

Abstract
Purpose:
We investigated 2-(5-fluoro-pentyl)-2-methyl-malonic acid (18F-ML-10) positron emission tomography (PET) imaging of apoptosis posttherapy to determine optimal timing for predicting chemotherapy response in a mouse head/neck xenograft cancer model.
Procedures:
BALB/c nude mice (4-8 weeks old) were implanted with UM-SCC-22B tumors. The treatment group received 2 doses of doxorubicin (10 mg/kg, days 0, 2). Small animal 18F-ML-10 PET/computed tomography was performed before and on days 1, 3, and 7 postchemotherapy. Using regions of interest around tumors, 18F-ML-10 uptake change was measured as %ID/g and uptake relative to liver. Terminal Uridine Nick-End Labeling (TUNEL) immunohistochemistry assay was performed using tumor samples of baseline and on days 1, 3, and 7 posttreatment.
Results:
Treated mice demonstrated increased 18F-ML-10 uptake compared to baseline and controls, and 10 of 13 mice showed tumor volume decreases. All control mice showed tumor volume increases. Tumor-to-liver (T/L) ratios from the control group mice did not show significant change from baseline (P > .05); however, T/L ratios of the treatment group showed significant 18F-ML-10 uptake differences from baseline compared to days 3 and 7 posttreatment (P < .05), but no significant difference at 1 day posttreatment.
Conclusion:
2-(5-Fluoro-pentyl)-2-methyl-malonic acid PET imaging has the potential for early assessment of treatment-induced apoptosis. Timing and image analysis strategies may require optimization, depending on the type of tumor and cancer treatment.
Keywords: molecular imaging, apoptosis, early response, 18F-ML-10, PET
Introduction
Apoptosis, also called programmed cell death, is an essential process for eliminating unwanted cells and maintenance of tissue homeostasis.1,2 After decades of intense research, apoptosis is now considered to be a fundamental process leading to the maintenance of normal tissues, but also as a form of cell death in response to oncolytic therapies.3 Tumors that are treated with a therapy that induces programmed cell death typically regress, which is attributable to apoptosis or senescence.4
The process of apoptosis is easily detected in vitro, relying on the measurement of intracellular or surface molecules, readily observed when phosphatidylserine is translocated to the outer leaflet of the plasma membrane.5 However, measurement of apoptosis in vivo is challenging. A noninvasive method to detect apoptosis in vivo can assist in monitoring the response to treatment and monitoring the efficacy of antitumor agents for cancer. Early assessment of response to chemotherapy may also prevent unnecessary toxicity and minimize cost of ineffective treatment, enabling the tailoring of treatment regimens to individual patients.6,7
Given the importance of being able to specifically image apoptosis, several potential imaging probes have been developed; however, no apoptosis positron emission tomography (PET) imaging agent has been approved for clinical use. Previously studied agents labeled with positron-emitting radionuclides include annexin V,5,8 synaptotagmin I,9 caspase-3 inhibitors based on isatins,10 [18F]-C-SNAT,11 as well as hydrophobic cations such as 18F-fluorobenzyl triphenylphosphonium cation12 and [18F]-CP18,13 all of which target different processes in the apoptotic cascade.14
The most widely studied agent that has been labeled with PET and single-photon emission computed tomography (SPECT) radionuclides is based on annexin-V, which binds phosphatidylserine head groups on the cell surface, an early and universal event in apoptosis.5,8 Annexin V has been labeled with Tc-99m for SPECT as well as with F-18 and Ga-68 for PET imaging.15-17 However, annexin-V labels both apoptotic and necrotic cells by binding to intracellular phosphatidylserine through disrupted plasma membranes18 and also has slow clearance from nontargeted tissues due to its large size (∼35 kDa).19
2-(5-Fluoro-pentyl)-2-methyl-malonic acid (18F-ML-10) is a low-molecular-weight PET probe (molecular weight 206 Da), a member of ApoSense (Aposense Ltd, Petach-Tikva, Israel) family of molecules, and was designed for selective detection of apoptosis in vivo in the clinical setting. The target of the probe is a unique set of alterations that take place on the cell membrane at the time when the cell commits to apoptosis. This selective set of alterations include the following elements: (1) activation of the phospholipid scrambling system that leads to exposure on the cell surface of the acidic phospholipid phosphatidylserine; (2) acidification of the membrane surface leading to monoprotonation of 18F-ML-10, which changes the molecule’s conformation and its interaction with the membrane interface; (3) subsequent flip-flopping of the molecule through the membrane’s hydrophobic hydrocarbon core to the inner membrane leaflet, driven by scramblase activation and the irreversible depolarization of the cell membrane during the apoptotic process; and (4) binding of the molecule to cytoplasmic proteins through electrostatic and hydrophobic interactions, augmented by the irreversible loss of cellular pH control that leads to reduction in the pH of the proteins to their isoelectric points and dehydration. This set of alterations acts in a concerted manner at the point of commitment of the cell to the death program and then acts as a selective transmembrane transporter of 18F-ML-10 into apoptotic cells and a driving force for its accumulation in the cytoplasm of the apoptotic cells, but not in viable or necrotic cells.20
In various preclinical models, both in vitro and in vivo, ranging from models of ischemic insults to anticancer therapy in cell models, selective accumulation of ML-10 was highly correlated with apoptosis observed using well-established in vitro markers such as annexin V, caspase activation, and the TUNEL assay for apoptotic DNA fragmentation.20 A human originated head/neck squamous cell carcinoma xenograft model, UM-SCC-22B cell line, was successfully used to document chemotherapy-induced apoptosis by various groups.21,22
Molecular imaging of early apoptosis with 18F-ML-10 PET/computed tomography (CT) may be very helpful in timely assessment of response to treatment cancer in order to improve clinical management. Oborski et al recently showed the first time use of 18F-ML-10 for early-therapy response assessment (ETA) in a patient with glioblastoma multiforme (GBM). In this case report, after 3 weeks of therapy, a change in 18F-ML-10 uptake between baseline and ETA was clearly visible.23 Selection of imaging time points for response assessment in clinical trials with 18F-fluoro-2-deoxy-D-glucose (18F-FDG) is typically based on an expected tumor metabolic response times of a few weeks; however, as apoptosis is a highly dynamic process, optimal timing for future clinical trials using 18F-ML-10 for apoptosis imaging requires further investigation.
