

Abstract
Linear oligomers equipped with complementary H-bond donor (D) and acceptor (A) sites can interact via intermolecular H-bonds to form duplexes or fold via intramolecular H-bonds. These competing equilibria have been quantified using NMR titration and dilution experiments for seven systems featuring different recognition sites and backbones. For all seven architectures, duplex formation is observed for homo-sequence 2-mers (AA·DD) where there are no competing folding equilibria. The corresponding hetero-sequence AD 2-mers also form duplexes, but the observed self-association constants are strongly affected by folding equilibria in the monomeric states. When the backbone is flexible (five or more rotatable bonds separating the recognition sites), intramolecular H-bonding is favored, and the folded state is highly populated. For these systems, the stability of the AD·AD duplex is 1–2 orders of magnitude lower than that of the corresponding AA·DD duplex. However, for three architectures which have more rigid backbones (fewer than five rotatable bonds), intramolecular interactions are not observed, and folding does not compete with duplex formation. These systems are promising candidates for the development of longer, mixed-sequence synthetic information molecules that show sequence-selective duplex formation.
Introduction
Macromolecules of defined sequence form the molecular basis for living systems. Selective base pairing allows the sequence information stored in nucleic acid polymers to be read and translated through template synthesis.1 The sequences of proteins and nucleic acids determine how they fold into three-dimensional structures, which in turn dictate the structural, recognition, and catalytic properties that govern the biochemistry of the cell.2 Synthetic supramolecular systems that recapitulate the sequence-selective recognition or folding properties of biomacromolecules are beginning to appear, and in the long term, synthetic information molecules may be expected to offer alternatives to proteins and nucleic acids, where sequence can be used to program function. Nucleic acids are unique in that they combine both duplex-forming and folding properties. The double-stranded structures are the basis for replication, and folded single strands act as receptors and catalysts, so it is possible to use nucleic acids in directed evolution experiments to obtain functional polymers.3 The balance between folding and duplex formation in nucleic acids is therefore thought to be critical in the evolution of living systems.4 In this Article, we examine the interplay of folding and duplex formation in prototype synthetic molecules.
Inspired by DNA, researchers have put considerable effort into synthetic supramolecular systems that form duplex structures held together by non-covalent interactions.5−7 Although programmable sequence-specific recognition of DNA has been achieved with synthetic molecules,8 the molecular features that allow nucleic acids to encode and replicate genetic information are still not available in any other material. In duplex-forming synthetic systems, the recognition sites are usually integrated into the backbone, which means that sequence variation is difficult to introduce without disrupting the complex. Synthetic oligomers that fold into well-defined secondary structures, i.e., foldamers, have also been widely studied, leading to synthetic receptors, nanomachines, and modulators of protein–protein interactions.9 Huc has shown that it is possible to program the recognition properties of foldamers by changing the sequence of building blocks.10
We recently reported a new class of synthetic molecules that form stable duplexes via multiple cooperative H-bonding interactions between recognition units appended to oligomeric backbones.11Figure 1 shows the systems reported to date. Four different backbones have been prepared using either reductive amination or photochemical thiol–ene coupling. We found that variations in the structure of the backbone were well-tolerated, and different backbones and recognition modules could be used interchangeably to form stable duplexes. However, all of these systems are homo-sequence oligomers, i.e., all H-bond donor (D) oligomers that form duplexes with length complementary all H-bond acceptor (A) oligomers. As a first step toward mixed-sequence oligomers, in this paper we describe the properties of several AD 2-mers based on the motifs shown in Figure 1.
Figure 1.
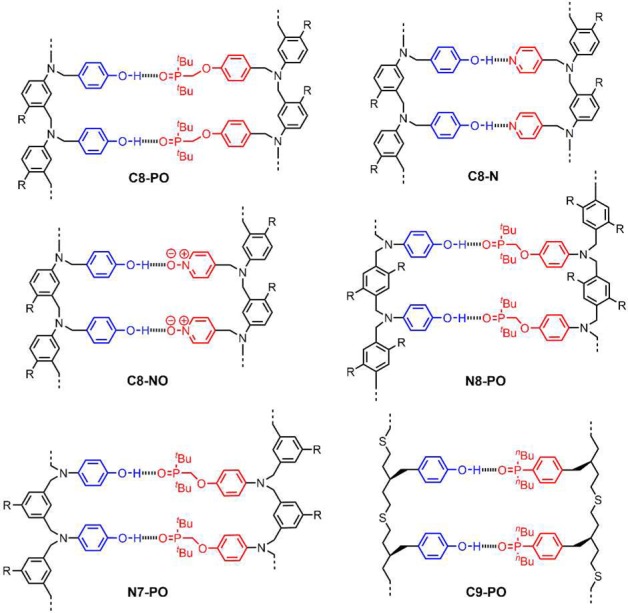
H-bonded duplexes formed by oligomers with different recognition units and backbones (R is a solubilizing group).11 The oligomers are labeled according the type of recognition unit (phosphine oxide = PO, pyridine = N, pyridine N-oxide = NO), the site of attachment of the recognition units (C or N), and the number of atoms separating neighboring recognition units (7, 8, or 9).
