

Abstract
Type I interferons are a cytokine family essential for antiviral defense. More recently, type I interferons have been shown to be important during bacterial infections. Here we show that, in addition to known cytokine functions, interferon-β (IFN-β) is also antimicrobial. Parts of the IFN-β molecular surface (especially helix 4) are cationic and amphipathic, both classic characteristics of antimicrobial peptides, and we have observed that IFN-β can directly kill Staphylococcus aureus. Further, a mutant S. aureus that is more sensitive to antimicrobial peptides was killed more efficiently by IFN-β than the wild-type S. aureus, and immunoblotting showed that IFN-β interacts with the bacterial cell surface. To determine whether specific parts of IFN-β are antimicrobial, we synthesized IFN-β helix 4 and found that it is sufficient to permeate model prokaryotic membranes using synchrotron x-ray diffraction and that it is sufficient to kill S. aureus. These results suggest that in addition to its well-known signaling activity, IFN-β may be directly antimicrobial and be part of a growing family of cytokines and chemokines, called kinocidins, that also have antimicrobial properties.
Introduction
The type I interferons (IFNs) are a pleiotropic family of cytokines. In particular, interferon-β (IFN-β) has been shown to be important in antiviral defense (1), antibacterial defense (2), cellular growth and apoptosis (3), autoimmune disorders (4). There is strong purifying selection for IFN-β in the human genome, suggesting it is an important host defense molecule (5).
Staphylococcus aureus is a gram-positive bacterium that naturally colonizes the nares of 30–50% of the world’s population (6). S. aureus can also cause disease in humans, especially as a hospital-acquired infection. In recent years, antibiotic-resistant strains, such as methicillin-resistant S. aureus (MRSA), have become increasingly prevalent (7). Consequently, it is becoming increasingly important that we understand how the host successfully combats S. aureus and how best to develop new therapies for treating MRSA infections.
Antimicrobial peptides (AMPs) are a large and diverse family of antimicrobial molecules that exhibit potent antibacterial activity. AMPs are present throughout the mammalian body and are found in especially high concentrations on mucosal surfaces (8). AMPs are evolutionarily ancient, being conserved from invertebrates onward (9). Overall AMPs are characterized by a net positive charge (cationic) and segregated regions of polar and non-polar residues (amphipathicity) (10). AMPs can selectively permeate bacterial membranes and kill via disruption of barrier function and/or binding to intracellular targets. The mechanism by which AMPs permeate bacterial membranes is the type of curvature topologically required for a variety of membrane disruption processes such as pore formation and blebbing, and is referred to as Negative Gaussian Curvature (NGC) (11, 12).
In this study, we found that IFN-β exhibits unexpected direct antimicrobial activity against S. aureus. Immunoblotting showed that IFN-β associates with the bacterial cell surface, and Sytox green staining suggested bacterial membrane permeabilization. This is suggestive of the activity of AMPs. In fact a S. aureus mutant that is specifically more susceptible to AMPs was killed more efficiently by IFN-β compared to the wild-type strain. Parts of the IFN-β molecular surface (especially IFN-β helix 4) are cationic and amphipathic and have amino acid compositional preferences similar to α-helical AMPs. We synthesized mouse and human IFN-β helix 4 and found using synchrotron small-angle X-ray scattering (SAXS) that they have the ability to selectively disrupt bacterial, but not eukaryotic, model membranes. Together, these results suggest that IFN-β can be classified as a kinocidin, a growing family of cytokines and chemokines with inherent antimicrobial activities (13, 14), and that its antimicrobial activity is cognate to that of AMPs.
Materials and Methods
Bacterial strains and culture
Staphylococcus aureus strains SA113, Newman, USA300, Romero and Staphylococcus epidermidis were grown in Luria broth (LB) or Todd Hewitt borth (THB). Escherichia coli DH5α, Listeria monocytogenes, P. aeurigenosa, S. typhimurium, and B. subtillis were grown in LB. Group B Streptococcus (GBS) was grown in THB + 0.5% yeast extract. All bacteria were grown overnight at 37°C with agitation except GBS, which was cultured without agitation at 37°C. Overnight bacterial cultures were sub-cultured and incubated until mid-log was reached, determined to be OD600=0.4. Cultures were washed in sterile PBS and renormalized to OD600=0.4 in culture media.
Bacterial killing assays
For killing assays using recombinant whole mouse IFN-β (mIFN-β) (PBL Interferon), bacteria were grown as described above and re-suspended in RPMI 1640 (Corning). 100 μl reactions (bacteria + IFN-β or vehicle) were added to sterile 1.5 mL tubes. Tubes were incubated at 37°C with shaking for 1, 3, or 24 hours. After specified incubation periods, ten-fold serial dilutions were plated on LB plates to quantify surviving CFU. We consistently noted significant differences in overall killing activity exhibited by different lots of recombinant IFN-β.
For pH-specific killing assays using recombinant whole human IFN-β (hIFN-β) (PBL Interferon), bacteria were grown as described above and re-suspended in either 2 mM MES, pH 5.5 or 10 mM PIPES, pH 7.5, buffers previously shown to be good for assessing antimicrobial peptide activity at a variety of pHs (15). Two types of human recombinant IFN-β were tested from E. coli or CHO sources (PBL Interferon). 100 μl reactions (bacteria + IFN-β or vehicle) were added to sterile 1.5 mL tubes. Tubes were incubated at 37°C with shaking for 3 hours. After specified incubation periods, ten-fold serial dilutions were plated on LB plates to quantify surviving CFU.
For killing assays using IFN-β helix 4 peptide or mutant IFN-β helix 4 peptide (synthesized by LifeTein or United Biosystems), bacteria were grown as described above and re-suspended in 100 mM NaCl, 20 mM HEPES, pH 7.4 and in some cases supplemented with 1% THB. 100 μl reactions (bacteria + IFN-β helix 4 peptides or vehicle) were added to sterile 1.5 mL tubes or 200 ul reactions were added to 96-well plates. Bacteria and treatments were incubated at 37°C with shaking for designated amounts of time. After specified incubation periods, ten-fold serial dilutions were plated on agar plates of the proper media type for each strain to quantify surviving CFU.
Sytox plate assay for bacterial survival
Bacteria were grown as described above and re-suspended in 10 mM Tris-HCL, pH 7.5. 1 mL of S. aureus was incubated with 1 μM Sytox Green (Thermo-Fisher) in the dark at 37°C with shaking for 20 minutes. 5 μl of each treatment was added to a black 96-well plate: vehicle, 1 mM nigericin, or 5×103 units/mL mIFN-β. 95 μl of Sytox Green-labelled bacteria were then added to each 96 well, and the fluorescence was read in a plate reader immediately and then every 15 minutes at 485 nm excitation and 520 nm emission. The plate was continuously incubated at 37°C with shaking in dark when not being read.
Sytox live-dead imaging of bacteria
Bacteria were grown as described above and re-suspended in RPMI 1640 (Corning). 100 μl of S. aureus were incubated with either vehicle, 1 mM nigericin, or 5×103 units/mL mIFN-β at 37°C with shaking for 10 minutes. Bacteria were then pelleted and washed in PBS. Bacteria were re-suspended in 10 mM Tris-HCL, pH 7.5. Bacteria were incubated in the dark with 1 μg/mL DAPI and 0.5 μM Sytox Green for 10 minutes. Labelled bacteria were applied to a slide by adding 2 μl and allowing it to dry (this process was repeated twice). Dried bacteria drops were then covered in ProLong Gold (Thermo-Fisher) mounting media and coverslips were added. Slides were dried for one hour in the dark and then imaged by fluorescence microscopy. Images were quantified by enumeration of blue and green bacteria.