In this study, we hypothesized that longitudinal imaging at multiple time points in a mouse model pre- and postchemotherapy might provide insight into the dynamic nature of drug-induced apoptosis. The timing of imaging apoptosis was investigated with 18F-ML-10 for the early prediction of therapy response in mice bearing human UM-SCC-22B head and neck squamous cell carcinoma cells.
Materials and Methods
Reagents and Instrumentation
Reagents
The XTT assay kit for calorimetric assay was purchased from Sigma-Aldrich (St Louis, Missouri). All other chemicals and reagents were obtained from Sigma-Aldrich. Female BALB/c nude mice (4-8 weeks old) were obtained from Charles River Laboratories.
Preparation of PET tracer
18F-ML-10 was produced at the University of Pittsburgh PET Facility Radiochemistry Laboratory (Pittsburgh, Pennsylvania) using precursor supplied by Aposense Ltd. [18F]Fluoride delivered from the cyclotron was trapped on a quaternary ammonium solid-phase extraction cartridge (Accell Plus-QMA initially treated with 10 mL of 0.1 M aqueous sodium bicarbonate followed by 5 mL anhydrous acetonitrile followed by air). An aqueous solution of 22 mg K2CO3 and 7 mg Kryptofix222 (1.0 mL total volume) was passed through the cartridge to elute the [18F]fluoride. Azeotropic drying at 90°C in a reaction vial under argon with acetonitrile (2 × 1 mL) furnished the dry [18F]fluoride complex to be used for the fluorination reaction.24
A solution of ML-10 precursor (2.5 mg) in anhydrous acetonitrile (1 mL) was heated at 100°C for 15 minutes. The crude reaction mixture was cooled and aqueous hydrochloric acid (2 N, 0.5 mL) was added. The solution was allowed to react at 110°C for 15 minutes. Isolation of 18F-ML-10 was achieved via purification using a semi-preparative reversed-phase High performance liquid chromatography (HPLC) system (mobile phase: water/acetonitrile/TFA 80/20/1 [vol/vol/vol], Phenomenex ODS column, flow rate: 4 mL/min for 0 to 5 minutes and then flow increased to 7 mL/min for remainder of purification) to provide purified 18F-ML-10. The HPLC product fraction containing 18F-ML-10 (retention time [RT] approximately 25 minutes) was diluted with 0.1 N hydrochloric acid (60 mL) and passed across a C-18 Sep-Pak. The final product was recovered into a sample vial by slowly flushing the C-18 Sep-Pak with absolute ethanol (1 mL).
Chemical purity, radiochemical purity, and specific activity were assessed by analytical HPLC (Phenomenex Prodigy ODS-3 5 micron [4.6 × 250 mm]; mobile phase 25/75/0.1 MeCN/H2O/TFA (vol/vol/vol); flow rate 1.5 mL/min, λ = 205 nm). To prepare a solution suitable for in vivo administration, the 1 mL ethanol preparation containing 18F-ML-10 was passed through a sterilization filter (Millipore FG, Bedford, MA, 0.22 µm, 25 mm) into a 20 mL sterile empty vial. The ethanol solution was diluted with saline for injection, United States Pharmacopeia (USP) to provide a final product formulation suitable for intravenous (IV) administration. 18F-ML-10 was obtained in overall yields of 35% ± 10% (decay corrected). Radiochemical and chemical purities were >95% and the end of synthesis-specific activity was >37 GBq/μmol (1000 Ci/mmol).
Cell line
The UM-SCC-22B human head and neck squamous cancer cell line transfected with luciferase was obtained from the University of Michigan. UM-SCC-22B/luc cells were cultured in Dulbecco Modified Eagle Medium supplemented with 10% fetal bovine serum, 2% l-glutamine, and 1% Pen-Strep.