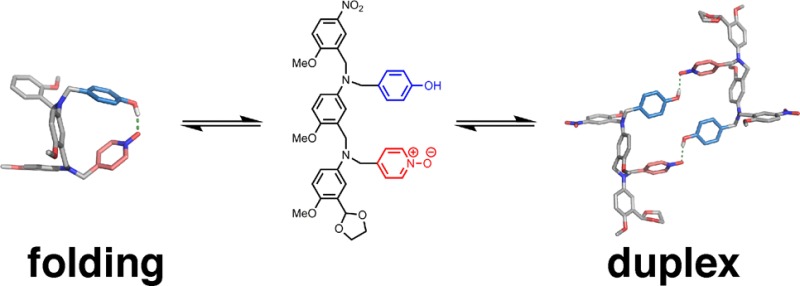
Mixed-sequence oligomers introduce the possibility of intramolecular H-bonds, which can lead to folding equilibria that compete with duplex formation (Figure 2). If two neighboring recognition units on an oligomer interact strongly, the folding channel will be favored over the duplex assembly channel. Thus, the selectivity of mixed-sequence duplex formation is limited by the extent to which intramolecular folding occurs in AD 2-mers. The key parameters that determine the distribution of the different species shown in Figure 2 are the effective molarities for folding (EMf) and duplex formation (EMd), the operating concentration (c), and the association constant for the intermolecular interaction between two complementary H-bonding sites (K). Figure 2 shows pathways where the first H-bond formed can be intramolecular or intermolecular: if EMf > c and KEMf > 1, then the folding channel will be preferred over intermolecular interactions. If the first H-bond formed is intramolecular, then further intramolecular interactions leading to folded hairpin loop structures will take place if the sequence is suitable and if KEMd > 1. If the first H-bond formed is intermolecular, there are two pathways for subsequent interactions: intramolecular interactions leading to duplex formation, or intermolecular interactions leading to aggregates. The duplex channel will be preferred over aggregation if EMd > c and KEMd > 1. Therefore, duplex formation will be the preferred self-assembly process if EMd is greater than all of c, 1/K, and EMf.
Figure 2.
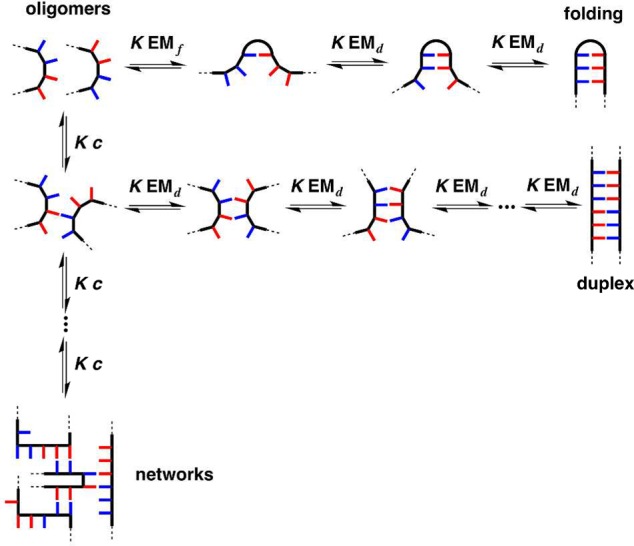
Competing equilibria in the formation of a duplex between two complementary oligomers. Intramolecular interactions between recognition sites lead to folding, and intermolecular interactions between multiple oligomers lead to networks of supramolecular aggregates. The effective molarities for folding (EMf) and duplex formation (EMd), the operating concentration (c), and the association constant for formation of a H-bond between two recognition sites (K) determine the distribution of oligomers between the three different assembly channels. The equilibrium constants have additional statistical factors that are not shown.
