

Abstract
Background
Glucose 6-phosphate dehydrogenase (G6PD) deficiency is the most common enzyme deficiency in the world. In failing hearts, G6PD is upregulated and generates NADPH that is used by the glutathione pathway to remove reactive oxygen species (ROS), but also as a substrate by ROS-generating enzymes. Therefore, G6PD deficiency might prevent heart failure by decreasing NADPH and ROS production.
Methods and Results
This hypothesis was evaluated in a mouse model of human G6PD deficiency (G6PDX mice, ~40% normal activity). Myocardial infarction with 3 months followup resulted in LV dilation and dysfunction in both WT and G6PDX mice, but significantly greater end diastolic volume and wall thinning in G6PDX mice. Similarly, pressure overload induced by transverse aortic constriction (TAC) for 6 weeks caused greater LV dilation in G6PDX mice than WT. We further stressed TAC mice by feeding a high fructose diet to increase flux through G6PD and ROS production, and again observed worse LV remodeling and a lower ejection fraction in G6PDX than WT mice. Tissue content of lipid peroxidation products was increased in G6PDX mice in response to infarction and aconitase activity was decreased with TAC, suggesting that G6PD deficiency increases myocardial oxidative stress and subsequent damage.
Conclusions
Contrary to our hypothesis, G6PD deficiency increased redox stress in response to infarction or pressure overload. However, we found only a modest acceleration of LV remodeling, suggesting that, in individuals with G6PD deficiency and concurrent hypertension or myocardial infarction, the risk for developing heart failure is higher, but limited by compensatory mechanisms.
Keywords: G6PD, reactive oxygen species, myocardial infarction, pressure overload, high fructose diet
Glucose 6-phosphate dehydrogenase (G6PD) controls the flux into the pentose phosphate pathway, and is the main cytoplasmic source of NADPH1, 2. Generation of NADPH by G6PD fuels antioxidant systems and thus decreases reactive oxygen species (ROS)3, 4. G6PD deficiency is the most common enzyme deficiency in the world, affecting over 400 million people worldwide and conferring resistance to malaria5, 6. It is prevalent in sub-Saharan Africa, Mediterranean countries and in Southeastern Asia. Alleles for mild to moderate severity deficiency result in 10-60% of normal G6PD activity7. The effects of G6PD deficiency on the risk for cardiovascular disease and subsequent outcomes are unclear. The limited available evidence suggests that G6PD deficiency may reduce the risk of heart disease and cardiovascular associated death, though a thorough evaluation in a large patient cohort has not been conducted8, 9.
At present, the impact of G6PD deficiency on the development and progression of heart failure is unclear, as G6PD has both pro- and anti-oxidant effects. Bing et al found that G6PD activity was increased in human myocardium following acute myocardial infarction10, and more recently we found that dogs and patients with heart failure have elevated myocardial G6PD activity and [NADPH]1, 11. This could be a compensatory mechanism necessary to oppose the increased generation of reactive oxygen species (ROS) in failing myocardium, as suggested by the observation that G6PD inhibition in cardiomyocytes compromises antioxidant capacity and increases sensitivity to ROS induced cell death3, 12, 13. G6PD deficient (G6PDX) mice develop normally, but present with moderate cardiac hypertrophy at 9 months of age3. Myocardial ischemia/reperfusion causes greater oxidative stress and injury in G6PDX compared to WT mice3, 4. On the other hand, pharmacological inhibition of G6PD decreased ROS generation in myocardial homogenates from failing hearts, suggesting that G6PD fuels NADPH dependent oxidant enzymes in advanced heart failure1, 11. Potential benefits of G6PD deficiency have also been reported on the vasculature14, 15, however no studies have addressed the effects of chronic suppression of G6PD on the development and/or progression of heart failure8, 9.
The goal of the present investigation was to test the hypothesis that G6PD deficiency would protect against the development of heart failure by decreasing NADPH and ROS production in myocardium subjected to injury or to chronic, severe stress. G6PDX mice, which have approximately 40% residual G6PD activity and recapitulate key aspects of clinical deficiency16, were subjected to myocardial infarction caused by left coronary artery ligation, or to transverse aortic constriction (TAC) to induce pressure overload. Alterations in LV mass, chamber remodeling, ROS production and lipid peroxidation were assessed. We further stressed these mice with a high fructose diet to increase flux through pro-oxidant NADPH dependent enzymes17-20. Recent studies from our lab and others have found that high fructose intake exacerbated the development of heart failure and accelerated mortality compared to a complex carbohydrate diet in salt-induced hypertension21, 22, TAC17, 23, and volume overload24 models of heart failure. Thus, we further hypothesized that G6PD deficiency would prevent the adverse cardiac effects of high fructose intake in mice subjected to TAC.
Methods
Experimental Design
Infarct Experimental Design
Sixteen week-old male mice underwent permanent coronary occlusion or sham surgery. Cardiac function was assessed by echocardiography at 11 weeks, and by intraventricular catheterization during euthanization at 12 weeks after surgery.
TAC Experimental Design
Ten week-old male mice underwent TAC or sham surgery. TAC was performed using a 28 gauge needle. Echocardiography was performed 1-2 days prior to euthanization at 6 weeks after surgery.
Fructose/TAC Experimental Design
Eight week-old male mice underwent sham or TAC surgery with a 27 gauge needle, and were placed on a high starch diet or high fructose diet which consisted of 60% of energy intake from fructose on the following day. Blood pressure was measured by tail cuff plethysmography at 10 weeks after surgery, echocardiography was performed at 16 weeks, and mice were euthanized at 17 weeks for biochemical analysis.