Cytotoxicity Assay
The cytotoxic effect of doxorubicin on UM-SCC-22B/luc cells was determined using the calorimetric XTT assay kit. Cells were seeded in a 96-well flat bottom plate with a density of 5 × 103 per well (n = 3) and incubated at 37°C. After 1 day, UM-SCC-22B/luc cells were treated with increasing concentrations of doxorubicin from 0.05 to 5 mg/mL for 24, 48, and 72 hours and incubated at 37°C. Optical density measurements at 450 nm reflected the viable cell population, and background was measured at 690 nm in each well using a microplate reader (Synergy H4 Hybrid Multi-Mode Microplate Reader; BioTek, Vermont, USA). Experiments were performed in triplicate. The ratio of the viability was determined by dividing the absorbance of treated cells to untreated cells.
Animal Model and Treatment Plan
Animal experiments were conducted in compliance with the guidelines for the care and use of research animals established by the Institutional Animal Care and Use Committee at the University of Pittsburgh. Mice were injected subcutaneously in the left or right shoulder with an average of 5.2 × 106 UM-SCC-22B/luc cells per site. Tumor volumes were measured by microCT by manually drawing contours on adjacent axial CT images. Volumetric calculations were performed with surface reconstruction method from parallel contours using Siemens Inveon Research Workplace (IRW) software (Knoxville, TN).
Mice were randomly divided into treated and control groups, respectively. Apoptosis was induced in the treatment group mice after intravenous administration of 2 doses of 10 mg/kg of doxorubicin via tail vein on the day of baseline imaging and then on day 2 (Figure 1). No treatment was given to control mice.
Figure 1.

Study design with small-animal PET/CT imaging using 18F-ML-10 in nude mice bearing UM-SCC-22B xenograft. IHC indicates immunohistochemical.
The 18F-ML-10 imaging study started with 26 nude mice bearing UM-SCC-22B xenografts. Two untreated mice and 6 mice from the treated group were killed (n = 8) for TUNEL assay. PET/CT images of 2 mice were excluded from the study due to skin contamination with urine activity and scatter correction errors. Mice that had a complete set of scans on all 4 days (n = 18) were included in the image analyses.
Positron emission tomography/CT Imaging
An Inveon Preclinical PET/CT scanner (Siemens, Knoxville, Tennessee) was used for small animal PET/CT imaging. Positron emission tomography and CT images were acquired on subsequent sequences without any movement of the animals. PET images were reconstructed with the maximum a posteriori (MAP) algorithm (16 iterations) and ordered subsets expectation maximization (3D-OSEM, 2 iterations) using the Siemens Acquisition Workspace software. Regions of interest (ROIs) were drawn on CT images and transferred to the simultaneously acquired PET images using the IRW software. Dynamic imaging was performed for 60 minutes with list mode acquisition after injection of ∼3.4 MBq (∼100 µCi) of 18F-ML-10 on healthy nude mice (n = 3). In UM-SCC-22B tumor-bearing mice, baseline PET imaging was performed 3 weeks after tumor implantation. Mice were anesthetized with 3% isoflurane and 97% oxygen, and each mouse was injected with 6.5 to 7.4 MBq (175-200 μCi) of 18F-ML-10 in a volume of 150 μL of 10% aqueous ethanol via tail vein. Positron emission tomography/CT scans (5-minute static) were acquired 30 minutes post-radiotracer injection. Baseline 18F-ML-10 PET/CT scans were performed on each mouse and repeated on 1, 3, and 7 days after initiation of doxorubicin treatment.
Image Analysis
Positron emission tomography images were reconstructed with MAP (16 iterations) and 3D-OSEM (2 iterations) using Siemens Acquisition Workspace software. Quantification of tracer uptake was performed by visually drawing ROI based on the fused PET/CT images, and the corresponding activity values were determined using the IRW software (Siemens). All values are represented as percentage injected dose per gram (%ID/g). Data were decay corrected from the time of 18F-ML-10 administration. Because of the consistent and low uptake in the liver for all scans, the liver uptake was used for normalization by calculating tumor-to-liver (T/L) ratios. Percentage change from baseline was calculated for each time point to compare groups.
TUNEL Immunohistochemistry Assay
At baseline (n = 2), day 1 (n = 3), and day 3 (n = 3), 8 mice were killed. Tumor tissues were excised, fixed in formalin, embedded in paraffin, sectioned into 5 μm slices, and processed for TUNEL staining, using the ApopTag Fluorescein In Situ Apoptosis Detection Kit (Millipore Corporation). The ProLong Gold Antifade mounting solution (Invitrogen, Woburn, MA) containing 4′,6-diamidino-2-phenylindole (DAPI) was added to tissue sections prior coverslips mounting. All assays were performed according to the manufacturer instructions, and sections were counterstained with hematoxylin and eosin staining. TUNEL assay staining was monitored using a Zeiss Apotome system and microscope (Peabody, MA). All histology image analysis was performed with Carl Zeiss ZEN software (Göttingen, Germany). To determine the amount of apoptosis, the DAPI counterstained cells (representing total cells) for an entire 10× field of view were counted using Zeiss Zen microscopy software. Then, the TUNEL-positive cells were counted. These counts were used to determine the percentage of apoptotic cells in the field. This was repeated for 3 fields of view for the 1- and 3-day groups and on 2 fields for the day 0 group.