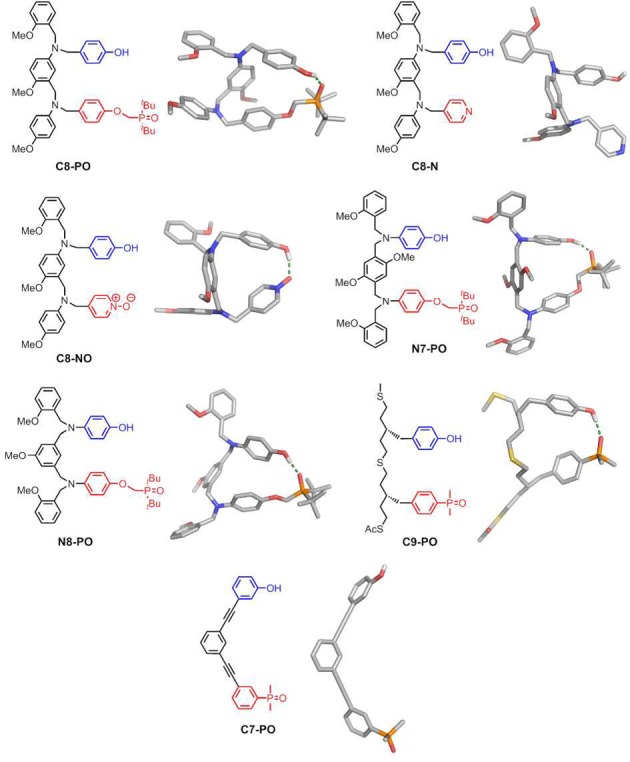
Before we embark on the synthesis of mixed-sequence oligomers, it is therefore important to investigate how K, EMd, and EMf vary with the chemical structures of the monomer units. The association constant for intermolecular H-bond formation (K) is straightforward to predict for any choice of recognition unit,12 but we do not yet have reliable methods for predicting effective molarities for intramolecular interactions.13−17 Here, we analyze the competing equilibria between folding and duplex formation for the seven different AD 2-mers shown in Figure 3. These compounds represent the six systems shown in Figure 1 for which the corresponding A·D and AA·DD complexes have been characterized previously and one additional system with a more rigid backbone (C7-PO), which provides a reference compound that cannot fold. The aim is to obtain experimental data for understanding the complex equilibria shown in Figure 2 in order to select suitable architectures for the development of mixed-sequence duplex-forming oligomers.
Figure 3.
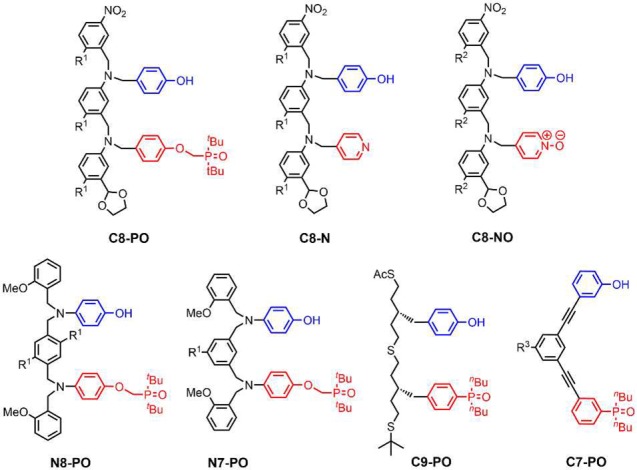
Chemical structures of the AD 2-mers (R1 = (2-ethylhexyl)oxy; R2 = (2-ethylhexyl)oxy or methoxy; R3 = H or S-β-citronelloxy).
