with DCF, indicates predominance of APPs only in the LPS treated co-culture medium.
Quantitative values of the selected APPs are listed in Table 1. The drug and LPS treatment samples were normalized to control samples. LPS with drug treatment samples were normalized to only LPS-treated conditioned medium. Treatment with LPS increased production of all the APPs, but most of the APP secretion was decreased by the addition of drug. Of the identified proteins in LPS-treated conditioned medium, approximately 50% were secreted into the medium due to the acute phase response after LPS treatment. Another 23% were associated with high-density lipoprotein particle clearance. The GO cellular component analysis indicated that these proteins are derived from the extracellular matrix and region, membrane, and organelle compartment of the cells.
LPS-Mediated Cytokine Profiles in Hepatocyte-Kupffer Cell Cocultures
By incorporating components of the innate immune system (i.e., Kupffer cells), we determined cytokine profiles of LPS-induced inflammation using an in vitro model that is more physiologically relevant than hepatocyte monocultures. To investigate the coherent “signature” of inflammation due to this specific stimulus, multiplexed bead-based immunoassays enabled the assessment of 62 unique cytokines, chemokines, growth factors, and metalloproteinases; 31 secreted factors were greater than the limit of detection (Supplemental Table 2). In the absence of LPS stimulation, fewer cytokines, chemokines, and growth factors were detectable in the culture medium and were therefore eliminated for further analyses (Supplemental Table 2). For proteins detectable in all conditions, concentrations (in picograms per milliliter) were normalized by total protein to account for well-to-well variability on the LiverChip; experiments (±1 μg/ml LPS) were carried out in duplicate.
Multivariate analysis identified a cohort of 11 proinflammatory cytokines that correlate with LPS-induced inflammation, including TNF-α, RANTES (regulated on activation normal T cell expressed and secreted), granulocyte colony-stimulating factor (G-CSF), IL-8, IL-6, macrophage colony-stimulating factor (M-CSF), IL-1β, macrophage inflammatory protein (MIP)-1β, IP-10, IL-5, and IL-1Ra. Unsupervised hierarchical clustering distinguishes these secreted factors while emphasizing reproducibility between experimental replicates (Fig. 9A). Consistent with recent findings, the treatment of cocultures (10:1, hepatocytes/Kupffer cells) with LPS for 24 hours resulted in substantially higher release of proinflammatory cytokines (TNF-α, IL-8, IL-6, and IL-1β) into the culture medium compared with untreated cocultures (Fig. 9B) (Nguyen et al., 2015). We further identified significant increases in secretion of RANTES, G-CSF, and Eotaxin-3 and, to a lesser degree, MIP-1α, M-CSF, MIP-1β, IL-5, and IP-10 (Fig. 9, B and C). This in-depth characterization of molecular signatures differentiated a distinct cytokine profile associated with decreased levels of select analytes that was reproducible among experimental replicates (Fig. 9D). Figure 9E shows the fold-change values for each analyte based on the LPS-stimulated or unstimulated results.
Fig. 9.

LPS-induced inflammation profiles of secreted factors in cocultures of Kupffer cells to hepatocytes (1:10). (A) Unsupervised hierarchical clustering of cytokines, chemokines, and growth factors assessed at 24 hours (day 7) in the presence or absence of 1 μg/ml LPS. Cultured medium of two replicates were sampled (rows) for 31 signaling factors (columns) detected above background levels. Secreted factor concentrations were mean-centered for comparison. This analysis confirmed elevated cytokine levels corresponding to LPS induction (red intensity values and dendrogram) and further identified a distinct profile associated with decreased levels of select analytes (blue intensity values and dendrogram) reproduced between replicates. (B) Secreted factors strongly elevated by LPS-induced inflammation (15.8 ≥ fold-change ≥ 6.2) due to LPS-induced inflammation. (C) Secreted factors mildly elevated by LPS-induced inflammation (2.7 ≥ fold-change ≥ 2.0). (D) Secreted factors decreased by LPS-induced inflammation (0.6 ≥ fold-change ≥ 0.3). (E) Fold-change presented for all 31 signaling factors detected. Full set of changes are shown in Supplemental Fig. 1. FGF, fibroblast growth factor; I-TAC, interferon-inducible T-cell alpha chemoattractant; MCP, monocyte chemoattractant protein; MDC, macrophage-derived chemokine; VEGF, vascular endothelial growth factor.