Statistical Analysis
Statistical analyses were performed using PRISM v6 software (Graphpad, La Jolla, California). P values were calculated using Student t test. Results with P < .05 were considered statistically significant.
Results
The treated UM22B cells showed significant inhibition with increasing concentration of doxorubicin compared to untreated cells (Figure 2). At lower concentrations of doxorubicin (0.005-0.05 mg/L), the viability ratio at 24 and 48 hours was low (0.3); however, the ratio increased to 0.8 at 72 hours. By increasing the concentration of doxorubicin to 5 mg/L, the viability ratio was low (0.2) at 24 and 48 hours and remained low (0.3) at 72 hours (Figure 2).
Figure 2.
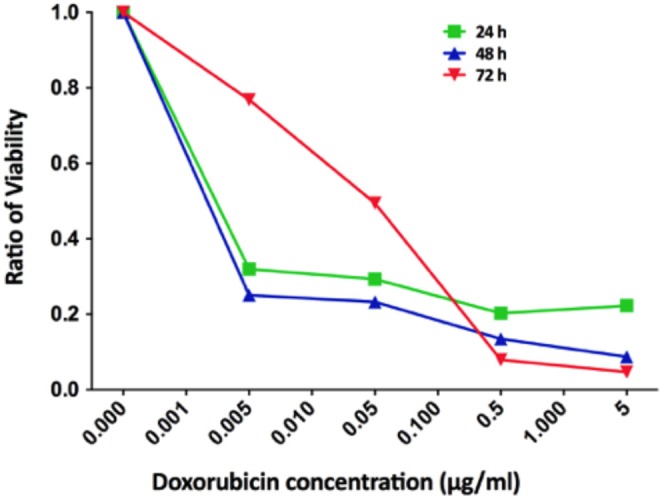
Cell viability ratio of UM-SCC-22B measured using the XTT proliferation assay after incubation with different concentrations of the doxorubicin out to 72 hours.
To determine the optimal imaging time point after radiotracer injection, dynamic PET imaging was performed in healthy nude mice (n = 3) after tail vein injection of 18F-ML-10 (∼3.4 MBq; ∼100 µCi). Images revealed a rapid clearance of the tracer from the blood into the kidneys, with no significant change in biodistribution from 30 to 60 minutes postinjection. Due to these results, static scans for tumor bearing mice were done at 30 minutes post-radiotracer injection. After the initial dynamic study in normal mice, we performed static scans on tumor-bearing mice at 30 minutes post-radiotracer injection, and 18F-ML-10 uptake in aorta, brain, kidneys, liver, and muscle was noted (Figure 3).
Figure 3.
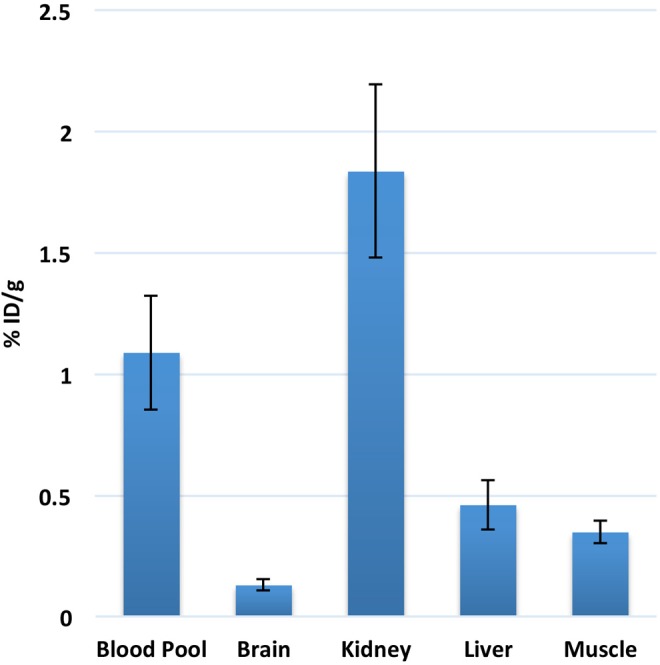
Radiotracer distribution based on imaging data of 18F-ML-10 in healthy nude female mice at 30 minutes post-injection. Low uptake was noted in brain and muscle. There was significant uptake of radiotracer in blood pool, which cleared out by 2 hours. Excretion of radiotracer was noted from kidneys and urinary bladder.
In tumor-bearing mice, baseline 18F-ML-10 PET/CT scans were acquired on day 0. Subsequent scans were obtained on days 1, 3, and 7. PET images demonstrated heterogeneous distribution of radiotracer in tumors. Uptake in the liver was relatively low. PET/CT imaging data in mice treated with doxorubicin (Figure 4) demonstrated an increase in 18F-ML-10 uptake by tumors compared to control mice as well as compared to baseline imaging at day 0 (Figure 4B).
Figure 4.