Results and Discussion
Synthesis
Two versions of the C8-NO AD 2-mer were prepared: one with (2-ethylhexyl)oxy solubilizing groups for binding studies, and one with methoxy groups for X-ray crystallography. Building blocks 4 and 7 required for synthesis of the methoxy analogue were prepared from 5-hydroxy-2-nitrobenzaldehyde 1 as shown in Scheme 1. After methylation of 1, the acetal-protected aldehyde 2 was obtained using ethylene glycol and catalytic tosic acid. Hydrogenation of the nitro group yielded aniline 3. The imines of 3 formed with 4-pyridinecarboxaldehyde N-oxide or 2-methoxy-5-nitrobenzaldehyde were reduced to give 4 and 5, respectively. Reduction of the imine derived from 5 and 6,11d followed by acid removal of the acetal protecting group, gave 7. The syntheses of the analogues of 4 and 7 bearing (2-ethylhexyl)oxy solubilizing groups instead of methoxy groups (8 and 9) were reported previously.11a,11c Reductive amination was used to couple the phenol and N-oxide building blocks in good yield to give 10 and 11 (Scheme 2). TBAF-mediated deprotection of the phenol groups afforded the C8-NO AD 2-mers 12 and 13.
Scheme 1. Synthesis of Building Blocks 4 and 7.

Scheme 2. Synthesis of C8-NO AD 2-mers.

The synthesis of the C8-PO 2-mer 16 is shown in Scheme 3. Reductive amination of aldehyde 8 with aniline 14(11a) yielded 15, and deprotection using TBAF gave the AD 2-mer 16. The route to the C8-N 2-mer 21 involves previously reported compounds 17, 18, and 20 (Scheme 4).11a,11c TBAF-mediated phenol deprotection of 17, followed by reductive amination with 18, afforded 19 in good yield. After the removal of the acetal protecting group, reductive amination with 20 gave 21.
Scheme 3. Synthesis of C8-PO AD 2-mer (R = (2-Ethylhexyl)oxy).

Scheme 4. Synthesis of C8-N AD 2-mer (R = (2-Ethylhexyl)oxy).

Scheme 5 shows the synthesis of the N8-PO 2-mer 26 from p-aminophenol and compounds 23 and 24, which have been reported previously.11b The statistical reaction between these three components gave 25 in modest yield after column chromatography. The NH H-bond donor sites of 25 could potentially compete with the phenol recognition units, so they were removed by capping with 2-methoxybenzaldehyde to give 26.
Scheme 5. Synthesis of N8-PO AD 2-mer (R = (2-Ethylhexyl)oxy).

The N7-PO AD 2-mer 29 differs from the N8-PO AD 2-mer 26 in the position of the substituents on the backbone aromatic ring. 29 was obtained using a synthetic strategy similar to that used for 26 but with the isophthalaldehyde derivative 27 in place of 23 (Scheme 6).11b The statistical reaction of 27 with 22 and 24 yielded 28 after column chromatography, and subsequent reductive amination with 2-methoxybenzaldehyde gave 29.
Scheme 6. Synthesis of N7-PO AD 2-mer (R = (2-Ethylhexyl)oxy).

Photochemically initiated thiol–ene coupling was used to synthesize the C9-PO 2-mer 33 from 30 and 32 (Scheme 7).11d Thiol–ene-mediated capping of 30 with tert-butyl mercaptan gave 31. Removal of the S-acetyl protecting group of 31 under basic conditions, and subsequent thiol–ene coupling with 32, afforded 33 in good yield.
Scheme 7. Synthesis of C9-PO AD 2-mer.

The C7-PO AD 2-mer is a new backbone architecture, so the corresponding AA and DD 2-mers were also synthesized. The AA·DD complex provides an important reference point for characterizing duplex formation (see Supporting Information for the synthesis of the corresponding A and D 1-mers). The C7-PO compounds were all prepared using Sonogashira coupling. The required phosphine oxide building blocks 35 and 36 were prepared as shown in Scheme 8. Palladium-mediated P-arylation of 3-bromoiodobenzene with di-n-butylphosphine 34(11d) gave 35 in excellent yield. Sonogashira coupling with trimethylsilylacetylene (TMSA) followed by TBAF-mediated deprotection gave 36. Scheme 9 shows the synthesis of the C7-PO AA 2-mer (40) and the C7-PO DD 2-mer (41). A Mitsunobu reaction between 3,5-dibromophenol and S-(−)-β-citronellol yielded 38 in excellent yield. A double Sonogashira coupling with TMSA followed by alkyne deprotection gave 39. Double Sonogashira coupling of 39 with 3-iodophenol or 35 gave 40 and 41.
Scheme 8. Synthesis of Building Blocks 35 and 36.

Scheme 9. Synthesis of C7-PO AA and DD 2-mers (R = S-β-Citronelloxy).