Discussion
As previously observed for this bioreactor (Wheeler et al., 2014; Long et al., 2016), albumin, CYP3A, total protein, glucose, and urea production were stable in LiverChip cultures, indicating viability, survival, and preservation of the hepatic CYP3A4 isoforms that were previously shown to be active on days 7–10 in cocultures (Sarkar et al., 2015). Since drug–plasma protein binding is a major factor influencing the bioavailability of DCF, the binding kinetics between DCF and albumin from media were measured, revealing significant effects on DCF half-life in the range of 1.25–25 mg/ml BSA.
We also confirmed that there was insignificant loss of DCF due to adsorption to the bioreactor components, in contrast with what was observed in a recent bioreactor study of DCF fate (Knöspel et al., 2016). This enabled quantitative analysis of DCF fate when measurements are combined with detailed models of plasma protein binding. As we and others have reviewed comprehensively elsewhere (LeCluyse et al., 2012; Godoy et al., 2013; Ebrahimkhani et al., 2014), the added complexity and expense of bioreactor culture make it desirable for relatively challenging problems in which long-term function and cocultures are required to illuminate responses. An earlier study of hepatocyte–Kupffer cell interactions in DCF toxicity, using standard two-dimensional culture, illustrated the rapid decline in function of primary cells and limited the ability to carry out coexposures and to examine the comprehensive range of responses we were able to study here (Messner et al., 2013).
DCF metabolites, including 4-hydroxy and 5-hydroxy DCF as major and minor phase I metabolites, respectively, and acylglucuronides of DCF, hydroxy-DCF, and DCF-sulfate as major phase II metabolites, were observed (Fig. 4). These metabolites were accounted for by CYP2C9, CYP2C8, and UGT2B7 metabolism; however, four glucuronide isomers of DCF and hydroxy-DCF were observed under our assay conditions, which may suggest the potential involvement of other isoform phase II enzymes. The formation of 4-hydroxy-DCF is mediated by CYP2C9; both DCF and its oxidative metabolites undergo glucuronidation and sulfation (Boelsterli, 2003; Tang, 2003).
LPS can alter gene expression, whereas combinations of LPS with DCF may further regulate concentrations of proinflammatory cytokines and markers of cell death. Under our conditions, DCF plus LPS exhibited a lower half-life; this supports the hypothesis that stress can induce oxidative stress–mediated pathways, which are independent of the formation of electrophilic metabolites, and play a role in drug metabolism.
Although polarized and well differentiated hepatocytes can synthesize physiologically relevant conjugated bile acids, their synthesis could be limited by oxidation and stress. Bile acids are synthesized primarily from cholesterol by CYP7A1, and all bile acids are conjugated and actively excreted (Schwarz et al., 2001). These conjugated bile acids can be up taken by hepatocytes as part of bile acid transport in a physiologically relevant in vitro model (Mörk et al., 2016). We characterized the level of bile acids as a function of DCF treatment and a glycine-conjugated bile acid (GCA) was found to be a marker for DCF dose-dependent early toxicity. The decrease in bile acid synthesis correlated with the suppression of cholesterol 7α-hydroxylase (CYP7A1).
To achieve greater depth in characterizing these models, we took a quantitative MS approach and used several methods to reduce the inherent complexity of the conditioned medium before processing the samples for proteomic analysis. Two high-abundant proteins (albumin and IgG) were removed prior to sample processing. Within the networks of closely associated proteins, differentially expressed genes were explored using Cytoscape analysis with ClueGO, especially those associated with toxicity and acute phase response. The number of proteins identified at 100 Cmax (Cmax = 4.4 µM for this study) in the early stage of drug treatment was relatively small (Fig. 7A). At day 7, however, increased numbers of proteins were identified in the DCF-treated samples (Fig. 7, B and C; Supplemental Table 1).