Immunoblotting for mIFN-β
S. aureus was incubated with increasing concentrations of mIFN-β. 2×107 S. aureus in 100 μl RPMI 1640 (Corning) in a 1.5 mL tube were incubated with 5 μl vehicle or mIFN-β for 1 hour. Bacteria were pelleted at maximum speed for 10 minutes on a microcentrifuge at room temperature. The supernatant was discarded, and bacteria were washed 3× in PBS and processed for immunoblotting using a NuPage system (Thermo-Fisher).
Washed bacterial pellets were lysed in NuPage LDS sample buffer (Thermo-Fisher) by vortexing and incubation at room temperature for 5 minutes. Samples were boiled and loaded onto a 4–12% SDS-polyacrylamide gel. Proteins were transferred to PVDF membranes (Millipore). Membranes were blocked with 5% BSA for 2 hours at room temperature. Blots were incubated with pimary antibodies either rat anti-mouse IFN-β (PBL, RMMB-1) or anti-GAPDH (MyBiosource, GA1R) at 1:1000 overnight at 4°C. IR secondary antibodies (Licor) were used at 1:10,000, and the membrane was processed for use with the Odyssey Imaging System (Licor).
Structural modeling of whole mouse and human IFN-β
The structural homology of mouse and human IFN-β were analyzed using Pymol (version 1.3). The protein structure of each molecule was downloaded as a PDB file from RCSB Protein Data Bank (http://www.pdb.org/pdb/home/home.do) and loaded into Pymol (Schrodinger, LLC. The PyMOL Molecular Graphics System, Version 1.3. 2011). Further analyses were facilitated using the APBS software plug-in (http://www.poissonboltzmann.org) (16). Mouse and human IFN-β were analyzed for regions of positive and negative amino acid residues, and the resulting graphs show regions of the molecule that most likely contribute to overall positive charge.
Mouse and human IFN-β helix 4 peptides
Mouse (3175.8 MW) and human IFN-β helix 4 (2587.1 MW) peptides and mouse mutant IFN-β helix 4 (QASSTALQLASYYWAVQTYLALMKY) (2912.4 MW) were synthesized (≥ 95% purity, LifeTein or United Biosystems) with N-terminal acetylation and C-terminal amidation. Wild type peptides were solubilized in ultrapure water. Helical wheel projections were used to assess the proximity of positively-charged amino acid residues in mouse and human IFN-β helix 4 peptides. Mutant IFN-β helix 4 was no longer soluble in water so mutant helix 4 and a portion of wild-type helix 4 were solubilized in DMSO for comparison. In killing assays, DMSO was diluted to less than 0.5% which was determined to have no effect on bacterial survival in 3 hour killing experiment.
Compositional comparison of mouse and human IFN-β helix 4 peptides with AMPs
We compare the amino acid compositions of mouse and human IFN-β helix 4 peptides with the compositions of known AMPs, which is a consequence of the required membrane curvature generation. A set of 1080 cationic AMP sequences were sourced from the antimicrobial peptide database (17) and analyzed using a procedure described previously (12, 18). We defined the average hydrophobicity of a given peptide, j, as:
where n is the number of amino acids in the peptide and wi is the hydrophobicity of the ith amino acid in the peptide using the Eisenberg consensus hydrophobicity scale (19). The minimum and maximum <hydrophobicity> values within the set of AMP sequences were used to define a <hydrophobicity> range. This range was then divided into 100 equal bins, into which the peptides were partitioned. For m peptides in a given bin, we define:
where NK/(NK + NR) represents the ratio of the number of lysines to the total number of lysines plus arginines. For each bin, NK/(NK + NR) versus <hydrophobicity> was plotted using Matlab. Mouse and human IFN-β helix 4 peptides were analyzed and plotted using the same procedure.
To compare the hydrophobicities of mouse and human IFN-β helix 4 peptides with those of AMPs, a histogram of <hydrophobicity> values for the set of AMPs was constructed with 50 bins using Matlab. The average hydrophobicities of both mouse and human IFN-β helix 4 peptides were superimposed over the AMP histogram.
Small-angle X-ray scattering experiments
Liposome preparation for X-ray measurements were carried out as follows. Lyophilized lipids (Avanti Polar Lipids) DOPG (1,2-dioleoyl-sn-glycero-3-[phospho-rac-(1-glycerol)] (sodium salt)), DOPS (1,2-dioleoyl-sn-glycero-3-phospho-L-serine (sodium salt)), DOPE (1,2-dioleoyl-sn-glycero-3-phosphoethanolamine), and DOPC (1,2-dioleoyl-sn-glycero-3-phosphocholine) were used without further purification to form small unilamellar vesicles (SUVs). Individual stock solutions of each lipid were prepared in chloroform at a concentration of 20 mg/mL. Lipid mixtures were prepared at specific molar ratios to yield each model membrane composition. Lipid mixture solutions were evaporated under N2, desiccated overnight under vacuum, and then resuspended to a concentration of 20 mg/mL in either aqueous 100 mM NaCl, 20 mM HEPES (pH 7.4) or 100 mM NaCl, 10 mM NaOAc (pH 5). Aqueous lipid solutions were incubated at 37°C overnight and then sonicated until clear. SUVs were obtained via extrusion of sonicated solution through a 0.2 μm pore Nucleopore filter (Whatman).
IFN-β helix 4 peptides and SUVs were mixed at specific peptide to lipid (P/L) molar ratios and hermetically sealed in quartz capillaries (Mark-tubes, Hilgenberg GmbH). All samples were prepared in either 100 mM NaCl, 20 mM HEPES (pH 7.4) or 100 mM NaCl, 10 mM NaOAc (pH 5) and incubated at 37°C. SAXS experiments were performed at the Stanford Synchrotron Radiation Lightsource (SSRL, beamline 4-2) using monochromatic X-rays with an energy of 9 keV. The scattered radiation was collected using a Rayonix MX225-HE detector (pixel size of 73.2 μm). 2D SAXS powder patterns were integrated using the Nika 1.50 (20) package for Igor Pro 6.31 and FIT2D.
SAXS data were analyzed by plotting the integrated scattering intensity I(Q) against Q using Origin Lab software. To determine the phase(s) present in each sample, the measured peak positions, Qmeas, were tabulated and their ratios compared with those of the permitted reflections for different liquid-crystalline phases. The lattice parameter of each identified phase was calculated from the slope of the linear regression through points corresponding to the peaks, with each point having coordinates of Qmeas and the assigned reflection in terms of Miller indices h, k, and l. For a powder-averaged cubic phase, , where a is the lattice parameter. Its average Gaussian curvature is defined as , where is the Euler characteristic and is the surface area per cubic unit cell specific to each cubic phase.
Results
Mouse interferon-β is directly antimicrobial to Staphylococcus aureus
In the course of studying the effects of IFN-β on host responses to S. aureus (21), we incubated the bacteria with a high dose of recombinant mIFN-β for three hours and for 24 hours. Surprisingly, after only three hours we saw reduced survival of S. aureus (Fig. 1A). A dose curve of IFN-β revealed that growth inhibition of S. aureus started at concentrations of 500 units/ml IFN-β after one hour of incubation (Fig. 1B). Such doses of IFN-β are not unusual in the literature and are thought to be biologically relevant (22–25). Having observed killing of S. aureus after incubation with IFN-β, we examined whether these antibacterial properties were effective against other species of bacteria. We also incubated Staphylococcus epidermis (Fig. S1) with IFN-β and observed killing. However, when we incubated Escherichia coli with increasing doses of IFN-β we did not observe any inhibitory or killing effect (Fig. 1C).
FIGURE 1. Mouse interferon-β (IFN-β) kills Staphylococcus aureus.