Animal Treatment
Male G6PDX mice were generated previously on a C3H/HeJ background by Pretsch et al16. G6PDX and WT littermates were transferred from Dr. Leopold’s laboratory at Harvard Medical School to the University of Maryland, Baltimore, genotyped as previously described4, and used as founders for separate colonies of G6PDX mice and WT mice. Subsequent offspring of the 3rd to 5th generations were used in our experiments. Animals were fed ad libitum and maintained on a 12 hour light/dark cycle. In the first two series, mice were fed a standard lab chow which consisted of 18% of energy from fat, 58% carbohydrate, and 24% protein (#2018SX, Teklad Diets, Indianapolis, IN). Animals in the fructose/TAC study were fed custom manufactured purified diets (See Supplementary Material) (Research Diets Inc, New Brunswick, NJ). All procedures were conducted in accordance with the Guidelines for the Care and Use of Laboratory Animals (NIH publication No. 85-23), and were approved by the University of Maryland Animal Care and Use Committee.
Mouse Surgery
For all surgical procedures, mice were anesthetized with 2% isoflurane in oxygen and ventilated at a tidal volume of 0.30 mL at 80 breaths / minute (Harvard Apparatus Inspira, Holliston, MA). Isoflurane was vaporized using a SurgiVet model 100 vaporizer from Atlantic Biomedical (Baltimore, MD).
Myocardial Infarction
Infarction was induced by permanent left anterior descending (LAD) occlusion. Anesthetized mice were placed in a position in which they were lying on their side on a warm heating pad. The ribs were separated, and a tapered 7-0 silk suture was used to tie off the LAD. The ribs and skin were then each closed with 6-0 silk. Sham animals were subjected to the same procedure except that the LAD was not occluded. Animals were allowed to recover for 3-5 minutes with 100% oxygen immediately after surgery, and then placed on a warm heating pad for full recovery.
Transverse Aortic Constriction
Pressure overload was induced by TAC. Anesthetized mice were placed in the supine position on a warm heating pad. A 27 gauge (17 week series) or 28 gauge needle (6 week series) was placed next to the transverse aorta, and a 6-0 silk suture was tied around both the aorta and the needle, and the needle was removed. The muscle and skin were closed separately with 6-0 silk suture. Sham animals were subjected to the same surgical procedure except that the aorta was not constricted. Animals were allowed to recover for 3-5 minutes with 100% oxygen immediately after surgery, and then placed on a warm heating pad.
Tissue Harvest
Mice were anesthetized with 2.5% isoflurane, and the rib cage was opened. A small 20-30mg piece of the LV free wall was quickly cut free and snap frozen, weighed, and kept at −80°C for later NADPH or glutathione measurement. Another portion was mounted for histological analysis. The remainder of the heart was dissected in ice-cold phosphate buffered saline, weighed, frozen in liquid nitrogen, and later ground to powder. Blood was drawn from the thorax and centrifuged for 15min at 1500g to obtain serum. The liver, right kidney, and epididymal fat pads were weighed, and the tibia length was measured. All samples were stored at −80°C until subsequent analysis.
Statistical Analysis
Continuous data are presented as mean±standard error, and categorical data is presented as percentages. Comparisons between strain and treatment groups were made using a two-way ANOVA. The Holm-Sidak test for multiple comparisons was used to test for difference between WT/Sham vs WT/infarct (or TAC), WT/Sham vs G6PDX/sham, G6PDX/sham vs G6PDX/infarct, and G6PDX/infarct vs WT/infarct. Recovery from surgery was assessed by a Chi-square analysis. Post-surgery survival was estimated using the Kaplan Meier method, and survival among groups was compared using the Log-Rank test. A two tailed t-test was used to compare the difference in infarct size between WT and G6PDX mice. P<0.05 was considered significant.
See Supplemental Material for additional methods.
Results
Myocardial Infarction
Recovery and Survival after Myocardial Infarction
We assessed the effect of G6PD deficiency on heart failure in response to myocardial infarction. A total of 32 WT and 46 G6PDX mice underwent LAD ligation to produce an infarct, while 10 WT and 10 G6PDX mice underwent sham surgery. Post-infarct 24 hour survival was 63% for WT and 61% for G6PDX mice. There was minimal mortality from 1 day to 12 weeks after infarction, with no statistically significant differences among groups (Figure S1). We found no significant differences in body mass among strain and treatment groups at the time of surgery or at the end of the study (Table S1).
G6PD Deficiency Increases LV dilation after Myocardial Infarction
G6PD mRNA and G6PD activity were decreased in G6PDX mice compared to WT mice, and infarction increased myocardial G6PD mRNA and G6PD activity in WT mice, but not in G6PDX mice (Figure 1A-B). Heart mass was increased by LAD ligation, indicating cardiac hypertrophy in both WT and G6PDX mice (Figure 1C). We did not find a statistically significant difference in infarct size between strains (WT: 14.2±1.4% of LV, G6PDX: 16.8±1.2% of LV, p=0.168). Myocardial infarction caused eccentric hypertrophy as seen in the increase in LV end diastolic volume, in both WT and G6PDX mice, however LV dilation was significantly greater with G6PD deficiency (Figure 1D-E; Table S2), reflected by a greater LV end diastolic volume in G6PDX mice compared to WT mice. G6PDX mice also displayed a decrease in relative wall thickness in G6PDX mice, further indicating that G6PD deficiency increased susceptibility to LV dilation (Figure 1F). This corresponded with an increase in myocyte size and a decrease in capillary density in G6PDX mice (Table). Myocardial infarction decreased systolic and diastolic function, as indicated by decreased ejection fraction and dP/dt min, and increased MPI and LV end diastolic pressure (Figure 1G-I; Figure S2). Taken together, G6PD deficiency exacerbated LV dilation, but did not accelerate systolic or diastolic dysfunction after myocardial infarction.
Figure 1. G6PD deficiency exacerbates LV dilation, but does not affect function after myocardial infarction.