This finding indicates a delay of the toxic response from day 5 to day 7, as measured by protein leakage. A toxic concentration of DCF caused significant cell death accompanied by leakage of numerous cytoplasmic proteins. GO annotation revealed that these proteins were distributed across different cellular components and were secreted due to oxidative stress (Fig. 7, D and E) and that few of them are involved in the metabolic process. Known markers of hepatotoxicity, such as alcohol dehydrogenase 4, aspartate aminotransferase, aldo-keto reductases, and Cu/Zn-superoxide dismutase, were detected at day 7 with higher doses of DCF. Some metabolic enzymes such as carbonic anhydrase, glucose-6-phosphate isomerase, glutathione-S-transferase, and protein disulfide isomerase were also identified on day 7. Our data also suggest that the hepatocytes were actively remodeling their environment, since we identified several structural extracellular matrix proteins as well as some proteins known to be secreted during liver regeneration.
LPS was added to the circulating media to elicit an inflammatory response resulting in increased protein levels of many acute-phase proteins. These proteins were initially confirmed by shotgun proteomics and then quantitated using iTRAQ labeling. Identified proteins belong to the medium-to-high abundance APPs and were used to compare protein synthesis in different experiments. iTRAQ analysis of coculture conditioned medium revealed the major reduction in overall APPs synthesis by the addition of DCF (Fig. 8, B and C; Table 1), with the exception of amyloid β A4 protein isoform b. Interestingly, there is significant difference between the LPS and drug treatment on the expression of the APPs (Table 1). We expected that the DCF exposure to the hepatocytes would increase the expression of APPs, but that was not the case. For example, exposure of carbon tetrachloride to rats decreases the expression of α2-macroglobulin (Fountoulakis et al., 2002), suggesting that a decrease in APP could be a marker of toxicity. It is possible that DCF causes toxicity through the downregulation of these proteins by leading to uncontrolled breakdown of liver tissue. That may be a reason why genetic deficiency in a1-antitrypsin is a risk factor in development of hepatocellular carcinoma (Blum 2002). Drug-treated cells did not resume production of these proteins even after exposure to LPS, supporting the results from other studies using proteomic-based measurements, where drug treatment affects the APP synthesis in hepatocytes cultured in collagen sandwiches. Nearly 50% of proteins released into the medium at day 7 are involved in the LPS-related acute phase response. The other half are involved in high-density lipoprotein particle clearance, negative regulation of endopeptidase and oxidoreductase activity, karyocyte differentiation, retinol homeostasis, and protein stabilization. Subcellular localization analyses revealed that most proteins are localized in the membrane and nucleus.
Proinflammatory cytokines such as TNF-α, 1L-1β, and IL-6 can induce acute and chronic liver damage. After high drug doses or long-term repeated therapeutic doses, TNF-α, 1L-1β, and IL-6 are released into the bloodstream from the liver during drug-induced hepatic injury. We evaluated the differential regulation of cytokine secretion in the culture media due to high dose of drug-induced injury and whether these profiles were potential biomarkers of in vivo human liver drug induced toxicity.
In the presence of Kupffer cells, we detected 31 of 62 cytokines whose profiles were assessed in the absence or presence of LPS at 24 hours. Eleven pro-inflammatory cytokines correlate with LPS-induced inflammation: TNF-α, RANTES, G-CSF, IL-8, IL-6, M-CSF, IL-1β, MIP-1β, IP-10, IL-5, and IL-1Ra. We also identified increases in the secretion of RANTES, G-CSF, and Eotaxin-3 and, to a lesser degree, MIP-1α, M-CSF, MIP-1β, IL-5, and IP-10.