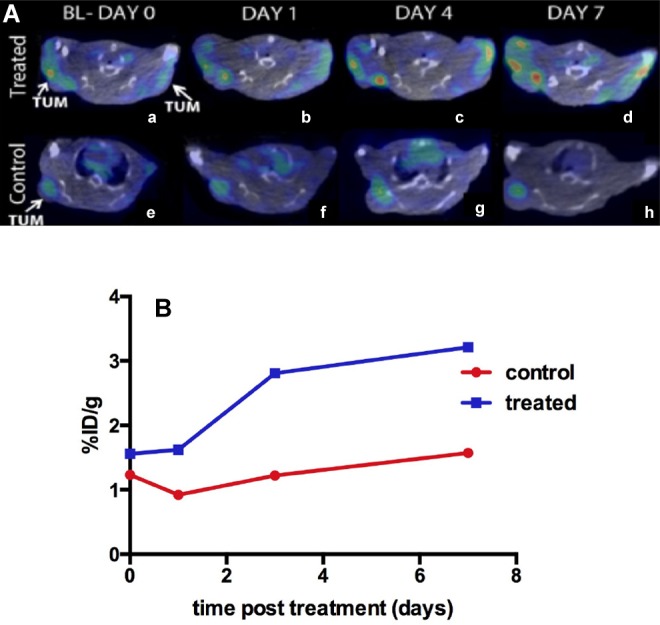
A, Example of coregistered PET/CT images of 2 mice with tumors in both shoulders (treated). The treated mouse (top row, a, b, c, and d) demonstrated increasing uptake of 18F-ML-10 over the 4 imaging sessions out to 7 days post-treatment. The control mouse (e, f, g, and h) showed minimal uptake in the tumor at all times post-treatment. B, Change in %ID/g of 18F-ML-10 in treated and control mice.
Image analysis was done for 18 mice that had a complete set of scans on all 4 days (5 controls and 13 treated mice). Regions of interest were drawn over the tumors. Liver uptake was used for normalization (T/L ratios). Analyses were performed by measuring change in T/L ratios. Percentage change in tumor volume from baseline to end of treatment was calculated in treated and control mice. The percentage change in T/L ratio of 18F-ML-10 was also calculated (Figure 5). Of the treatment group mice (n = 13), 10 mice responded as shown by the decrease in tumor volume (Figure 5A). On analysis of T/L ratios of the treatment in the group (Figure 5B), the ratios started to increase at 1 day posttreatment, with the most significant increase noted at 3 days compared to baseline (P < .05). The increase in radiotracer uptake was also statistically significant at day 7 when compared to baseline uptake in treated mice (P < .05).
Figure 5.
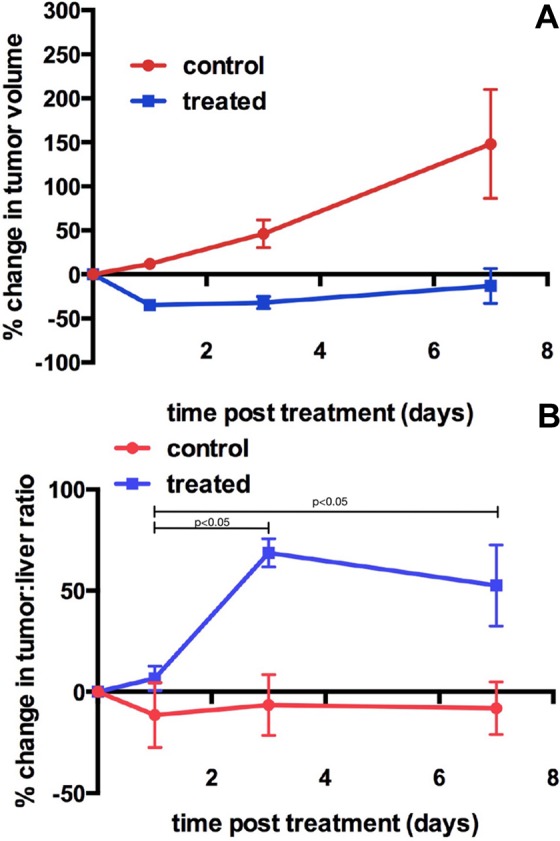
A, Percent change in tumor volume in control (n = 5; red symbols) and treated mice (n = 13; blue symbols; mean ± SD) showing that tumor volume of control mice increased over time, whereas the tumor volume of the treated mice decreased over the same time period. B, Percent change in tumor-to-liver ratio in control (n = 5; red symbols) and treated mice (n = 13; blue symbols; mean ± SD) showing that the percentage change in tumor-to-liver ratio of treated mice increased over time, with the most significant change at day 3, indicating high levels of apoptosis occurring post-treatment. SD indicates standard deviation.
DNA fragmentation was analyzed by the TUNEL assay. Tumors from untreated mice (day 0) were compared to mice that received 1 dose of 10 mg/kg doxorubicin (day 1) and tumors in mice 1 day after receiving a second dose of 10 mg/kg doxorubicin (day 3). The tumors from mice that had 1 or 2 treatments exhibited more apoptosis (P < .01 and P < .1 respectively; Figure 6).
Figure 6.