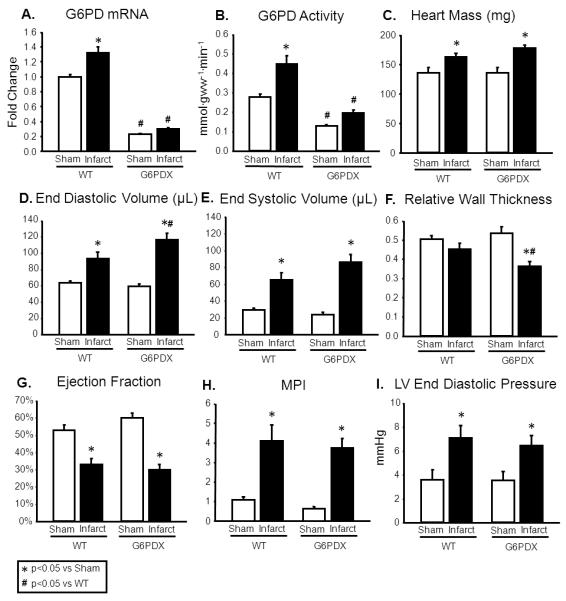
A-B. Myocardial G6PD mRNA expression and enzymatic activity were increased by LAD ligation in WT mice, but not in G6PD deficient mice. C. Myocardial infarction increased heart mass. D-F. Myocardial infarction increased LV end diastolic and end systolic volume. LV end diastolic volume was increased further by G6PD deficiency, and myocardial infarction resulted in decreased relative wall thickness in G6PDX mice, indicating that G6PD deficiency increased susceptibility to dilation. G-I. Myocardial infarction decreased ejection fraction indicating systolic dysfunction, and increased MPI and LV end diastolic pressure indicating diastolic dysfunction. These functional parameters were unaffected by G6PD deficiency. Data were obtained using mice at 11-12 weeks after LAD ligation or sham surgery. For echocardiography measurements, mice were anesthetized with 1.5% isoflurane. *P <0.05 vs. Sham; #P <0.05 vs. WT; Sham n=9-10/group, WT infarct n=19, G6PDX infarct n=27-28.
Table.
Histology - 12 Weeks Post-Infarct
| Parameter | Wild Type | G6PD Deficient | ||
|---|---|---|---|---|
| Sham | Infarct | Sham | Infarct | |
| Myocyte Cross Sectional Area (μm2) | 347 ± 15 | 364 ± 11 | 349 ± 15 | 398 ± 9 * # |
| Capillaries/mm2 | 2275 ± 109 | 2111 ± 79 | 2256 ± 109 | 1928 ± 68 * |
| Volume Fraction Interstitial Fibrosis % | 12.4 ± 0.7 | 12.3 ± 0.5 | 11.2 ± 0.7 | 12.0 ± 0.4 |
Data were obtained using myocardium from mice at 12 weeks after LAD ligation or sham surgery;
P <0.05 vs. Sham;
P <0.05 vs. WT; Sham n=10/group, WT Infarct n=19, G6PDX n=26.
Effects of G6PD Deficiency on Biochemical Parameters after Myocardial Infarction
Myocardial [NADPH] is increased in severe heart failure1, 11, and we saw an increase in myocardial [NADPH] in infarcted G6PDX mice (Figure 2A). Although myocardial superoxide production was unaffected by infarction or G6PD deficiency, infarction increased myocardial lipid peroxidation products in G6PDX mice suggesting increased oxidative stress with G6PD deficiency (Figure 2B-C). Heart failure decreases the capacity for oxidative metabolism in myocardium, as reflected in a decrease in the activities of aconitase, citrate synthase, and MCAD. The activities of these enzymes were decrease by myocardial infarction but were unaffected by G6PD deficiency (Figure 2D-F). Heart failure increases myocardial mRNA expression of the fetal genes ANP and MHCβ, and expression of these genes was increased by infarction but unaffected by G6PD deficiency (Figure 2G-H; Table S3). Taken together, these results suggest that G6PD deficiency increased oxidative stress, but not other indicators of heart failure in response to myocardial infarction.
Figure 2. Infarct - Biochemical Data.
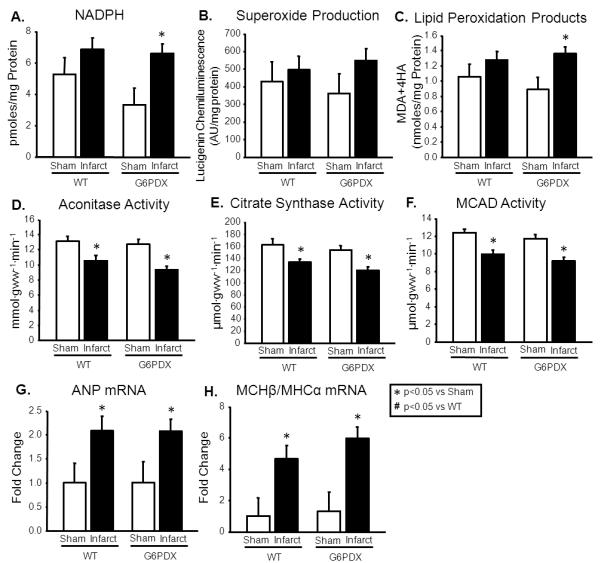
A. Infarction increased myocardial [NADPH] in G6PDX mice. B. Myocardial superoxide production was unaffected by infarction or G6PD deficiency. C. Infarction increased myocardial lipid peroxidation products in G6PDX mice. D-F. Infarction decreased myocardial aconitase activity, citrate synthase activity, and MCAD activity to the same extent in G6PDX and WT mice. G-H. Myocardial ANP and MHCβ/MHCα mRNA were also increased by infarction and unaffected by G6PD deficiency. Data were obtained using myocardium from mice at 12 weeks after LAD ligation or sham surgery. *P <0.05 vs. Sham; #P <0.05 vs. WT; Sham n=9-10/group, WT infarct n=19, G6PDX infarct n=27-28.
Transverse Aortic Constriction
Recovery and Survival after TAC
In addition to myocardial infarction, we assessed the effect of G6PD deficiency on heart failure in response to pressure overload. A total of 28 WT mice and 24 G6PDX mice underwent TAC with a 28 gauge needle to produce severe pressure overload, while 7 WT mice and 7 G6PDX mice underwent sham surgery. TAC 24 hour survival was 39% for WT mice and 54% for G6PDX mice (p=0.43 for difference). Minimal mortality was again observed from 1 day to 6 weeks after TAC, with no significant differences among groups (Figure S3). There were no statistically significant differences in body mass among strain and treatment groups (Table S4).