The proinflammatory effect of IL-1β is likely due to its synergism with Toll-like receptor signaling, which markedly amplifies inflammation via LPS-inducible cytokines (Szabo and Petrasek, 2015). LPS signals through Toll-like receptor 4 and appears to be the initial signal that induces IL-1β expression (Miura et al., 2010; Petrasek et al., 2012). The secretion of IL-1β is specific to Kupffer cells (Petrasek et al., 2012) and mediates cytokines and chemokines including TNF-α and monocyte Chemoattractant Protein-1 (Granowitz et al., 1992; Dinarello 2009; Mandrekar et al., 2011), respectively, and recruits inflammatory cells to the liver during disease progression (Mehal and Imaeda, 2010). In addition to IL-1β, elevated signals produced by Kupffer cells were observed for MIP-1β and IL-8. Taken together, cytokine release data confirmed that Kupffer cells are present and functional at day 7 (i.e., 24 hours after LPS induction).
Collectively, noninvasive functional analyses (e.g., LDH release, total protein per well, albumin secretion, phase I and II biotransformation, cytokine profiling, and proteomics-based toxicity results) demonstrated that our system is capable of recapitulating DCF metabolism and escalated dose-induced toxicity in the human liver. This platform can be a valuable tool in the different phases of the drug development processes (fit for purpose utility) to predict in vivo drug biotransformation, PK, and drug-induced hepatotoxicity (i.e., adverse effects). Each small molecule, however, may behave differently in vivo from the perspectives of PK, pharmacodynamics, and types of diseases. Additional experimental optimizations, based on low-, moderate-, and high-clearance compounds, are consequently needed to develop a more accurate and predictive human cell–based in vitro microphysiological system.
Acknowledgments
The authors thank the Defense Advance Research Project Agency Microphysiological Barrier-Immune-Organ: Microphysiology, Microenvironment Engineered Tissue Construct Systems team for general technical advice. The authors acknowledge Dr. Douglas Ferguson from AstraZeneca for helpful discussion on the in vitro/in vivo clearance of DCF.
Abbreviations
- 3D
three-dimensional
- ABC
ammonium bicarbonate
- ACN
acetonitrile
- APP
acute phase protein
- BSA
bovine serum albumin
- CAS
Chemical Abstracts Service
- CL
clearance
- DCF
diclofenac
- DMSO
dimethylsulfoxide
- DTT
dl-dithiothreitol
- EIC
extracted ion chromatogram
- ESI
electrospray ionization
- FA
formic acid
- G-CSF
granulocyte colony-stimulating factor
- GCA
glycocholic acid
- GO
Gene Ontology
- HPLC
high-performance liquid chromatography
- IAA
iodoacetamide
- IL
interleukin
- LC
liquid chromatography
- LDH
lactate dehydrogenase
- LPS
lipopolysaccharide
- m/z
mass-to-charge ratio
- M-CSF
macrophage colony-stimulating factor
- MeOH
methanol
- MIP
macrophage inflammatory protein
- MS
mass spectrometry
- MS/MS
tandem mass spectrometry
- PK
pharmacokinetics
- QTOF
quadrupole time of flight
- RANTES
regulated on activation normal T cell expressed and secreted
- t1/2
half-life
- TNF
tumor necrosis factor
- UGT
UDP-glucuronosyltransferase
- WEM
William’s E medium
Authorship Contributions
Participated in research design: Sarkar, Ravindra, Large, Young, Yu, Cirit, Hughes, Wishnok, Lauffenburger, Griffith, Tannenbaum.
Conducted experiments: Sarkar, Ravindra, Large, Young.
Performed data analysis: Sarkar, Ravindra, Young, Rivera-Burgos, Yu, Cirit, Hughes, Wishnok.
Wrote or contributed to the writing of the manuscript: Sarkar, Ravindra, Young, Rivera-Burgos, Yu, Cirit, Hughes, Wishnok, Griffith, Tannenbaum.
Footnotes
This research was supported by the U.S. Defense Advanced Research Projects Agency [Grant W911NF-12-2-0039], the National Institutes of Health National Center for Advancing Translational Sciences [Grant 5-UH2-TR000496], and the Massachusetts Institute of Technology Center for Environmental Health Sciences via the National Institutes of Health National Institute of Environmental Health Sciences [Grant P30-ES002109].