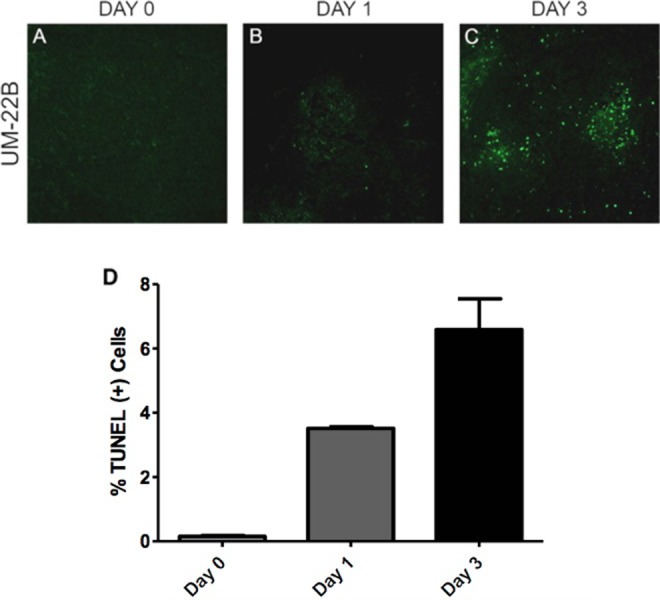
Representative TUNEL-stained images of tumor specimen samples from different time points: (A) baseline, (B) 1 day after 1 dose of 10 mg/kg doxorubicin treatment, and (C) 3 days after a second dose of doxorubicin treatment in the UM-22B group. Apoptotic nuclei are shown in green. Normal cell nuclei are shown in blue, stained by DAPI. (D) TUNEL-positive cells counted and these counts were used to determine the percentage of apoptotic cells in the field. This was repeated for 3 fields of view for the 1- and 3-day groups, and on 2 fields for the day 0 group. DAPI indicates 4′,6-diamidino-2-phenylindole.
Discussion
For clinicians and oncologists, an early response assessment to chemotherapy is of utmost importance. An important effect of all anticancer therapy is to kill tumor cells, through proapoptotic signaling, such as those triggered by induction of DNA damage, by inhibition of antiapoptotic activity, or through stimulation of apoptotic effectors.25,26 Studies have shown that induction of apoptosis after chemotherapy is associated with a good treatment response.27 The ability to directly and noninvasively image apoptosis in solid tumors could provide an effective means to evaluate early therapeutic response to cancer treatment.
A noninvasive way to evaluate treatment-induced apoptosis is attractive, as it can be used to determine and predict early response effectiveness of an anticancer regimen. Different preclinical and clinical studies have demonstrated that 18F-ML-10 has potential as a PET agent to detect tumor apoptosis using PET imaging.23,28-30 However, a major challenge to apoptosis imaging includes the need to specifically image a downstream signal from apoptosis. Another significant challenge is that apoptosis is a transient process of relatively short duration between the time of initiation and ultimate cell death. During the process of apoptosis, at baseline imaging, a subset of cells will be undergoing apoptosis, which typically has proceeded to complete death and cell elimination at the time of follow-up imaging. Subsequently, at the time of follow-up imaging, a new population of cells will be undergoing apoptosis, as a result of effective therapy, but the net change in macroscopic signal may be minimal. Therefore, novel approaches to analyzing data from apoptosis imaging will be required to assess therapy response. For example, measuring the change in temporally integrated tracer uptake from baseline to some time interval after therapeutic intervention. In addition, the timing of imaging apoptosis posttreatment is critical, and this timing will depend both on the type of tumor cell and the nature of the drug and the treatment regimen.
Recently, Oborski et al23 demonstrated the first use of 18F-ML-10 to assess apoptosis change in a patient with GBM before and 3 weeks after whole brain radiation therapy with concomitant temozolomide. This group31 further investigated challenges and approaches to quantitative therapy response assessment in GBM using 18F-ML-10, highlighting the importance of imaging timing after therapy, given both the ephemeral nature of the apoptotic process and the dynamic changes in tumor cell populations over the time from baseline to response assessment. As a result, their study demonstrated heterogeneous increases in radiotracer activity as compared to baseline imaging. In the present study as well, most of the tumors demonstrated heterogeneous radiotracer uptake. Allen et al30 also evaluated the performance of 18F-ML-10 in brain metastases as a tool for early detection of response of brain metastases to whole brain radiation. They correlated their findings with magnetic resonance imaging, the gold standard for imaging brain metastases, and found a significant correlation between early changes on the 18F-ML-10 scan and later changes in tumor anatomical dimensions.
These examples underscore that a crucial factor in imaging apoptosis is the timing of scanning after the start of treatment. In this study, PET/CT imaging of apoptosis with 18F-ML-10 was performed at multiple time points after chemotherapy (doxorubicin) administration in UM-SCC-22B xenograft-bearing mice. Doxorubicin, an intercalating agent that inhibits topoisomerase II, has been shown to cause apoptosis in cells cultured in vitro.32 Doxorubicin is applied through intravenous administration to achieve higher concentrations in the tumor.33 PET imaging showed increasing radiotracer uptake starting around day 1, with the most significant change in uptake day 3, which was 1 day after second doxorubicin treatment. Consistent with the increase in apoptosis shown by 18F-ML-10 imaging, the tumors in the treatment group grew more slowly and showed increased uptake of tracer than the control group tumors.