G6PD Deficiency Increases Propensity for LV Dilation after TAC
TAC increased LV mass to the same extent in WT and G6PDX mice (Figure 3C), however only G6PDX mice had a statistically significant increase in LV end diastolic and end systolic volumes, and absolute wall thickness, (Figure 3D-F; Table S5). This indicates that G6PD deficiency increased the propensity for LV dilation. TAC decreased systolic function, as indicated by decreased ejection fraction, but with no effect of G6PD deficiency (Figure 3G). TAC increased G6PD mRNA and G6PD activity in WT mice, but not in G6PDX mice (Figure 3A-B).
Figure 3. G6PD deficiency increases propensity for LV dilation after TAC.
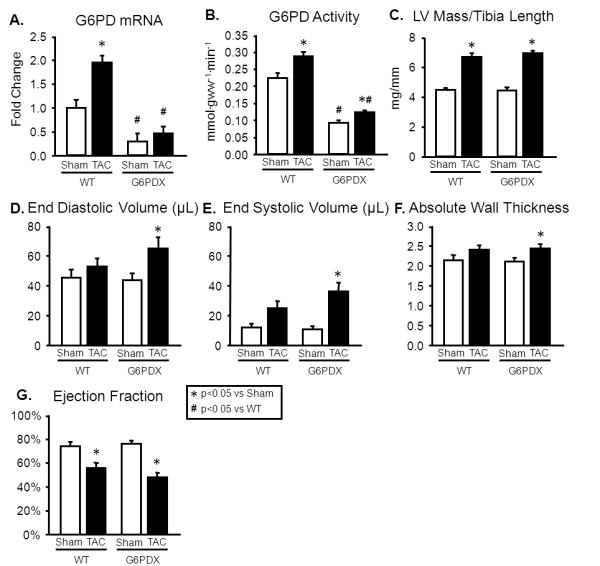
A-B. TAC increased G6PD mRNA and G6PD activity in WT mice, but not in G6PDX mice. C. TAC increased LV mass to the same extent in WT and G6PDX mice. D-F. TAC increased LV end diastolic volume, end systolic volume, and absolute wall thickness in G6PDX mice. G. TAC decreased systolic function to the same extent in G6PDX and WT mice. Data were obtained using mice at 6 weeks after Sham or TAC surgery with a 28 gauge needle. For echocardiography measurements, mice were anesthetized with 1% isoflurane.*P <0.05 vs. Sham; #P <0.05 vs. WT; Sham n=7/group, TAC=10-12/group.
Effects of G6PD Deficiency on Biochemical Parameters after TAC
As with myocardial infarction, [NADPH] was increased in G6PDX mice in response to TAC (Figure 4A). G6PD deficiency decreased superoxide production, but this did not prevent a decrease in GSH in response to TAC in G6PDX mice (Figure 4B-C). There was no effect of G6PD deficiency or TAC on lipid peroxidation products (Figure 4D). TAC decreased myocardial aconitase activity, citrate synthase activity, and MCAD activity to the same extent in G6PDX and WT mice (Figure 4E-G), and these changes corresponded with an increase in ANP and MHCβ/MHCα mRNA (Figure 4H-I; Table S6).
Figure 4. TAC - Biochemical Data.
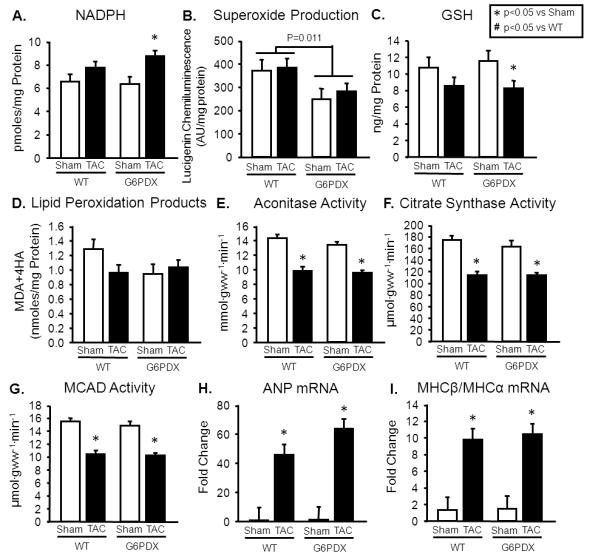
A. LV NADPH was increased by TAC in G6PDX mice. B. Superoxide production was decreased by G6PD deficiency. C. GSH was decreased in response to TAC in G6PDX mice. D. Lipid peroxidation products were unaffected by G6PD deficiency or TAC. E-G. TAC decreased myocardial aconitase activity, citrate synthase activity, and MCAD activity to the same extent in G6PDX and WT mice H-I. TAC increased ANP and MHCβ/MHCα in WT and G6PDX mice to the same extent. Data were obtained using LV myocardium from mice at 6 weeks after Sham or TAC surgery with a 28 gauge needle. *P <0.05 vs. Sham; #P <0.05 vs. WT; Sham n=7/group, TAC=10-12/group.
High Fructose Intake and Transverse Aortic Constriction
Recovery and Survival after Fructose/TAC
We assessed whether the effect of G6PD deficiency on heart failure in response to pressure overload with the additional stress of high fructose intake. A total of 55 WT mice and 47 G6PDX mice underwent moderate TAC with a 27 gauge needle, while 28 WT mice and 29 G6PDX mice underwent sham surgery. Immediately following surgery, mice were assigned to either a high starch or a high fructose diet (Table S7). TAC 24 hour survival was 65% for WT mice and 72% for G6PDX mice (p=0.594). There was minimal mortality from 1 day to 17 weeks after TAC, with no significant differences among groups (Figure S4). There were no statistically significant differences among strain and treatment groups in body mass (Table S8).