 This article has supplemental material available at dmd.aspetjournals.org.
This article has supplemental material available at dmd.aspetjournals.org.
References
- Axelson M, Ellis E, Mörk B, Garmark K, Abrahamsson A, Björkhem I, Ericzon BG, Einarsson C. (2000) Bile acid synthesis in cultured human hepatocytes: support for an alternative biosynthetic pathway to cholic acid. Hepatology 31:1305–1312. [DOI] [PubMed] [Google Scholar]
- Bindea G, Mlecnik B, Hackl H, Charoentong P, Tosolini M, Kirilovsky A, Fridman WH, Pagès F, Trajanoski Z, Galon J. (2009) ClueGO: a Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics 25:1091–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blum HE. (2002) Molecular targets for prevention of hepatocellular carcinoma. Dig Dis 20:81–90. [DOI] [PubMed] [Google Scholar]
- Boelsterli UA. (2003) Diclofenac-induced liver injury: a paradigm of idiosyncratic drug toxicity. Toxicol Appl Pharmacol 192:307–322. [DOI] [PubMed] [Google Scholar]
- Bort R, Macé K, Boobis A, Gómez-Lechón MJ, Pfeifer A, Castell J. (1999) Hepatic metabolism of diclofenac: role of human CYP in the minor oxidative pathways. Biochem Pharmacol 58:787–796. [DOI] [PubMed] [Google Scholar]
- Brogden RN, Heel RC, Pakes GE, Speight TM, Avery GS. (1980) Diclofenac sodium: a review of its pharmacological properties and therapeutic use in rheumatic diseases and pain of varying origin. Drugs 20:24–48. [DOI] [PubMed] [Google Scholar]
- Brown HS, Griffin M, Houston JB. (2007) Evaluation of cryopreserved human hepatocytes as an alternative in vitro system to microsomes for the prediction of metabolic clearance. Drug Metab Dispos 35:293–301. [DOI] [PubMed] [Google Scholar]
- Dash A, Inman W, Hoffmaster K, Sevidal S, Kelly J, Obach RS, Griffith LG, Tannenbaum SR. (2009) Liver tissue engineering in the evaluation of drug safety. Expert Opin Drug Metab Toxicol 5:1159–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies B, Morris T. (1993) Physiological parameters in laboratory animals and humans. Pharm Res 10:1093–1095. [DOI] [PubMed] [Google Scholar]
- Dinarello CA. (2009) Immunological and inflammatory functions of the interleukin-1 family. Annu Rev Immunol 27:519–550. [DOI] [PubMed] [Google Scholar]
- Domansky K, Inman W, Serdy J, Dash A, Lim MH, Griffith LG. (2010) Perfused multiwell plate for 3D liver tissue engineering. Lab Chip 10:51–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutta SK, Basu SK, Sen KK. (2006) Binding of diclofenac sodium with bovine serum albumin at different temperatures, pH and ionic strengths. Indian J Exp Biol 44:123–127. [PubMed] [Google Scholar]
- Ebrahimkhani MR, Neiman JA, Raredon MS, Hughes DJ, Griffith LG. (2014) Bioreactor technologies to support liver function in vitro. Adv Drug Deliv Rev 69-70:132–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finney DJ. (1987) Statistical Method in Biological Assay, 3rd ed, Griffin, London. [Google Scholar]
- Fountoulakis M, de Vera MC, Crameri F, Boess F, Gasser R, Albertini S, Suter L. (2002) Modulation of gene and protein expression by carbon tetrachloride in the rat liver. Toxicol Appl Pharmacol 183:71–80. [DOI] [PubMed] [Google Scholar]
- Godoy P, Hewitt NJ, Albrecht U, Andersen ME, Ansari N, Bhattacharya S, Bode JG, Bolleyn J, Borner C, Böttger J, et al. (2013) Recent advances in 2D and 3D in vitro systems using primary hepatocytes, alternative hepatocyte sources and non-parenchymal liver cells and their use in investigating mechanisms of hepatotoxicity, cell signaling and ADME. Arch Toxicol 87:1315–1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gómez-Lechón MJ, Donato MT, Castell JV, Jover R. (2004) Human hepatocytes in primary culture: the choice to investigate drug metabolism in man. Curr Drug Metab 5:443–462. [DOI] [PubMed] [Google Scholar]