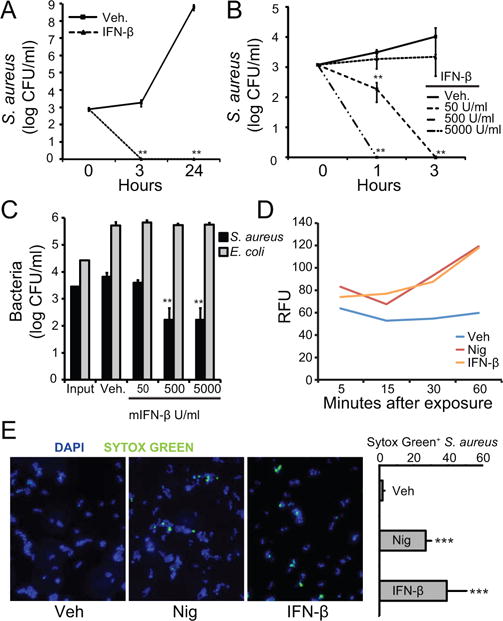
(A) S. aureus were incubated with vehicle (VEH) or 500 units/ml recombinant mIFN-β. Bacteria and treatments were incubated 3 and 24 hours. Remaining bacteria were serially-diluted and plated for enumeration. (B) S. aureus were incubated with VEH or mIFN-β (50, 500, or 5000 units/ml which is equal to 0.0006, 0.006, or 0.06 μM). Bacteria were treated for 1 and 3 hours. Remaining bacteria were serially-diluted and plated for enumeration. (C) S. aureus or E. coli were incubated with VEH or mIFN-β at indicated concentrations for 3 hours. Remaining bacteria were serially-diluted and plated for enumeration. (D) S. aureus were incubated with Sytox Green for 20 min. Bacteria were added to a 96-well plate pre-loaded with VEH, 1 mM nigericin (NIG), or 5×103 units/ml mIFN-β. The plate was imaged at 485/520 nm. Relative fluorescent units (RFU) are shown. (E) S. aureus were incubated with VEH, 1 mM NIG, or 5×103 units/ml mIFN-β for 10 min. Bacteria were stained with DAPI (blue) or Sytox (green) and imaged on a confocal microscope. Five separate images from each treatment were enumerated for blue versus green bacteria and results are plotted. Data are shown as mean ± SD, * p ≤ 0.05, ** p ≤ 0.01, *** p ≤ 0.001 (unpaired, two-tailed t test).
To determine if the killing effect of IFN-β was correlated with permeation of the bacterial cell membrane, we employed a Sytox Green assay. Sytox Green dye is cationic and adheres to the negative surface of bacteria. The dye becomes fluorescent upon binding intracellular nucleic acids and can therefore be used to determine bacterial membrane disruption by external compound (26). We incubated S. aureus with Sytox Green and added IFN-β or the pore-forming toxin nigericin and monitored fluorescence initially and then every 15 minutes. We observed a marked increase above vehicle in fluorescent signal when bacteria were exposed to IFN-β or nigericin (Fig. 1D), suggesting that IFN-β disrupts the bacterial membrane. We also used Sytox Green for imaging the effect of both IFN-β and nigericin on S. aureus by confocal microscopy (Fig. 1E).
IFN-β antimicrobial activity correlates with cell-binding activity
There are many known naturally-occurring antimicrobial molecules found in mammals that aid in host defense (8). A large number of these molecules are AMPs, which are commonly characterized by an overall positive charge and amphipathicity. The positive charge of AMPs promotes their electrostatic attraction to the negative membranes of bacteria and less to host molecules, which are not as negatively-charged (9). To determine whether IFN-β was capable of binding to the S. aureus, we incubated bacteria with the cytokine and processed the S. aureus for immunoblotting. We observed dose-dependent association of IFN-β with the bacteria (Fig. 2A).
FIGURE 2. IFN-β has properties resembling antimicrobial peptides.
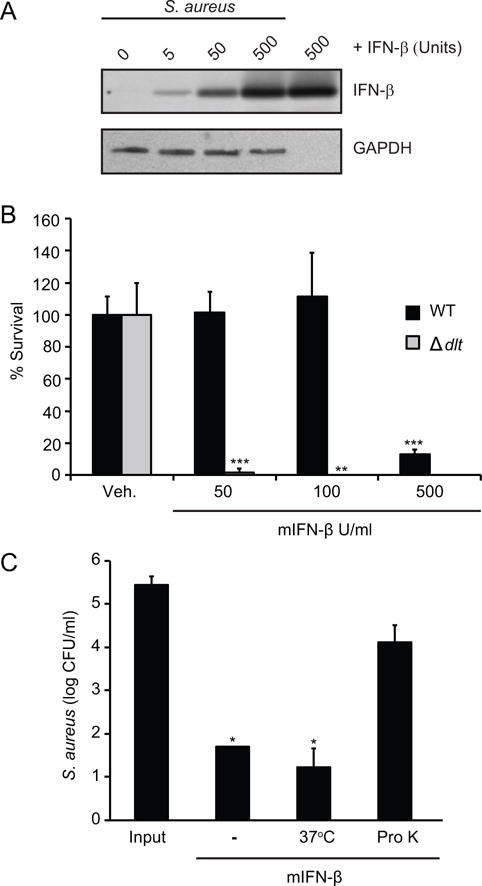
(A) S. aureus were incubated with VEH or indicated units of mIFN-β for 1 hour. Bacteria were washed 2X to remove any mIFN-β not adhered to bacterial surface. mIFN-β interacting with S. aureus cell wall were detected by immunoblotting and 500 units mIFN-β was used as a positive control. Detection of bacterial GAPDH was used to control for the loading of equal amounts of bacteria. (B) Wild-type (WT) or DLT-deficient (ΔDLT) S. aureus were incubated with VEH or mIFN-β at indicated concentrations for 3 hours. Remaining bacteria were serially-diluted and plated for enumeration. (C) S. aureus were incubated with VEH, 5×102 units/ml mIFN-β (−), 5×102 units/ml mIFN-β incubated at 37°C for 60 min (37C), or 5×102 units/ml mIFN-β incubated with proteinase K for 60 min (Pro K). Bacteria were incubated with variably-treated mIFN-βs for 3 hours. Remaining bacteria were serially-diluted and plated for enumeration. Data are shown as mean ± SD, * p ≤ 0.05 (unpaired, two-tailed t test).
Multiple species of gram-positive bacteria subvert host AMPs by altering the lipoteichoic acids in their cell wall, effectively masking negative charges to result in a more positive overall charge (27). The dlt gene is one gene responsible for cell wall alterations that increase bacterial resistance to AMPs. S. aureus strains in which the dlt gene has been deleted are more sensitive than wild-type strains to AMPs but not to antibiotics with other modes of action (28). We incubated wild-type S. aureus (WT) and an isogenic mutant lacking the dlt gene (Δdlt) with increasing concentrations of IFN-β. We observed increased killing of Δdlt bacteria compared to WT, consistent with the hypothesis that IFN-β exhibits binding behavior cognate to those of AMPs (Fig. 2B).
To investigate whether the antibacterial effects of IFN-β are intrinsic to the cytokine and not due to, for example, a contaminant introduced during purification of the commercially-prepared recombinant cytokine, we pretreated the IFN-β with proteinase K to degrade the protein. Degradation diminished the ability of the of the cytokine to kill S. aureus, while killing was not lost after similar incubation at 37°C (Fig. 2C). These results indicated that the IFN-β protein, or portion of it, must be intact to function in an antimicrobial manner against S. aureus.
Human IFN-β kills S. aureus, but only at acidic pH
Having observed killing of S. aureus by mIFN-β, we investigated whether hIFN-β had similar properties. We incubated S. aureus with the same quantity of either mouse or human IFN-β and quantified surviving bacteria. The results show that hIFN-β did not kill bacteria as well as mIFN-β (Fig. 3A). The nucleic acid and amino acid sequences of mouse and human IFN-β are homologous, and the tertiary structures of mouse and human IFN-β are quite similar, with an RMSD over 0.4 (Fig. 3B). Thus, it was not immediately clear why they should have different killing abilities. Many AMPs have improved function in an acidic pH environment (8, 29). Depending on the amino acid composition of the AMP, an acidic pH can contribute to a higher positive charge. There are many microenvironments in the body that are characterized by an acidic pH, including the intracellular compartments of phagocytes (phagolysosomes), the surface of the skin, and abscesses. We evaluated the effect of pH on hIFN-β antimicrobial activity by incubating the protein with S. aureus at an acidic pH, and observed a marked increase in bacterial killing at lower pH (Fig. 3C). We compared S. aureus survival after treatment with different concentrations of hIFN-β at an acidic pH of 5, relative to vehicle (Fig. 3D). These results indicate that hIFN-β also has antimicrobial activity against S. aureus, but at lower pH. Furthermore, recombinant hIFN-β produced in either a bacterial (E. coli) or a mammalian cell expression system (CHO cells) both had killing activity at acidic pH (Fig. 3D), suggesting that post-translational modifications are not required for antimicrobial activity.