Two versions of the C7-PO AD 2-mer were prepared: one with a (S)-β-citronelloxy solubilizing group for binding studies (44), and one with no solubilizing groups for X-ray crystallography (47). The C7-PO AD 2-mer (44) was synthesized from compound 38 in three steps (Scheme 10). Lithium–halogen exchange using n-BuLi followed by reaction with iodine gave the bromoiodobenzene derivative 42 in good yield. A selective Sonogashira coupling between the iodide substituent and alkyne 36 gave 43 in excellent yield, and subsequent Sonogashira coupling between the bromine substituent and 3-hydroxyphenylacetylene gave 44. The C7-PO AD 2-mer 47, which does not have the solubilizing group, was prepared from a statistical Sonogashira coupling of an equimolar mixture of 36, 45, and 46 followed by column chromatography (Scheme 11).
Scheme 10. Synthesis of C7-PO AD 2-mer for Binding Studies (R = S-β-Citronelloxy).

Scheme 11. Synthesis of C7-PO AD 2-mer for X-ray Crystallography.

NMR Binding Studies
NMR titration and dilution experiments were performed to measure the association constants for the A·D, AA·DD, and AD·AD complexes (Table 1). For the A·D and AA·DD complexes, association constants were determined by fitting 1H and 31P NMR titration data to a 1:1 binding isotherm.11 Self-association constants for the AD·AD complexes were determined by fitting 1H and 31P NMR dilution data to a dimerization isotherm.
Table 1. Association Constants (Kobs) and Effective Molarities (EM) Measured by NMR Titrations and Dilutions in Toluene-d8 at 298 Ka.
| complex | Kobs (M–1) | Kfold | EMf (mM) | EMd (mM) | χfoldb |
|---|---|---|---|---|---|
| C8-PO | |||||
| A·D | 350 (3) | ||||
| AA·DD | 1900 (470) | 8 (3) | |||
| AD·ADc | 45 (15) | 2.2 (0.5) | 7 (1) | 0.69 (0.06) | |
| C8-N | |||||
| A·D | 34 (1) | ||||
| AA·DD | 130 (16) | 57 (8) | |||
| AD·AD | 60 (20) | n.d. | n.d. | 0 | |
| C8-NO | |||||
| A·D | 330 (80) | ||||
| AA·DD | 5400 (1600) | 30 (10) | |||
| AD·AD | 2400 (500) | n.d. | n.d. | 0 | |
| N8-PO | |||||
| A·D | 250 (10) | ||||
| AA·DD | 2500 (200) | 20 (2) | |||
| AD·AD | 225 (13) | 0.7 (0.1) | 3 (1) | 0.40 (0.03) | |
| N7-PO | |||||
| A·D | 250 (10) | ||||
| AA·DD | 1200 (400) | 10 (3) | |||
| AD·AD | 14 (5) | 3.6 (0.9) | 15 (4) | 0.78 (0.05) | |
| C9-PO | |||||
| A·D | 560 (210) | ||||
| AA·DD | 9000 (4000) | 14 (12) | |||
| AD·AD | 85 (22) | 4.1 (1.1) | 7 (3) | 0.81 (0.05) | |
| C7-POd | |||||
| A·D | 30 (1) | ||||
| AA·DD | 240 (80) | 130 (40) | |||
| AD·ADe | 130 (30) | n.d. | n.d. | 0 | |
Average values with errors (reported in parentheses) at the 95% confidence limit based on the variation between different signals and experiments.
AD 2-mer 16.
Experiments carried out in CDCl3 due to the high stability of the complexes in toluene-d8.
AD 2-mer 44. n.d. = not detected.
Figure 4 shows the equilibria that are possible in these systems. Under dilute conditions, the higher order complexes are not expected to be significantly populated. The equilibrium constants for the A·D complexes measure the intrinsic H-bond strength of the recognition units (K), which is similar for the phenol·phosphine oxide and phenol·N-oxide systems and an order of magnitude lower for the phenol·pyridine system. The phenol·phosphine oxide recognition system was also studied in chloroform, where the association constant is an order of magnitude lower than in toluene (see C7-PO entry in Table 1). For the AA·DD complexes, the product KEMd is greater than 1 in all cases, which means that the duplex shown in Figure 4a is the most populated state for concentrations lower than EMd. The values of Kduplex in Figure 4 are expected to be similar for the AA·DD and corresponding AD·AD duplexes. If there were no competing equilibria, then the observed self-association constants for the AD systems would be similar to the association constants measured for formation of the corresponding AA·DD duplexes. For most of the systems in Table 1, this is not the case, which means that the competing equilibria shown in Figure 4b play an important role in the self-association of the AD 2-mers.