G6PD Deficiency Exacerbates the Cardiac Effects of Pressure Overload with High Fructose Intake
TAC increased LV mass in all groups, but to a greater extent in G6PDX mice compared to WT mice after high fructose intake (Figure 5B). Atrial and right ventricular mass were also increased by TAC, with a greater increase in right ventricular mass in G6PDX mice compared to WT mice after high fructose intake (Table S9). TAC resulted in decreased stroke volume and concentric hypertrophy indicated by increased absolute and relative wall thickness (Table S10). G6PD deficient animals displayed systolic dysfunction, as seen by an increase in LV end systolic volume and a decrease in ejection fraction in response to TAC after high fructose intake (Figure 5C). TAC increased LV fetal gene expression, as indicated by increased MHCβ/MHCα and ANP mRNA in all groups (Figure 5D; Table S11). The greatest ANP expression was in G6PDX mice with the high fructose diet. TAC also increased left ventricular G6PD activity in WT mice on the high starch diet (Figure 5A). G6PD activity was unaffected by TAC in WT mice fed the high fructose diet, and decreased in G6PDX mice. Taken together, the results indicate G6PD deficiency exacerbates cardiac hypertrophy and dysfunction in response to pressure overload after high fructose intake.
Figure 5. G6PD deficiency exacerbates the cardiac effects of pressure overload with high fructose intake.
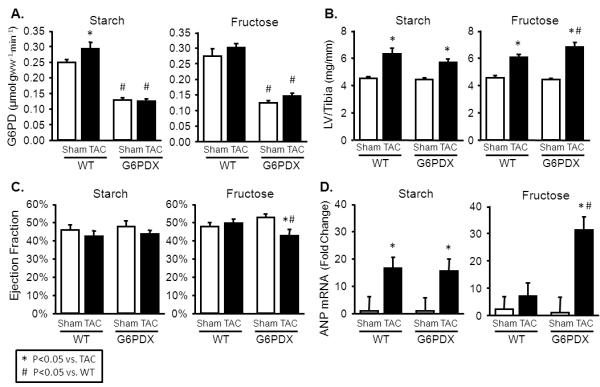
A. LV G6PD activity was increased by TAC in the high starch diet, and was decreased in G6PDX mice. B. LV mass was increased by TAC in all groups, and was increased to the greatest extent in G6PDX mice with high fructose intake. C. Ejection Fraction was decreased in G6PDX mice with the high fructose diet. D. LV ANP mRNA expression was increased by TAC to the greatest extent in G6PD deficient animals with high fructose intake. Data were obtained 16-17 weeks post-TAC. For echocardiography measurements, mice were anesthetized with 2.5% isoflurane.*P <0.05 vs. TAC; #P <0.05 vs. WT; Sham n=9-16/group, TAC n=12-16/group.
Effects of G6PD Deficiency on Metabolic Parameters after High Fructose Intake and TAC
We hypothesized that a combination of pressure overload and high fructose increases flux through G6PD in WT mice to increase [NADPH], superoxide production and oxidative stress. In accordance with this hypothesis, [NADPH] was increased in response to TAC in WT mice with high fructose intake (Figure 6A). However, contrary to our hypothesis, this increase in [NADPH] did not correspond with increases in superoxide production or lipid peroxidation (Figure 6B; Table S11). On the other hand, decreased aconitase activity is indicative of oxidative stress25, and aconitase activity was decreased in response to TAC in G6PDX mice after high fructose intake (Figure 6C). These results suggest that G6PD deficiency may increase rather than decrease oxidative stress in response to high fructose intake and TAC.
Figure 6. Fructose/TAC - Biochemical and Metabolic Data.

A. TAC increased LV [NADPH] after high fructose intake in WT mice, but not in G6PDX mice. B. Superoxide production was unchanged by TAC or G6PD deficiency among animals that received the high fructose diet or the high starch diet. C. Aconitase activity, an index of oxidative stress, was decreased by TAC in G6PDX mice after high fructose intake. D. G6PD deficiency increased serum triglycerides compared to WT mice with high fructose intake. Data were obtained 17 weeks post-TAC. *P <0.05 vs. TAC; #P <0.05 vs. WT; Sham n=9-16/group, TAC n=12-16/group.
High sugar intake is associated with increased circulating triglycerides, and adverse metabolic effects may interact with pressure overload to adversely affect heart failure26. G6PDX mice had increased serum triglycerides after high sugar intake, indicating that G6PD deficiency increases propensity for hypertriglceridemia (Figure 6D). G6PD deficiency also decreased serum insulin in animals with high fructose intake, consistent with a decrease in β-cell function as reported by others (Table S12)27. Serum glucose and free fatty acids were unaffected by G6PD deficiency or TAC.
Discussion
The main novel finding of this study is that the progression of heart failure was modestly accelerated by a clinically relevant level of G6PD deficiency. In response to myocardial infarction or chronic pressure overload, G6PDX mice displayed modest but significantly worsening of LV dilation and/or contractile dysfunction, associated with decreased GSH or increased lipid peroxidation products in cardiac tissue. Thus, contrary to our hypothesis, G6PD deficiency did not attenuate development of myocardial dysfunction and ventricular remodeling in response to injury or pressure overload.
Our hypothesis was based on the observation that pharmacological inhibition of G6PD decreases ROS generation in myocardial homogenates from canine and human failing hearts, suggesting that G6PD fuels NADPH dependent oxidant enzymes in advanced heart failure1, 11. On the other hand, given the critical role attributed to the oxidative pentose phosphate pathway in the maintenance of redox homeostasis, one may argue that an alternative hypothesis could have been that G6PD deficiency will exacerbate myocardial pathology in response to chronic stress. This is suggested by the observation that inhibition of G6PD increases the sensitivity of cells to oxidative damage and death, while G6PD overexpression protects cells against oxidative damage13. Further, the activity of G6PD is enhanced in isolated cardiomyocytes in response to oxidative stress, and inhibition of G6PD increased oxidative stress and impaired cardiomyocyte calcium handling and contractility3.