FIGURE 3. Human IFN-β kills S. aureus, but only at low pH.
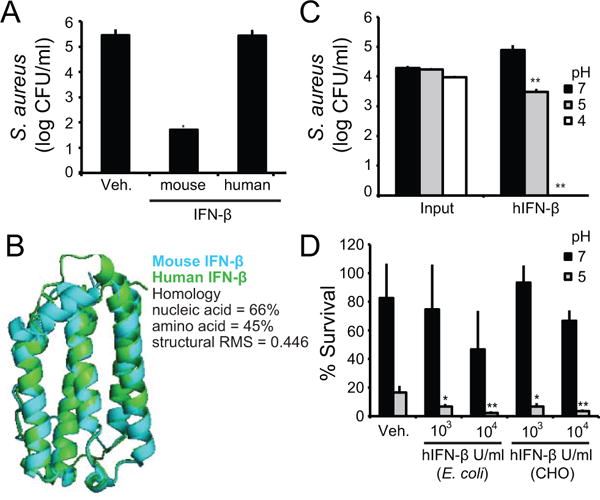
(A) S. aureus were incubated with VEH, mIFN-β, or recombinant hIFN-β at the same concentration of 5×102 units/ml for 3 hours. Remaining bacteria were serially-diluted and plated for enumeration. (B) Mouse and human IFN-β were compared at the nucleic acid, amino acid and structural levels. Structural RMS is shown for overlay. (C) S. aureus were incubated with VEH) or 5×103 units/ml hIFN-β in varying pHs for 3 hours. Remaining bacteria were serially-diluted and plated for enumeration. (D) S. aureus were incubated with VEH, hIFN-β produced in E. coli (E. coli), or hIFN-β produced in CHO cells (CHO) at indicated concentrations. Bacteria were incubated with hIFN-β in neutral and low pH for 3 hours. Remaining bacteria were serially-diluted and plated for enumeration. Data are shown as % surviving bacteria based on initial CFU. Data are shown as mean ± SD, * p ≤ 0.05, ** p ≤ 0.01 (unpaired, two-tailed t test).
Structural and compositional comparison of mouse and human IFN-β with AMPs
Similar to many cytokines, the tertiary structure of IFN-β is a bundle of five α-helices, (helix 1–5, in order of the N to C termini) (Fig. 4A). We compared the electrostatic potentials at both neutral and acidic pH mapped onto space-filling models of both mouse and human IFN-β (Fig. 4B). With cationic charge shown in blue, mIFN-β is highly positively-charged at both pHs, while the net charge of hIFN-β doubles at acidic pH (Fig. 4B). In addition, charge calculations of all type I interferon family members for both mouse and human showed a similar trend; many mouse type I IFNs are positively-charged at both neutral and acidic pH, while only a few human type I IFNs are positively-charged at a neutral pH (Fig. S2). Overall, the pH-dependent cationic charge of IFN-β may explain in part the killing ability we observed experimentally.
FIGURE 4. Structural comparison of mouse and human IFN-β.
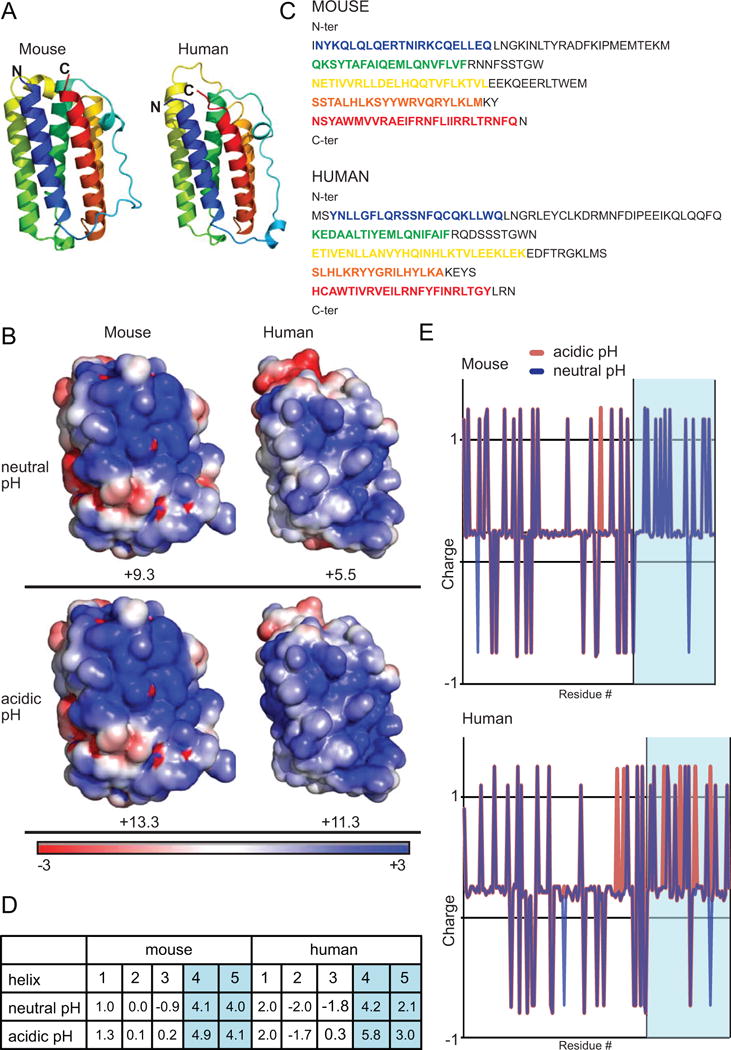
(A) Mouse and human IFN-β ribbon structures are shown. N and C termini are labeled. The five α-helices are color coded from N to C terminus blue, green, yellow, orange, and red. (B) Mouse and human IFN-β electrostatic space filling models are shown for neutral (pH 7) and acidic (pH 5) pH. Regions of positive charge are shown in blue, regions of neutral charge are shown in white, and regions of negative charge are shown in red. (C) Mouse and human IFN-β amino acid sequences are shown. The α-helices are color-coded as in (A). (D) Charge was calculated for all five helices for mouse and human IFN-β. The high positive charge of helices 4 and 5 are highlighted in blue. (E) Mouse and human IFN-β amino acid plots were generated showing approximate charge of each amino acid residue. Values for neutral (blue) and acidic (red) pH are shown. Blue region shows helices 4 and 5.
To determine whether we could identify a specific region of IFN-β that confers killing activity, we examined the individual component helices (Fig. 4C) and calculated the net charge for each helix (Fig. 4D). For both mouse and human IFN-β, the largest contribution to the net positive charge comes from the last two helices, 4 and 5 (Fig. 4D). In fact, when the individual charges of each amino acid were plotted, it was clear that the majority of positive residues were localized near the C-terminus of the molecule in helices 4 and 5 (Fig. 4E). These charge plots also showed that at an acidic pH, the net charge of hIFN-β C-terminal region increases dramatically (Fig. 4E).