- Granowitz EV, Clark BD, Vannier E, Callahan MV, Dinarello CA. (1992) Effect of interleukin-1 (IL-1) blockade on cytokine synthesis: I. IL-1 receptor antagonist inhibits IL-1-induced cytokine synthesis and blocks the binding of IL-1 to its type II receptor on human monocytes. Blood 79:2356–2363. [PubMed] [Google Scholar]
- Hallifax D, Foster JA, Houston JB. (2010) Prediction of human metabolic clearance from in vitro systems: retrospective analysis and prospective view. Pharm Res 27:2150–2161. [DOI] [PubMed] [Google Scholar]
- Huang W, Sherman BT, Lempicki RA. (2009) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4:44–57. [DOI] [PubMed] [Google Scholar]
- Kegel V, Pfeiffer E, Burkhardt B, Liu JL, Zeilinger K, Nüssler AK, Seehofer D, Damm G. (2015) Subtoxic concentrations of hepatotoxic drugs lead to Kupffer cell activation in a human in vitro liver model: an approach to study DILI. Mediators Inflamm 2015:640631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knöspel F, Jacobs F, Freyer N, Damm G, De Bondt A, van den Wyngaert I, Snoeys J, Monshouwer M, Richter M, Strahl N, et al. (2016) In vitro model for hepatotoxicity studies based on primary human hepatocyte cultivation in a perfused 3D bioreactor system. Int J Mol Sci 17:584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kostrzewski T, Cornforth T, Snow SA, Ouro-Gnao L, Rowe C, Large EM, Hughes DJ. (2017) Three-dimensional perfused human in vitro model of non-alcoholic fatty liver disease. World J Gastroenterol 23:204–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeCluyse EL, Witek RP, Andersen ME, Powers MJ. (2012) Organotypic liver culture models: meeting current challenges in toxicity testing. Crit Rev Toxicol 42:501–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li AP. (2007) Human hepatocytes: isolation, cryopreservation and applications in drug development. Chem Biol Interact 168:16–29. [DOI] [PubMed] [Google Scholar]
- Long TJ, Cosgrove PA, Dunn RT, 2nd, Stolz DB, Hamadeh H, Afshari C, McBride H, Griffith LG. (2016) Modeling therapeutic antibody-small molecule drug-drug interactions using a three-dimensional perfusable human liver coculture platform. Drug Metab Dispos 44:1940–1948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandrekar P, Ambade A, Lim A, Szabo G, Catalano D. (2011) An essential role for monocyte chemoattractant protein-1 in alcoholic liver injury: regulation of proinflammatory cytokines and hepatic steatosis in mice. Hepatology 54:2185–2197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehal W, Imaeda A. (2010) Cell death and fibrogenesis. Semin Liver Dis 30:226–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messner S, Agarkova I, Moritz W, Kelm JM. (2013) Multi-cell type human liver microtissues for hepatotoxicity testing. Arch Toxicol 87:209–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miura K, Kodama Y, Inokuchi S, Schnabl B, Aoyama T, Ohnishi H, Olefsky JM, Brenner DA, Seki E. (2010) Toll-like receptor 9 promotes steatohepatitis by induction of interleukin-1beta in mice. Gastroenterology 139:323–34.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mörk LM, Strom SC, Mode A, Ellis EC. (2016) Addition of dexamethasone alters the bile acid composition by inducing CYP8B1 in primary cultures of human hepatocytes. J Clin Exp Hepatol 6:87–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen TV, Ukairo O, Khetani SR, McVay M, Kanchagar C, Seghezzi W, Ayanoglu G, Irrechukwu O, and Evers R (2015) Establishment of a hepatocyte-Kupffer cell co-culture model for assessment of proinflammatory cytokine effects on metabolizing enzymes and drug transporters. Drug Metab Dispos 43:774–785. [DOI] [PubMed] [Google Scholar]