In the light of this alternative hypothesis, our observation of a relatively modest effect of G6PD deficiency on LV remodeling and contractile function, and a lack of effect on survival, suggests that G6PD deficiency does not trigger catastrophe in heart failure. Although LV dilation was enhanced in response to G6PD deficiency, accelerated contractile dysfunction was not evident. There may be a complex and evolving balance between negative and positive effects of G6PD deficiency during the progression of heart failure. Previous studies found that G6PDX mice display worse contractile recovery following an acute bout of myocardial ischemia that appeared to be mediated by accelerated oxidative stress4. Nonetheless, there might be a transient detrimental consequence of G6PD deficiency in acute stress that is not sufficient to affect long-term outcome.
We expected G6PD deficiency to decrease NADPH concentration, as seen by others in the kidneys of G6PDX mice and in cardiomyocytes with pharmacological inhibition of G6PD12, 28. The myocardial concentration of NADPH reflects the NADPH set point, rather than the rate of NADPH production. Further, the G6PD enzymatic activity is an index of the maximal capacity for the reaction, rather than the actual flux through G6PD, which is dependent on cellular location and the concentrations of substrates and products. However, we found that NADPH levels were not significantly different in G6PDX compared to WT mice, and we were surprised to find a significant increase in NADPH concentration in response to either myocardial infarction or TAC in G6PDX. The reason for the increase is unclear, but suggests that there are compensatory mechanisms for increasing NADPH29.
High fructose intake may accelerate heart failure by increasing superoxide production17. In the liver, fructose bypasses key regulatory step of glycolysis and causes a transient increase in circulating triglycerides, which could inhibit glycolysis in the heart and increase myocardial glucose flux through non-oxidative pathways such as the pentose phosphate pathway to increase NADPH dependant ROS production1, 30, 31. High glucose increased NADPH concentration, NADPH oxidase activity, and ROS, and the glucose-induced increase in ROS was prevented by G6PD knockdown in cultured adipose cells20, and a high fructose diet previously accelerated heart failure via increased ROS17. Thus, we hypothesized that high fructose intake would exacerbate heart failure by increasing flux through G6PD to increase NADPH and superoxide production, and predicted that G6PD deficiency would have a beneficial effect on the heart in the context of a high fructose diet. We saw an increase in NADPH in WT mice with TAC and high fructose, but not in G6PDX mice, supporting the hypothesis that high sugar intake affects flux through the pentose phosphate pathway. However, this increase in NADPH concentration in WT mice did not correspond to any changes in superoxide production, and G6PD deficiency was not beneficial. Rather, G6PD deficiency resulted in increased triglycerides, and a decrease in aconitase activity, a marker of oxidative stress, which corresponded with greater LV hypertrophy and dysfunction with TAC and high fructose intake. Our results are similar to those seen by Zhang et al, who reported that high glucose decreased G6PD activity and increased oxidative stress in cultured endothelial cells32. Our results also support recent findings that pharmacological activation of G6PD suppresses heart failure after myocardial infarction and diabetes33. Thus, G6PD deficiency made the myocardium susceptible to adverse effects of high sugar intake in heart failure.
Study Limitations
The underlying signaling pathways responsible for the increased hypertrophy seen in G6PDX myocardium following TAC with high fructose intake are unclear. Potential pathways include changes in advanced glycation end products, neurohormonal responses, VEGF, Pim-1, and Akt, as seen by others with pharmacological modulation of the pentose phosphate pathway33. Further, it is unclear what effect the increase in [NADPH] has on NADPH oxidases. These potential mechanisms were not investigated in the present investigation, and this should be addressed in future work. A further limitation to our approach is the use of severe and abrupt pressure overload and coronary ligation in young healthy mice. This differs greatly from clinical heart failure, which generally occurs with advanced age and takes years to develop. Our models might not adequately recapitulate the redox and metabolic conditions in human myocardium that would warrant rescue by G6PD deficiency. This limitation further points toward the importance of human studies to provide clear answers regarding the effects of G6PD deficiency on heart failure.
Conclusion
In summary, we assessed the impact of G6PD deficiency on the development of heart failure using an established genetic mouse model G6PD deficiency combined with pressure overload or infarct-induced failure. Cardiac dysfunction and/or LV remodeling was modestly worse with G6PD deficiency, which was associated with greater redox stress. These findings suggest that the risk for developing heart failure is higher, but still limited by compensatory mechanisms, when hypertension or myocardial infarction occur in individuals with G6PD deficiency. Given that G6PD deficiency is the most common enzyme deficiency in the world, our findings suggest that further investigation in humans is warranted.
Supplementary Material
Clinical Perspective.
G6PD deficiency affects about 400 million people worldwide, particularly in sub-Saharan Africa, the Mediterranean region, and Southeast Asia, and it is the most common enzyme deficiency in the world. G6PD deficiency controls the entry of glucose into the pentose phosphate pathway and the production of cytoplasmic [NADPH], and affects oxidative stress. While severe G6PD deficiency increases severity of neonatal jaundice and hemolytic anemia in response to broad beans or antimalarial drugs, little information is available regarding the cardiac effects of G6PD deficiency. Limited clinical evidence indicates that G6PD deficiency may be associated with hypertension, however data also support a protective role of G6PD deficiency for risk of heart disease and cardiovascular-associated death. Thus it is unclear how G6PD affects the response of the heart to chronic stress. Here, we used a mouse model to address the effects of G6PD deficiency on the development of heart failure. In response to myocardial infarction or chronic aortic hypertension we found that the increase in end diastolic volume and eccentric ventricular hypertrophy in G6PD deficient mice was modestly enhanced compared to normal mice. Further, G6PD deficiency increased left ventricular hypertrophy and dysfunction in response to pressure overload with high sugar intake. These effects corresponded with increased oxidative stress in G6PD deficient myocardium. Our results indicate that G6PD deficiency adversely impacts the development of heart failure in response to chronic cardiac stress, and suggest that this may be due to increased oxidative stress in G6PD deficient myocardium.