While vastly diverse in sequence and structure, most AMPs share several common features; they are generally short (< 50 amino acids), have a net positive charge (+2 to +9) and a substantial proportion (≥ 30%) of hydrophobic residues. The fundamental motif common among AMPs is the ability to adopt an amphipathic secondary structure that clusters hydrophobic and cationic amino acids into distinct domains (30). Indeed, the cationic and amphipathic nature of AMPs has been found to be associated with their activity (30, 31).
The 21- and 17- residue long regions corresponding to helix 4 of mouse and human IFN-β, respectively, have the highest proportion of cationic polar residues and hydrophobic residues relative to all helices present in the cytokine (Fig. 5A, 5B, S3A). The two sequences also exhibit facially amphipathic character when adopting α-helical conformations, with cationic and hydrophobic residues segregated to opposite faces along the helical axis (Fig. 5A). These characteristics are similar to the structural motif common to a large class of AMPs (32, 33).
FIGURE 5. Mouse and human IFN-β helix 4 are similar to antimicrobial peptides.
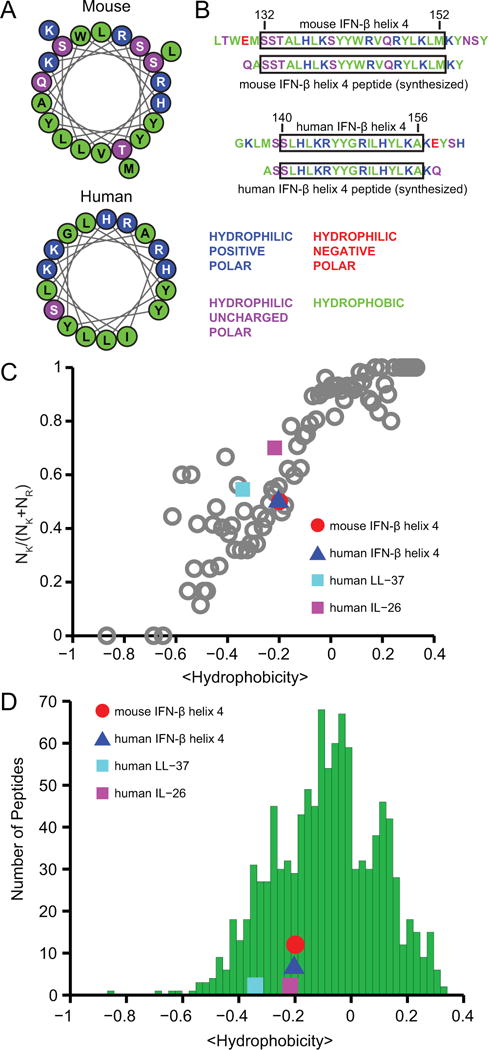
(A) Sequences of both mouse and human IFN-β helix 4 plotted as helical wheel projections to illustrate their facially amphipathic character. Positively-charged hydrophilic residues are indicated in blue, uncharged hydrophilic residues in violet, and hydrophobic residues in green. (B) Mouse and human IFN-β helix 4 sequences; native sequences are shown alongside synthesized peptides. Residues are color-coded as in (A). (C) Relationship between positively-charged residues (NK/(NK + NR)) and average peptide hydrophobicity for 1080 cationic AMPs in the antimicrobial peptide database (grey open circles). Mouse IFN-β helix 4 (red circle), human IFN-β helix 4 (blue triangle), human LL-37 (cyan square), and human IL-26 (magenta square). (D) The hydrophobicity of IFN-β helix 4 is comparable to that of antimicrobial peptides. Histogram depicting the distribution of average hydrophobicities among 1080 cationic AMPs in the antimicrobial peptide database (green bars) compared with the hydrophobicities of mouse IFN-β helix 4, human IFN-β helix 4, human LL-37, and human IL-26.
We also observed similarities between the amino acid compositions of IFN-β helix 4 and those of AMPs in general. In relation to the previously observed trend between NK/(NK + NR) (the ratio of the number of lysines to the total number of lysines and arginines) and average peptide hydrophobicity based on the Eisenberg consensus scale (19) for 1080 cationic AMPs from the antimicrobial peptide database (17), we found both mouse and human IFN-β helix 4 to contain similar proportions of cationic and hydrophobic residues (Fig. 5C). In fact, their positions relative to the AMP trendline (Fig. 5C) suggest high compositional resemblance to AMPs. Among the five helices of both mouse and human IFN-β, helix 4 exhibited the greatest correspondence to the AMP trendline (Fig. S3B). Moreover, when evaluating the amounts of hydrophobic residues independently, we found that the hydrophobicities of both mouse and human IFN-β helix 4 also fall within the range of hydrophobicities of AMPs (Fig. 5D and S3C). In both comparisons, we included human LL-37, a well-characterized α-helical AMP, and human IL-26, a cytokine recently described as having antimicrobial properties (14), for reference. Taken together, these results suggest that mouse and human IFN-β helix 4 may interact with membranes in a manner similar to AMPs.
Mouse IFN-β helix 4 induces membrane curvature in bacterial membranes and kills S. aureus
Earlier studies have identified a strong correlation between the formation of NGC and AMP-induced membrane destabilization mechanisms (11, 12, 34–37). NGC is also known as saddle-splay curvature, on which a surface curves upwards in one direction and downward in the perpendicular direction. NGC is topologically necessary for membrane destabilizing processes, including pore formation, budding, and blebbing, and thus, is said to be the primary mode by which AMPs compromise the barrier function of membranes (11, 12). For instance, NGC can be observed along the curved surface of a transmembrane pore (Fig. 6A).
FIGURE 6. Mouse IFN-β helix 4 induces membrane curvature in bacterial membranes and kills S. aureus.
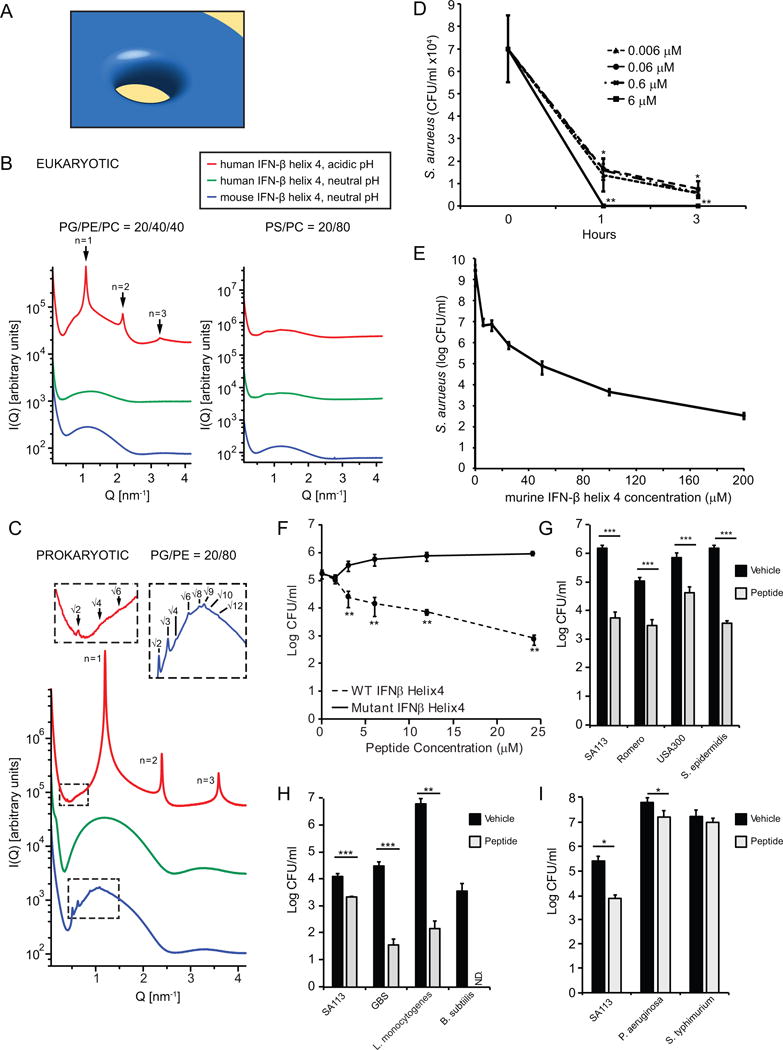
(A) Model of negative Gaussian curvature (NGC) required for membrane destabilization processes such as pore formation by AMPs. (B) IFN-β helix 4 does not generate NGC in eukaryotic-like membranes. SAXS spectra for mouse and human IFN-β helix 4 with eukaryotic model membranes DOPG/DOPE/DOPC = 20/40/40 and DOPS/DOPC = 20/80 at P/L = 1/50 peptide to lipid (P/L) molar ratio. (C) IFN-β helix 4 generates NGC in prokaryotic-like membranes. SAXS spectra from prokaryotic model membrane, DOPG/DOPE = 20/80, vesicles incubated with either mouse or human IFN-β helix 4 at P/L = 1/50 molar ratio. Insets show expanded view of cubic phase reflections. (D) S. aureus were incubated with VEH or mIFN-β helix 4 at 0.006, 0.06, 0.6, and 6 μM. Bacteria and treatments were incubated for 1 or 3 hours. Remaining bacteria were serially-diluted and plated for enumeration. (E) S. aureus were incubated with VEH or mouse IFN-β helix 4 at 6.25, 12.5, 25, 50, 100, and 200 μM. Bacteria and treatments were incubated for 3 hours. Remaining bacteria were serially-diluted and plated for enumeration. (F) Wild-type (WT) and mutant IFN-β helix 4 peptide were incubated with 1×105 SA113/ml in buffer + 1%THB for 3 hours. Remaining bacteria were serially-diluted and plated for enumeration. (G–I) Bacteria (1×105/ml) were incubated with vehicle or 6 μM mouse hexlix 4 peptide for 3 hours at 37°C. Remaining bacteria were serially-diluted and plated for enumeration. Data are shown as mean ± SD, * p ≤ 0.05,** p ≤ 0.01, ***p≤0.001 (unpaired, two-tailed t test). N.D.- below limit of detection.