Recently 99mTc-Annexin V, 18F-ML-10, and 18F-C-SNAT were compared in a preclinical study for cell death imaging.11 In this study, authors performed PET scans 1 day posttreatment, and 18F-ML-10 failed to detect apoptosis at this time point. These results are consistent with our data, since 18F-ML-10 detected statistically increased chemotherapy-induced cell death at 3 and 7 days posttreatment, with minimal change at 1 day.
In another recent study, 18F-ML-10 PET/CT was studied on animal models using 2 types of human nasopharyngeal carcinoma cell lines, highly and poorly differentiated variants. After irradiation, tumors not only had statistically different levels of 18F-ML-10 uptake at 24 and 48 hours but they also showed a changing pattern of radiotracer uptake.34 Similarly, Bauwens used 68Ga-Annexin-V to demonstrate therapy-induced apoptosis 4 to 6 hours after irradiation using a Burkitt lymphoma mouse model.35 These studies and our results suggest that it is important to consider using different time points for apoptosis imaging for different tumor types and different types of therapeutic regimens.
Conclusion
The data presented here demonstrate that 18F-ML-10 PET/CT can detect doxorubicin-induced apoptosis in UM-SCC-22B tumor-bearing mice at 3 and 7 days posttreatment. However, single time point imaging with 18F-ML-10 PET/CT may not be reliable to assess treatment outcome, since different tumor types show variable levels of apoptosis, and the type of treatment regimen will also likely play a role in when apoptosis occurs.
Acknowledgments
The authors would like to thank Ms Kathryn Day for technical help during the PET/CT imaging and Ms Jalpa Modi for assistance with the cell cultures. The authors thank Aposense Ltd for supplying the ML-10 precursor.
Footnotes
Declaration of Conflicting Interests: The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the US National Institutes of Health research grant U01 CA140230 and the Department of Energy Grant DE SC0008833. The preclinical PET/CT imaging was supported in part by the NCI Cancer Center Support Grant P30CA047904 to UPCI.
References
- 1. Kerr JF, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer. 1972;26(4): 239–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lheureux S, Lecerf C, Briand M, et al. (18)F-FDG is a surrogate marker of therapy response and tumor recovery after drug withdrawal during treatment with a dual PI3K/mTOR inhibitor in a preclinical model of cisplatin-resistant ovarian cancer. Transl Oncol. 2013;6(5):586–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Yang TJ, Haimovitz-Friedman A, Verheij M. Anticancer therapy and apoptosis imaging. Exp Oncol. 2012;34(3):269–276. [PubMed] [Google Scholar]
- 4. Serrano M. Cancer regression by senescence. N Engl J Med. 2007;356(19):1996–1997. [DOI] [PubMed] [Google Scholar]
- 5. Vermes I, Haanen C, Steffens-Nakken H, Reutelingsperger C. A novel assay for apoptosis. Flow cytometric detection of phosphatidylserine expression on early apoptotic cells using fluorescein labelled Annexin V. J Immunol Methods. 1995;184(1):39–51. [DOI] [PubMed] [Google Scholar]
- 6. Song S, Xiong C, Lu W, Ku G, Huang G, Li C. Apoptosis imaging probe predicts early chemotherapy response n preclinical models: a comparative study with 18F-FDG PET. J Nucl Med. 2013. 54(1):104–110. [DOI] [PubMed] [Google Scholar]
- 7. Wang JW, Zheng W, Liu JB, et al. Assessment of early tumor response to cytotoxic chemotherapy with dynamic contrast-enhanced ultrasound in human breast cancer xenografts. PLoS One. 2013;8(3):e58274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Koopman G, Reutelingsperger CP, Kuijten GA, Keehnen RM, Pals ST, van Oers MH. Annexin V for flow cytometric detection of phosphatidylserine expression on B cells undergoing apoptosis. Blood. 1994. 84(5):1415–1420. [PubMed] [Google Scholar]
- 9. Zhu X, Li Z, Zhao M. Imaging acute cardiac cell death: temporal and spatial distribution of 99mTc-labeled C2A in the area at risk after myocardial ischemia and reperfusion. J Nucl Med. 2007;48(6):1031–1036. [DOI] [PubMed] [Google Scholar]
- 10. Chen DL, Zhou D, Chu W, et al. Comparison of radiolabeled isatin analogs for imaging apoptosis with positron emission tomography. Nucl Med Biol. 2009;36(6):651–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Witney TH, Hoehne A, Reeves RE, et al. A systematic comparison of 18F-C-SNAT to established radiotracer imaging agents for the detection of tumor response to treatment. Clin Cancer Res. 2015;21(17):3896–3905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Madar I, Huang Y, Ravert H, et al. Detection and quantification of the evolution dynamics of apoptosis using the PET voltage sensor 18F-fluorobenzyl triphenyl phosphonium. J Nucl Med. 2009;50(5):774–780. [DOI] [PubMed] [Google Scholar]