Acknowledgments
Sources of Funding This work was supported by the National Institutes of Health [grants P01 HL074237,T32 HL072751].
Footnotes
Disclosures None.
This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Gupte SA, Levine RJ, Gupte RS, Young ME, Lionetti V, Labinskyy V, Floyd BC, Ojaimi C, Bellomo M, Wolin MS, Recchia FA. Glucose-6-phosphate dehydrogenase-derived NADPH fuels superoxide production in the failing heart. J Mol Cell Cardiol. 2006;41:340–9. doi: 10.1016/j.yjmcc.2006.05.003. [DOI] [PubMed] [Google Scholar]
- 2.Frederiks WM, Kummerlin IP, Bosch KS, Vreeling-Sindelarova H, Jonker A, Van Noorden CJ. NADPH production by the pentose phosphate pathway in the zona fasciculata of rat adrenal gland. J Histochem Cytochem. 2007;55:975–80. doi: 10.1369/jhc.7A7222.2007. [DOI] [PubMed] [Google Scholar]
- 3.Jain M, Brenner DA, Cui L, Lim CC, Wang B, Pimentel DR, Koh S, Sawyer DB, Leopold JA, Handy DE, Loscalzo J, Apstein CS, Liao R. Glucose-6-phosphate dehydrogenase modulates cytosolic redox status and contractile phenotype in adult cardiomyocytes. Circ Res. 2003;93:e9–16. doi: 10.1161/01.RES.0000083489.83704.76. [DOI] [PubMed] [Google Scholar]
- 4.Jain M, Cui L, Brenner DA, Wang B, Handy DE, Leopold JA, Loscalzo J, Apstein CS, Liao R. Increased myocardial dysfunction after ischemia-reperfusion in mice lacking glucose-6-phosphate dehydrogenase. Circulation. 2004;109:898–903. doi: 10.1161/01.CIR.0000112605.43318.CA. [DOI] [PubMed] [Google Scholar]
- 5.Luzzatto L, Notaro R. Malaria. Protecting against bad air. Science. 2001;293:442–3. doi: 10.1126/science.1063292. [DOI] [PubMed] [Google Scholar]
- 6.Cappellini MD, Fiorelli G. Glucose-6-phosphate dehydrogenase deficiency. Lancet. 2008;371:64–74. doi: 10.1016/S0140-6736(08)60073-2. [DOI] [PubMed] [Google Scholar]
- 7.Glucose-6-phosphate dehydrogenase deficiency. WHO Working Group. Bull World Health Organ. 1989;67:601–11. [PMC free article] [PubMed] [Google Scholar]
- 8.Cocco P, Todde P, Fornera S, Manca MB, Manca P, Sias AR. Mortality in a cohort of men expressing the glucose-6-phosphate dehydrogenase deficiency. Blood. 1998;91:706–9. [PubMed] [Google Scholar]
- 9.Meloni L, Manca MR, Loddo I, Cioglia G, Cocco P, Schwartz A, Muntoni S, Muntoni S. Glucose-6-phosphate dehydrogenase deficiency protects against coronary heart disease. J Inherit Metab Dis. 2008;31:412–7. doi: 10.1007/s10545-008-0704-5. [DOI] [PubMed] [Google Scholar]
- 10.Bing RJ, Gudbjarnason S, Tschopp H, Braasch W. Molecular changes in myocardial infarction in heart muscle. Ann N Y Acad Sci. 1969;156:583–93. doi: 10.1111/j.1749-6632.1969.tb16753.x. [DOI] [PubMed] [Google Scholar]
- 11.Gupte RS, Vijay V, Marks B, Levine RJ, Sabbah HN, Wolin MS, Recchia FA, Gupte SA. Upregulation of glucose-6-phosphate dehydrogenase and NAD(P)H oxidase activity increases oxidative stress in failing human heart. J Card Fail. 2007;13:497–506. doi: 10.1016/j.cardfail.2007.04.003. [DOI] [PubMed] [Google Scholar]
- 12.Tian WN, Braunstein LD, Pang J, Stuhlmeier KM, Xi QC, Tian X, Stanton RC. Importance of glucose-6-phosphate dehydrogenase activity for cell growth. J Biol Chem. 1998;273:10609–17. doi: 10.1074/jbc.273.17.10609. [DOI] [PubMed] [Google Scholar]
- 13.Tian WN, Braunstein LD, Apse K, Pang J, Rose M, Tian X, Stanton RC. Importance of glucose-6-phosphate dehydrogenase activity in cell death. Am J Physiol. 1999;276:C1121–C1131. doi: 10.1152/ajpcell.1999.276.5.C1121. [DOI] [PubMed] [Google Scholar]
- 14.Matsui R, Xu S, Maitland KA, Hayes A, Leopold JA, Handy DE, Loscalzo J, Cohen RA. Glucose-6 phosphate dehydrogenase deficiency decreases the vascular response to angiotensin II. Circulation. 2005;112:257–63. doi: 10.1161/CIRCULATIONAHA.104.499095. [DOI] [PubMed] [Google Scholar]
- 15.Matsui R, Xu S, Maitland KA, Mastroianni R, Leopold JA, Handy DE, Loscalzo J, Cohen RA. Glucose-6-phosphate dehydrogenase deficiency decreases vascular superoxide and atherosclerotic lesions in apolipoprotein E(−/−) mice. Arterioscler Thromb Vasc Biol. 2006;26:910–6. doi: 10.1161/01.ATV.0000205850.49390.3b. [DOI] [PubMed] [Google Scholar]
- 16.Pretsch W, Charles DJ, Merkle S. X-linked glucose-6-phosphate dehydrogenase deficiency in Mus musculus. Biochem Genet. 1988;26:89–103. doi: 10.1007/BF00555491. [DOI] [PubMed] [Google Scholar]
- 17.Chess DJ, Xu W, Khairallah R, O’Shea KM, Kop WJ, Azimzadeh AM, Stanley WC. The antioxidant tempol attenuates pressure overload-Induced cardiac hypertrophy and contractile dysfunction in mice fed a high-fructose diet. Am J Physiol Heart Circ Physiol. 2008;295:H2223–H2230. doi: 10.1152/ajpheart.00563.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mellor K, Ritchie RH, Meredith G, Woodman OL, Morris MJ, Delbridge LM. High-fructose diet elevates myocardial superoxide generation in mice in the absence of cardiac hypertrophy. Nutrition. 2010;26:842–8. doi: 10.1016/j.nut.2009.08.017. [DOI] [PubMed] [Google Scholar]