We synthesized mouse and human IFN-β helix 4 peptides and used SAXS to quantitatively characterize the membrane curvature deformations they induce. We systematically examined a range of membranes with varying lipid compositions, modeled from the differing compositions of bacterial and eukaryotic cell membranes (30, 33, 38, 39). For example, in bacterial membranes, phosphatidylglycerol (PG) and phosphatidylethanolamine (PE) are the main anionic and zwitterionic lipids, respectively. In eukaryotic membranes, however, phosphatidylserine (PS) is the primary anionic lipid and phosphatidylcholine (PC) is the primary zwitterionic lipid. We prepared small unilamellar vesicles (SUVs) with fixed anionic charge typical of bacterial membranes (20%) but with varying concentrations of PE to represent both bacterial membranes (DOPG/DOPE = 20/80) and more eukaryotic-like membranes (DOPG/DOPE/DOPC = 20/40/40 and PS/PC = 20/80). Additional lipid compositions DOPG/DOPE/DOPC = 20/60/20 and DOPS/DOPE = 20/80 were included to assess the roles of PE content and distinctions between anionic lipids PG and PS, respectively. SUVs were incubated with mouse and human IFN-β helix 4 peptides at specific peptide/lipid (P/L) molar ratios and pHs, and the resulting structures were characterized by SAXS.
SAXS spectra for two eukaryotic-like membrane compositions, DOPG/DOPE/DOPC = 20/40/40 and DOPS/DOPC = 20/80, incubated with the mouse and human IFN-β helix 4 peptides at P/L = 1/50 are shown in Figure 6B. Synchrotron SAXS spectra for control SUVs each exhibited characteristic features consistent with a single lipid bilayer (Fig. S4A). Both mouse and human IFN-β helix 4 peptides did not restructure either of the two eukaryotic membrane compositions at neutral pH as shown by the form factors without correlation peaks. Similarly, hIFN-β helix 4 peptide at acidic pH (Fig. 6B) did not restructure DOPS/DOPC = 20/80 vesicles. For DOPG/DOPE/DOPC = 20/40/40, hIFN-β helix 4 peptide at an acidic pH, induced a lamellar (Lα) phase ( nm) with correlation peaks at integral Q-ratios of 1:2:3. This result reveals intermembrane attraction without the generation of significant curvature. Overall, we find that both mouse and human IFN-β helix 4 peptides do not induce NGC in eukaryotic-like membranes.
Figure 6C shows SAXS spectra for prokaryotic-like membrane DOPG/DOPE = 20/80 with IFN-β helix 4 peptides at P/L = 1/50. Upon exposure to mIFN-β helix 4 peptide at neutral pH, the lipid vesicles underwent a structural transition, as revealed by correlation peaks with Q-ratios , which indexed to a Pn3m “double-diamond” cubic (QII) lattice, with lattice parameter nm (Fig. S4B) and an average Gaussian curvature of (See Methods for indexing procedures). In contrast, hIFN-β helix 4 peptide at neutral pH, did not restructure the DOPG/DOPE = 20/80 vesicles, as evidenced by a broad form factor characteristic of isolated spherical vesicles and the absence of correlation peaks (Fig. 6C). However, hIFN-β helix 4 peptide at an acidic pH resulted in two distinct sets of correlation peaks. One set of peaks has Q-ratios , which indexed to an Im3m QII phase with a lattice parameter nm and . The second set of correlation peaks shows integral Q-ratios 1:2:3 consistent with an Lα phase of periodicity nm, indicating intermembrane attraction. Both the Pn3m and Im3m are bicontinuous cubic phases that consist of two non-intersecting aqueous regions separated by a lipid bilayer. The center of the bilayer traces a minimal surface characterized by NGC at every point. We find that both mouse and human IFN-β helix 4 peptides are able to induce this membrane destabilizing saddle-splay curvature in model bacterial membranes but not in model eukaryotic membranes. This type of behavior has been broadly observed for AMPs, which suggest that the ability to selectively induce NGC is a general property of AMPs (12). In fact, the NGC induced by both mouse and human IFN-β helix 4 peptides are quantitatively of similar magnitude to those of typical AMPs (12). Moreover, we found that the generation of NGC by both mouse and human IFN-β helix 4 peptides recapitulates the bactericidal activity of mouse and human IFN-β proteins, respectively, and the pH-dependent generation of NGC by hIFN-β helix 4 peptide tracks exactly with the pH-dependent killing activity by hIFN-β against S. aureus.
Similar to results shown in Figure 1 using the whole cytokine, we observed antimicrobial activity against S. aureus with mIFN-β helix 4 starting at nM concentrations (Figs. 6D and 6E). To define the minimum bactericidal concentration (MBC) of mIFN-β helix 4 against S. aureus we quantified killing of S. aureus in response to 6.25–200 μM mIFN-β helix 4 (Fig. 6E). The mIFN-β helix 4 exhibits an MBC of 12.5–25 μM, which is comparable to that of other AMPs, such as LL-37. Taken together, these results demonstrate that mIFN-β helix 4 exhibits antimicrobial activity against S. aureus and are mutually consistent with our SAXS data. We found that the ability of mIFN-β helix 4 to generate NGC was similar to natural AMPs and tracked with its bactericidal activity, which together suggests membrane permeation as at least one of its modes of its antimicrobial activity.
To examine the specificity of mIFN-β helix 4’s ability to kill bacteria, we generated a mutated version of helix 4 in which the charged amino acids hypothesized to be important for killing were neutralized. We observed that this mutated peptide was no longer able to kill S. aureus (Fig. 6F). When we incubated mIFN-β helix 4 with different strains of Staphylococcus bacteria we found that mIFN-β helix 4 displays antimicrobial activity against multiple stains of Staphylococcus bacteria (Fig. 6G). In addition, the antimicrobial activity of mIFN-β was found to be effective against multiple strains of gram-positive bacteria (Fig. 6H), but was significantly less effective against the gram-negative strains of bacteria that we tested (Fig. 6I).