Figure 4.
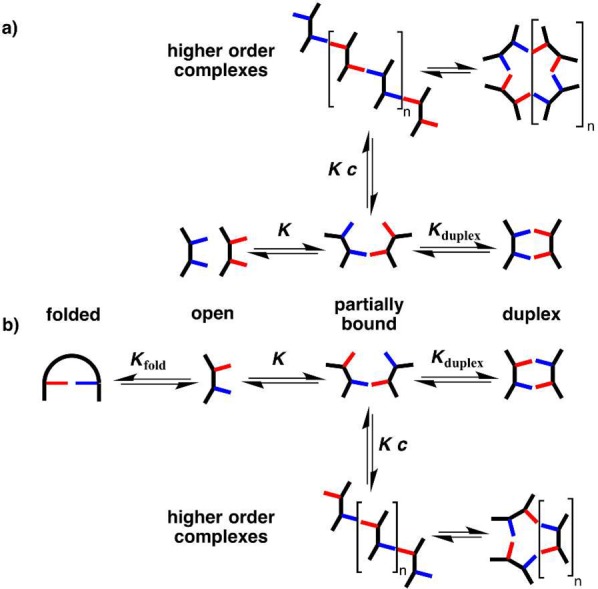
Competing equilibria in the assembly of (a) AA·DD and (b) AD·AD duplexes. The equilibrium constants have additional statistical factors that are not shown.
Figure 4 shows that the equilibria involving the partially bound 1:1 complex and the closed duplex are the same for the AA·DD and AD·AD systems, so we assume that the relative populations of these states are similar in both cases; i.e., the closed duplex dominates. The major competing equilibrium that leads to the lower self-association constants observed for the AD systems must therefore be due to folding of the monomer to form an intramolecular A·D H-bond. This conclusion is supported by examination of the 31P NMR chemical shifts of the free (δfree) and bound (δbound) states from the dilution experiments (Table 2). With the exception of the C7-PO system, the free chemical shifts for the AD 2-mers are much higher than the values observed for the corresponding A 1-mers and AA 2-mers. The free chemical shift measured in the AD dilution experiments is the population-weighted average over all monomeric states, so the higher values of δfree observed for the AD 2-mers indicate that the folded state with an intramolecular H-bond is significantly populated.
Table 2. 31P NMR Chemical Shifts (ppm) Measured in Toluene-d8 at 298 K.
| complex | δfree | δbound | Δδ | χfolda |
|---|---|---|---|---|
| C8-PO | ||||
| A·D | 53.7 | 58.7 | 5.0 | |
| AA·DD | 53.6 | 58.8 | 5.2 | |
| AD·ADb | 57.0 | 58.1 | 1.1 | 0.78 (0.01) |
| N8-PO | ||||
| A·D | 53.5 | 58.5 | 5.0 | |
| AA·DD | 53.5 | 57.5 | 4.0 | |
| AD·AD | 55.1 | 57.5 | 2.4 | 0.46 (0.08) |
| N7-PO | ||||
| A·D | 53.5 | 58.5 | 5.0 | |
| AA·DD | 53.4 | 60.3 | 6.9 | |
| AD·AD | 56.5 | 57.9 | 1.4 | 0.76 (0.05) |
| C9-PO | ||||
| A·D | 35.4 | 41.8 | 6.4 | |
| AA·DD | 35.6 | 42.0 | 6.4 | |
| AD·AD | 40.6 | 41.1 | 0.5 | 0.92 (0.01) |
| C7-POc | ||||
| A·D | 40.3 | 45.8 | 5.5 | |
| AA·DD | 40.2 | 44.4 | 4.2 | |
| AD·ADd | 40.2 | 44.1 | 3.9 | 0.18 (0.16) |
The data in Tables 1 and 2 can used to determine the equilibrium constant for folding, Kfold, in these systems. Equations 1–3 show the relationship between the measured self-association constant for dimerization of AD·AD, Kobs, and the equilibrium constants, Kduplex and Kfold, illustrated in Figure 4b.
| 1 |
where [AD] is the total concentration of monomeric AD 2-mer as defined in eq 2, i.e., the sum of the open, [AD]open, and folded, [AD]fold, states.