- 13. Su H, Chen G, Gangadharmath U, et al. Evaluation of [(18)F]-CP18 as a PET imaging tracer for apoptosis. Mol Imaging Biol. 2013;15(6):739–747. [DOI] [PubMed] [Google Scholar]
- 14. Reshef A, Shirvan A, Akselrod-Ballin A, Wall A, Ziv I. Small-molecule biomarkers for clinical PET imaging of apoptosis. J Nucl Med. 2010;51(6):837–840. [DOI] [PubMed] [Google Scholar]
- 15. Cai J, Li F. Single-photon emission computed tomography tracers for predicting and monitoring cancer therapy. Curr Pharm Biotechnol. 2013;14(7):693–707. [DOI] [PubMed] [Google Scholar]
- 16. Li X, Link JM, Stekhova S, et al. Site-specific labeling of annexin V with F-18 for apoptosis imaging. Bioconjug Chem. 2008;19(8):1684–1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bauwens M, De Saint-Hubert M, Devos E, et al. Site-specific 68Ga-labeled Annexin A5 as a PET imaging agent for apoptosis. Nucl Med Biol. 2011;38(3):381–392. [DOI] [PubMed] [Google Scholar]
- 18. Blankenberg FG. In vivo detection of apoptosis. J Nucl Med. 2008;49(suppl 2):81S–95S. [DOI] [PubMed] [Google Scholar]
- 19. Vermeersch H, Loose D, Lahorte C, et al. 99mTc-HYNIC Annexin-V imaging of primary head and neck carcinoma. Nucl Med Commun. 2004;25(3):259–263. [DOI] [PubMed] [Google Scholar]
- 20. Cohen A, Shirvan A, Levin G, Grimberg H, Reshef A, Ziv I. From the Gla domain to a novel small-molecule detector of apoptosis. Cell Res. 2009;19(5):625–637. [DOI] [PubMed] [Google Scholar]
- 21. Zhang F, Zhu L, Liu G, et al. Multimodality imaging of tumor response to doxil. Theranostics. 2011;1:302–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hu S, Kiesewetter DO, Zhu L, et al. Longitudinal PET imaging of doxorubicin-induced cell death with 18F-Annexin V. Mol Imaging Biol. 2012;14(6):762–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Oborski MJ, Laymon CM, Lieberman FS, Drappatz J, Hamilton RL, Mountz JM. First use of (18)F-labeled ML-10 PET to assess apoptosis change in a newly diagnosed glioblastoma multiforme patient before and early after therapy. Brain Behav. 2014;4(2):312–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Aloya R, Shirvan A, Grimberg H, et al. Molecular imaging of cell death in vivo by a novel small molecule probe. Apoptosis. 2006;11(12):2089–2101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Call JA, Eckhardt SG, Camidge DR. Targeted manipulation of apoptosis in cancer treatment. Lancet Oncol. 2008;9(10):1002–1011. [DOI] [PubMed] [Google Scholar]
- 26. Meiler J, Schuler M. Therapeutic targeting of apoptotic pathways in cancer. Curr Drug Targets. 2006;7(10):1361–1369. [DOI] [PubMed] [Google Scholar]
- 27. Evan GI, Vousden KH. Proliferation, cell cycle and apoptosis in cancer. Nature. 2001;411(6835):342–348. [DOI] [PubMed] [Google Scholar]
- 28. Höglund J, Shirvan A, Antoni G, et al. 18F-ML-10, a PET tracer for apoptosis: first human study. J Nucl Med. 2011;52(5):720–725. [DOI] [PubMed] [Google Scholar]
- 29. Kadirvel M, Fairclough M, Cawthorne C, et al. Detection of apoptosis by PET/CT with the diethyl ester of [(1)(8)F]ML-10 and fluorescence imaging with a dansyl analogue. Bioorg Med Chem. 2014;22(1):341–349. [DOI] [PubMed] [Google Scholar]
- 30. Allen AM, Ben-Ami M, Reshef A, et al. Assessment of response of brain metastases to radiotherapy by PET imaging of apoptosis with 18F-ML-10. Eur J Nucl Med Mol Imaging. 2012;39(9):1400–1408. [DOI] [PubMed] [Google Scholar]
- 31. Oborski MJ, Laymon CM, Qian Y, Lieberman FS, Nelson AD, Mountz JM. Challenges and approaches to quantitative therapy response assessment in glioblastoma multiforme using the novel apoptosis positron emission tomography tracer F-18 ML-10. Transl Oncol. 2014;7(1):111–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kaufmann SH, Earnshaw WC. Induction of apoptosis by cancer chemotherapy. Exp Cell Res. 2000;256(1):42–49. [DOI] [PubMed] [Google Scholar]
- 33. Johansen PB. Doxorubicin pharmacokinetics after intravenous and intraperitoneal administration in the nude mouse. Cancer Chemother Pharmacol. 1981;5(4):267–270. [DOI] [PubMed] [Google Scholar]
- 34. Bao X, Yang Z, Wang S, et al. The preclinical study of predicting radiosensitivity in human nasopharyngeal carcinoma xenografts by 18F-ML-10 animal-PET/CT imaging. Oncotarget. 2016;7(15):20743–20752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bauwens M. In vivo apoptosis imaging using site-specifically (68)Ga-labeled annexin V. Methods Mol Biol. 2016;1419:17–26. [DOI] [PubMed] [Google Scholar]