Discussion
We have identified a previously unrecognized antimicrobial function of the cytokine IFN-β. We showed that mIFN-β is able to inhibit growth and directly kill S. aureus. In fact, the antibacterial activity of IFN-β resembles that of natural AMPs in several respects, as demonstrated by its ability to bind and disrupt bacterial membranes. Examining this further, we found that the component helix 4 of IFN-β had structural and compositional properties, as well as net charge, that were similar to known AMPs. Using SAXS, we then observed that, like typical AMPs, both synthesized mouse and human IFN-β helix 4 are capable of generating membrane NGC, the type of curvature required for membrane disruption events. In fact, the generation of NGC by both mouse and human IFN-β helix 4 peptides recapitulates the bactericidal activity of mouse and human IFN-β proteins, respectively, and the pH-dependent generation of NGC by human IFN-β helix 4 peptide tracks exactly with the pH-dependent killing activity by human IFN-β against S. aureus. Together these results suggest that the antimicrobial activity we observed experimentally with whole IFN-β is likely attributed, at least in part, to its component helix 4 having AMP-like properties. Thus, in addition to its well-documented cytokine activity, IFN-β has direct antimicrobial activity. Both activities are likely relevant to efficient clearance of bacterial infections, although their relative contributions likely depend on the type of bacteria, the site(s) of infection and the duration of the infection. Future studies will be required to try to tease these roles apart.
Recent observations have shown that proteins with AMP-like sequence trends exhibit antimicrobial activity (40), which is likely correlated with the ability of that protein surface to induce similar types of membrane curvatures as AMPs. Another possibility is IFN-β degradation. Many AMPs are secreted in pro- or even pre-pro- forms, requiring cleavage events before the active peptide is released. For example, human cathelicidin is cleaved into the AMP LL-37 by serine proteases (10). Consistent with the degradation or cleavage hypothesis, we observed that a synthetic peptide consisting of helix 4 of IFN-β is sufficient to kill bacteria.
Kinocidins are defined as chemokines and cytokines that also have antimicrobial function, many of which function as AMPs. A few notable examples include the α-chemokine family CXCL-1, 4, 6, 7, 8, 9, 10, and 14; the β-chemokine family CCL- 1, 2, 5, and 8; and the cytokine IL-26 (13, 14). The findings here suggest that IFN-β is another member of the kinocidin family. Type I interferons are increasingly recognized as important for host defense against bacteria. A protective role for this cytokine family has been found for both environmental and host-adapted bacterial pathogens (41). The new evidence presented here supports the notion that IFN-β has complementary antimicrobial activity, and offers new insights into the multifunctional role and evolution of IFN-β in host defense.
Supplementary Material
Acknowledgments
We thank Dr. Jeff Miller for his edits of the manuscript.
This work was supported by grants from the National Institutes of Health (T32 AI007323 to A. K., R01 GM085796 to D.M.U., T32 AI089553-01 to A.J.W., R01 AI074832 to G. Y. L., and NSF-DMR-1411329 to M.W.L. and G.C.L.W.
References
- 1.Platanias LC. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat Rev Immunol. 2005;5:375–386. doi: 10.1038/nri1604. [DOI] [PubMed] [Google Scholar]
- 2.Monroe KM, McWhirter SM, Vance RE. Induction of type I interferons by bacteria. Cell Microbiol. 2010;12:881–890. doi: 10.1111/j.1462-5822.2010.01478.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rizza P, Moretti F, Belardelli F. Recent advances on the immunomodulatory effects of IFN-α: Implications for cancer immunotherapy and autoimmunity. Autoimmunity. 2010;43:204–209. doi: 10.3109/08916930903510880. [DOI] [PubMed] [Google Scholar]
- 4.González-Navajas JM, Lee J, David M, Raz E. Immunomodulatory functions of type I interferons. Nat Rev Immunol. 2012;12:125–135. doi: 10.1038/nri3133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang SY, Boisson-Dupuis S, Chapgier A, Yang K, Bustamante J, Puel A, Picard C, Abel L, Jouanguy E, Casanova J-L. Inborn errors of interferon (IFN)-mediated immunity in humans: insights into the respective roles of IFN-α/β, IFN-γ, and IFN-λ in host defense. Immunol Rev. 2008;226:29–40. doi: 10.1111/j.1600-065X.2008.00698.x. [DOI] [PubMed] [Google Scholar]
- 6.Wertheim H, Melles DC, Vos MC, Leeuwen Wvan, Belkum Avan, Verbrugh HA, Nouwen JL. The role of nasal carriage in. Lancet Infectious Disease. 2005;5:751–762. doi: 10.1016/S1473-3099(05)70295-4. [DOI] [PubMed] [Google Scholar]
- 7.Miller LS, Cho JS. Immunity against Staphylococcus aureus cutaneous infections. Nat Rev Immunol. 2011;11:505–518. doi: 10.1038/nri3010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ganz T. Defensins: antimicrobial peptides of innate immunity. Nat Rev Immunol. 2003;3:710–720. doi: 10.1038/nri1180. [DOI] [PubMed] [Google Scholar]
- 9.Peschel A, Sahl H-G. The co-evolution of host cationic antimicrobial peptides and microbial resistance. Nat Rev Micro. 2006;4:529–536. doi: 10.1038/nrmicro1441. [DOI] [PubMed] [Google Scholar]
- 10.Gallo RL, Hooper LV. Epithelial antimicrobial defence of the skin and intestine. Nat Rev Immunol. 2012;12:503–516. doi: 10.1038/nri3228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schmidt NW, Wong GCL. Antimicrobial peptides and induced membrane curvature: Geometry, coordination chemistry, and molecular engineering. Current Opinion in Solid State and Materials Science. 2013;17:151–163. doi: 10.1016/j.cossms.2013.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schmidt NW, Mishra A, Lai GH, Davis M, Sanders LK, Tran D, Garcia A, Tai KP, McCray PB, Jr, Ouellette AJ, Selsted ME, Wong GCL. Criterion for Amino Acid Composition of Defensins and Antimicrobial Peptides Based on Geometry of Membrane Destabilization. J Am Chem Soc. 2011;133:6720–6727. doi: 10.1021/ja200079a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yount NY, Yeaman MR. Emerging Themes and Therapeutic Prospects for Anti-Infective Peptides. Annu Rev Pharmacol Toxicol. 2012;52:337–360. doi: 10.1146/annurev-pharmtox-010611-134535. [DOI] [PubMed] [Google Scholar]