- Obach RS. (1999) Prediction of human clearance of twenty-nine drugs from hepatic microsomal intrinsic clearance data: an examination of in vitro half-life approach and nonspecific binding to microsomes. Drug Metab Dispos 27:1350–1359. [PubMed] [Google Scholar]
- Obach RS, Baxter JG, Liston TE, Silber BM, Jones BC, MacIntyre F, Rance DJ, Wastall P. (1997) The prediction of human pharmacokinetic parameters from preclinical and in vitro metabolism data. J Pharmacol Exp Ther 283:46–58. [PubMed] [Google Scholar]
- Petrasek J, Bala S, Csak T, Lippai D, Kodys K, Menashy V, Barrieau M, Min SY, Kurt-Jones EA, Szabo G. (2012) IL-1 receptor antagonist ameliorates inflammasome-dependent alcoholic steatohepatitis in mice. J Clin Invest 122:3476–3489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravindra KC, Ho WE, Cheng C, Godoy LC, Wishnok JS, Ong CN, Wong WS, Wogan GN, Tannenbaum SR. (2015) Untargeted proteomics and systems-based mechanistic investigation of artesunate in human bronchial epithelial cells. Chem Res Toxicol 28:1903–1913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts RA, Ganey PE, Ju C, Kamendulis LM, Rusyn I, Klaunig JE. (2007) Role of the Kupffer cell in mediating hepatic toxicity and carcinogenesis. Toxicol Sci 96:2–15. [DOI] [PubMed] [Google Scholar]
- Sarkar U, Rivera-Burgos D, Large EM, Hughes DJ, Ravindra KC, Dyer RL, Ebrahimkhani MR, Wishnok JS, Griffith LG, Tannenbaum SR. (2015) Metabolite profiling and pharmacokinetic evaluation of hydrocortisone in a perfused three-dimensional human liver bioreactor. Drug Metab Dispos 43:1091–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz M, Russell DW, Dietschy JM, Turley SD. (2001) Alternate pathways of bile acid synthesis in the cholesterol 7alpha-hydroxylase knockout mouse are not upregulated by either cholesterol or cholestyramine feeding. J Lipid Res 42:1594–1603. [PubMed] [Google Scholar]
- Skoutakis VA, Carter CA, Mickle TR, Smith VH, Arkin CR, Alissandratos J, Petty DE. (1988) Review of diclofenac and evaluation of its place in therapy as a nonsteroidal antiinflammatory agent. Drug Intell Clin Pharm 22:850–859. [DOI] [PubMed] [Google Scholar]
- Szabo G, Petrasek J. (2015) Inflammasome activation and function in liver disease. Nat Rev Gastroenterol Hepatol 12:387–400. [DOI] [PubMed] [Google Scholar]
- Tang W. (2003) The metabolism of diclofenac--enzymology and toxicology perspectives. Curr Drug Metab 4:319–329. [DOI] [PubMed] [Google Scholar]
- Tsamandouras N, Kostrzewski T, Stokes CL, Griffith LG, Hughes DJ, and Cirit M (2017) Quantitative assessment of population variability in hepatic drug metabolism using a perfused three-dimensional human liver microphysiological system. J Pharmacol Exp Ther 360:95–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler SE, Clark AM, Taylor DP, Young CL, Pillai VC, Stolz DB, Venkataramanan R, Lauffenburger D, Griffith L, Wells A. (2014) Spontaneous dormancy of metastatic breast cancer cells in an all human liver microphysiologic system. Br J Cancer 111:2342–2350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Lee P, Liang S, Zhou Z, Wu X, Yang F, Liang H. (2015) Structural basis of non-steroidal anti-inflammatory drug diclofenac binding to human serum albumin. Chem Biol Drug Des 86:1178–1184. [DOI] [PubMed] [Google Scholar]