- 19.Mellor KM, Ritchie RH, Davidoff AJ, Delbridge LM. Elevated dietary sugar and the heart: experimental models and myocardial remodeling. Can J Physiol Pharmacol. 2010;88:525–40. doi: 10.1139/y10-005. [DOI] [PubMed] [Google Scholar]
- 20.Han CY, Umemoto T, Omer M, Den Hartigh LJ, Chiba T, Leboeuf R, Buller CL, Sweet IR, Pennathur S, Abel ED, Chait A. NADPH oxidase-derived reactive oxygen species increases expression of monocyte chemotactic factor genes in cultured adipocytes. J Biol Chem. 2012;287:10379–93. doi: 10.1074/jbc.M111.304998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sharma N, Okere IC, Duda MK, Johnson J, Yuan CL, Chandler MP, Ernsberger P, Hoit BD, Stanley WC. High fructose diet increases mortality in hypertensive rats compared to a complex carbohydrate or high fat diet. Am J Hypertens. 2007;20:403–9. doi: 10.1016/j.amjhyper.2006.09.022. [DOI] [PubMed] [Google Scholar]
- 22.Sharma N, Okere IC, Barrows BR, Lei B, Duda MK, Yuan CL, Previs SF, Sharov VG, Azimzadeh AM, Ernsberger P, Hoit BD, Sabbah H, Stanley WC. High-sugar diets increase cardiac dysfunction and mortality in hypertension compared to low-carbohydrate or high-starch diets. J Hypertens. 2008;26:1402–10. doi: 10.1097/HJH.0b013e3283007dda. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chess DJ, Lei B, Hoit BD, Azimzadeh AM, Stanley WC. Deleterious effects of sugar and protective effects of starch on cardiac remodeling, contractile dysfunction, and mortality in response to pressure overload. Am J Physiol Heart Circ Physiol. 2007;293:H1853–H1860. doi: 10.1152/ajpheart.00544.2007. [DOI] [PubMed] [Google Scholar]
- 24.Bouchard-Thomassin AA, Lachance D, Drolet MC, Couet J, Arsenault M. A high-fructose diet worsens eccentric left ventricular hypertrophy in experimental volume overload. Am J Physiol Heart Circ Physiol. 2011;300:H125–H134. doi: 10.1152/ajpheart.00199.2010. [DOI] [PubMed] [Google Scholar]
- 25.Cantu D, Schaack J, Patel M. Oxidative inactivation of mitochondrial aconitase results in iron and H2O2-mediated neurotoxicity in rat primary mesencephalic cultures. PLoS One. 2009;4:e7095. doi: 10.1371/journal.pone.0007095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Welsh JA, Sharma A, Abramson JL, Vaccarino V, Gillespie C, Vos MB. Caloric sweetener consumption and dyslipidemia among US adults. JAMA. 2010;303:1490–7. doi: 10.1001/jama.2010.449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang Z, Liew CW, Handy DE, Zhang Y, Leopold JA, Hu J, Guo L, Kulkarni RN, Loscalzo J, Stanton RC. High glucose inhibits glucose-6-phosphate dehydrogenase, leading to increased oxidative stress and beta-cell apoptosis. FASEB J. 2010;24:1497–505. doi: 10.1096/fj.09-136572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xu Y, Zhang Z, Hu J, Stillman IE, Leopold JA, Handy DE, Loscalzo J, Stanton RC. Glucose-6-phosphate dehydrogenase-deficient mice have increased renal oxidative stress and increased albuminuria. FASEB J. 2010;24:609–16. doi: 10.1096/fj.09-135731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Merritt TJ, Kuczynski C, Sezgin E, Zhu CT, Kumagai S, Eanes WF. Quantifying interactions within the NADP(H) enzyme network in Drosophila melanogaster. Genetics. 2009;182:565–74. doi: 10.1534/genetics.109.100677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Matsushima S, Kinugawa S, Yokota T, Inoue N, Ohta Y, Hamaguchi S, Tsutsui H. Increased myocardial NAD(P)H oxidase-derived superoxide causes the exacerbation of postinfarct heart failure in type 2 diabetes. Am J Physiol Heart Circ Physiol. 2009;297:H409–H416. doi: 10.1152/ajpheart.01332.2008. [DOI] [PubMed] [Google Scholar]
- 31.Stanley WC, Recchia FA, Lopaschuk GD. Myocardial substrate metabolism in the normal and failing heart. Physiol Rev. 2005;85:1093–129. doi: 10.1152/physrev.00006.2004. [DOI] [PubMed] [Google Scholar]
- 32.Zhang Z, Apse K, Pang J, Stanton RC. High glucose inhibits glucose-6-phosphate dehydrogenase via cAMP in aortic endothelial cells. J Biol Chem. 2000;275:40042–7. doi: 10.1074/jbc.M007505200. [DOI] [PubMed] [Google Scholar]
- 33.Katare R, Caporali A, Emanueli C, Madeddu P. Benfotiamine improves functional recovery of the infarcted heart via activation of pro-survival G6PD/Akt signaling pathway and modulation of neurohormonal response. J Mol Cell Cardiol. 2010;49:625–38. doi: 10.1016/j.yjmcc.2010.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.