- 14.Meller S, Domizio JDi, Voo KS, Friedrich HC, Chamilos G, Ganguly D, Conrad C, Gregorio J, Roy DLe, Roger T, Ladbury JE, Homey B, Watowich S, Modlin RL, Kontoyiannis DP, Liu Y-J, Arold ST, Gilliet M. TH17 cells promote microbial killing and innate immune sensing of DNA via interleukin 26. Nat Immunol. 2015;16:970–979. doi: 10.1038/ni.3211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tang YQ, Yeaman MR, Selsted ME. Antimicrobial Peptides from Human Platelets. Infection and Immunity. 2002;70:6524–6533. doi: 10.1128/IAI.70.12.6524-6533.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA. Electrostatics of nanosystems: Application to microtubules and the ribosome. Proc Natl Acad Sci. 2001;98:10037–10041. doi: 10.1073/pnas.181342398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang G, Li X, Wang Z. APD2: the updated antimicrobial peptide database and its application in peptide design. Nucleic Acids Research. 2009;37:D933–D937. doi: 10.1093/nar/gkn823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schmidt NW, Mishra A, DeGrado WF, Wang J, Wong GCL. Influenza Virus A M2 Protein Generates Negative Gaussian Membrane Curvature Necessary for Budding and Scission. J Am Chem Soc. 2013;135:13710–13719. doi: 10.1021/ja400146z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Eisenberg D, Schmidt NW, Terwilliger TC, Weiss RM, Wong GCL, Wilcox W. Hydrophobic moments and protein structure. Faraday Symp Chem Soc. 1982;17:109–120. [Google Scholar]
- 20.Ilavsky J. Nika: software for two-dimensional data reduction. J Appl Crystallogr. 2012;45:324–328. [Google Scholar]
- 21.Kaplan A, Kaplan A, Ma J, Ma J, Kyme P, Kyme P, Wolf AJ, Wolf AJ, Becker CA, Becker CA, Tseng CW, Tseng CW, Liu GY, Liu GY, Underhill DM, Underhill DM. Failure To Induce IFN- Production during Staphylococcus aureus Infection Contributes to Pathogenicity. J Immunol. 2012;189:4537–4545. doi: 10.4049/jimmunol.1201111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Inoue M, Chen P-H, Siecinski S, Li Q-J, Liu C, Steinman L, Gregory SG, Benner E, Shinohara ML. An interferon-β-resistant and NLRP3 inflammasome-independent subtype of EAE with neuronal damage. Nat Neurosci. 2016;19:1599–1609. doi: 10.1038/nn.4421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Andrade WA, Firon A, Schmidt T, Hornung V, Fitzgerald KA, Kurt-Jones EA, Trieu-Cuot P, Golenbock DT, Kaminski P-A. Group B Streptococcus Degrades Cyclic-di-AMP to Modulate STING-Dependent Type I Interferon Production. Cell Host Microbe. 2016;20:49–59. doi: 10.1016/j.chom.2016.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mayer-Barber KD, Andrade BB, Oland SD, Amaral EP, Barber DL, Gonzales J, Derrick SC, Shi R, Kumar NP, Wei W, Yuan X, Zhang G, Cai Y, Babu S, Catalfamo M, Salazar AM, Via LE, Barry CE, Sher A. Host-directed therapy of tuberculosis based on interleukin-1 and type I interferon crosstalk. Nature. 2014;511:99–103. doi: 10.1038/nature13489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Teles RMB, Graeber TG, Krutzik SR, Montoya D, Schenk M, Lee DJ, Komisopoulou E, Kelly-Scumpia K, Chun R, Iyer SS, Sarno EN, Rea TH, Hewison M, Adams JS, Popper SJ, Relman DA, Stenger S, Bloom BR, Cheng G, Modlin RL. Type I Interferon Suppresses Type II Interferon-Triggered Human Anti-Mycobacterial Responses. Science. 2013;339:1448–1453. doi: 10.1126/science.1233665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Saar-Dover R, Bitler A, Nezer R, Shmuel-Galia L, Firon A, Shimoni E, Trieu-Cuot P, Shai Y. D-Alanylation of Lipoteichoic Acids Confers Resistance to Cationic Peptides in Group B Streptococcus by Increasing the Cell Wall Density. PLoS Pathog. 2012;8:e1002891. doi: 10.1371/journal.ppat.1002891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Peschel A. How do bacteria resist human antimicrobial peptides? Trends in Microbio. 2002;10:179–186. doi: 10.1016/s0966-842x(02)02333-8. [DOI] [PubMed] [Google Scholar]
- 28.Peschel A, Otto M, Jack RW, Kalbacher H, Jung G, Götz F. Inactivation of the dlt operon in Staphylococcus aureus confers sensitivity to defensins, protegrins, and other antimicrobial peptides. J Biol Chem. 1999;274:8405–8410. doi: 10.1074/jbc.274.13.8405. [DOI] [PubMed] [Google Scholar]
- 29.Yount NY, Yeaman MR. Multidimensional signatures in antimicrobial peptides. Proc Natl Acad Sci. 2004;101:7363–7368. doi: 10.1073/pnas.0401567101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zasloff M, Schmidt NW, Mishra A, Lai GH, Davis M, Sanders LK, Tran D, Garcia A, Tai KP, McCray PB, Jr, Ouellette AJ, Selsted ME, Wong GCL. Antimicrobial peptides of multicellular organisms. Nature. 2002;415:389–395. doi: 10.1038/415389a. [DOI] [PubMed] [Google Scholar]
- 31.Hancock REW, Sahl H-G. Antimicrobial and host-defense peptides as new anti-infective therapeutic strategies. Nat Biotechnol. 2006;24:1551–1557. doi: 10.1038/nbt1267. [DOI] [PubMed] [Google Scholar]
- 32.Hancock REW, Lehrer R, Schmidt NW, Tai KP, Kamdar K, Mishra A, Lai GH, Zhao K, Ouellette AJ, Wong GCL. Cationic peptides: a new source of antibiotics. Trends in Biotechnology. 1998;16:82–88. doi: 10.1016/s0167-7799(97)01156-6. [DOI] [PubMed] [Google Scholar]
- 33.Brogden KA. Antimicrobial peptides: pore formers or metabolic inhibitors in bacteria? Nat Rev Micro. 2005;3:238–250. doi: 10.1038/nrmicro1098. [DOI] [PubMed] [Google Scholar]
- 34.Schmidt NW, Hu K, Zhu R, Jiang Y, Wong GCL, Lai GH, Wei G, Palermo EF, Kuroda K, Yang L. A Critical Evaluation of Random Copolymer Mimesis of Homogeneous Antimicrobial Peptides. Macromolecules. 2013;46:1908–1915. doi: 10.1021/ma302577e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xiong M, Lee MW, Song Z, Mansbach RA, Yao C, Bao Y, Wong GCL, Peek RM, Jr, Chen L-F, Ferguson AL, Cheng J. Helical antimicrobial polypeptides with radial amphiphilicity. Proc Natl Acad Sci. 2015;112:13155–13160. doi: 10.1073/pnas.1507893112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schmidt NW, Tai KP, Kamdar K, Mishra A, Lai GH, Zhao K, Ouellette AJ, Wong GCL. Arginine in α-defensins: differential effects on bactericidal activity correspond to geometry of membrane curvature generation and peptide-lipid phase behavior. J Biol Chem. 2012;287:21866–21872. doi: 10.1074/jbc.M112.358721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lee MW, Chakraborty S, Schmidt NW, Murgai R, Gellman SH, Wong GCL. Two interdependent mechanisms of antimicrobial activity allow for efficient killing in nylon-3-based polymeric mimics of innate immunity peptides. Biochimica et Biophysica Acta (BBA) - Biomembranes. 2014;1838:2269–2279. doi: 10.1016/j.bbamem.2014.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.van Meer G, Voelker DR, Feigenson GW. Membrane lipids: where they are and how they behave. Nat Rev Mol Cell Biol. 2008;9:112–124. doi: 10.1038/nrm2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Epand RM, Epand RF. Lipid domains in bacterial membranes and the action of antimicrobial agents. Biochimica et Biophysica Acta (BBA) - Biomembranes. 2009;1788:289–294. doi: 10.1016/j.bbamem.2008.08.023. [DOI] [PubMed] [Google Scholar]
- 40.Realegeno S, Kelly-Scumpia KM, Kelly-Scumpia KM, Dang AT, Dang AT, Lu J, Lu J, Teles R, Teles RMB, Liu PT, Liu PT, Schenk M, Schenk M, Lee EY, Lee EY, Schmidt NW, Schmidt NW, Wong GCL, Wong GCL, Sarno EN, Sarno EN, Rea TH, Rea TH, Ochoa MT, Ochoa MT, Pellegrini M, Pellegrini M, Modlin RL, Modlin RL. S100A12 Is Part of the Antimicrobial Network against Mycobacterium leprae in Human Macrophages. PLoS Pathog. 2016;12:e1005705. doi: 10.1371/journal.ppat.1005705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McNab F, Mayer-Barber K, Sher A, Wack A, O’Garra A. Type I interferons in infectious disease. Nat Rev Immunol. 2015;15:87–103. doi: 10.1038/nri3787. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.