| 2 |
Combining eqs 1 and 2 allows Kobs to be expressed as a function of Kfold and Kduplex, the equilibrium constant for the formation of the AD·AD duplex from the open monomeric state (eq 3).
| 3 |
Rearranging eq 3 allows Kfold to be expressed in terms of Kobs and Kduplex (eq 4).
| 4 |
If we assume that the AD·AD and AA·DD duplexes make H-bonds of similar stability, then it is possible to use the equilibrium constant for formation of the AA·DD duplex to estimate Kduplex. In systems where multiple H-bonding sites are constrained in very close proximity, local secondary electrostatic interactions can alter the relative stabilities of different H-bonding patterns, but the recognition sites in the compounds described here are too far apart for such effects to be significant.18 However, there is a difference in symmetry between the AA·DD and AD·AD duplexes, which leads to a statistical contribution to the value of Kduplex. For AA·DD, there is one unbound state, but there are two degenerate states for the duplex, because two different H-bonding arrangements are possible.19 The AD·AD duplex is made from two identical molecules, so there are two degenerate unbound states, but there is one bound state, because only one H-bonding arrangement is possible. Thus, the observed association constant for formation of the AA·DD duplex, Kobs(AA·DD), can be used to estimate Kduplex for formation of the AD·AD duplex from the open monomeric state (eq 5), and the resulting values were used in eq 4 to calculate values of Kfold.
| 5 |
The effective molarity for intramolecular H-bonding in the monomeric state, EMf, is given by the ratio of Kfold to the association constant for the corresponding intermolecular interaction, KA·D (eq 6).
| 6 |
The values of Kfold can also be used to estimate the relative populations of the open (χopen) and folded states (χfold) for the monomeric AD 2-mers (eq 7).
| 7 |
Rearranging eq 7 allows χfold to be calculated from the value of Kfold (eq 8).
| 8 |
The values of Kfold, EMf, and χfold are tabulated in Table 1 for each of the seven AD 2-mers. Four systems fold to a significant extent, and the other three systems show no evidence of folding. No folding was observed for C7-PO, which has a rigid backbone that holds the recognition sites apart. The other two systems that do not fold are C8-N and C8-NO, which have a relatively flexible backbone. A common feature of the three AD 2-mers that do not fold is that the recognition units are connected by fewer than five rotatable bonds. Although C8-PO shares the same backbone as C8-N and C8-NO, this system does fold due to the additional flexibility associated with the two atom linker that connects the phosphine oxide to the aromatic ring. For C8-PO, C9-PO, and N7-PO, the folded state is highly populated (χfold = 70–80%). N8-PO also folds, but the effective molarity for folding (EMf) is lower, so the open and folded states are equally populated (χfold ≈ 40%). The values of EMf and EMd are similar in all four cases and fall in the range 3–20 mM. For the systems that do not fold, the values of EMd are somewhat higher (30–130 mM).
The 31P NMR chemical shift data in Table 2 can also be used to estimate values of χfold. The values of δbound for the A·D, AA·DD, and AD·AD complexes are similar in all cases, indicating that the phosphine oxides are fully H-bonded. However, there are significant differences between the values of δfree for the AD·AD complexes compared with the corresponding A·D and AA·DD complexes, because the values represent the population-weighted average of the chemical shifts of the open and folded states of the AD monomer. If we assume that the open AD monomer has the same chemical shift as the free state of A (δA) and that the folded AD monomer has the same chemical shift as the bound state A·D (δA·D), the free chemical shift observed for the AD 2-mer (δfree) can be used to quantify the population of the folded state via eq 9.
| 9 |
The limiting complexation-induced change in chemical shift observed in dilution experiments for the AD 2-mers (ΔδAD·AD) can be expressed as eq 10.
| 10 |
Rearranging eq 10 allows χfold to be obtained from the values of ΔδAD·AD and ΔδA·D (or ΔδAA·DD) listed in Table 2 (eq 11).
| 11 |
Figure 5 shows that there is a good correlation between the values of χfold calculated from the association constants (eq 8) and those obtained from the complexation-induced changes in 31P NMR chemical shift (eq 11). For systems with phosphine oxide H-bond acceptors, the 31P NMR chemical shift therefore provides a convenient and straightforward method for assessing folding. For systems where this chemical shift information is not available, the correlation in Figure 5 provides confidence that the method for assessing folding on the basis of measurement of association constants is robust.
Figure 5.
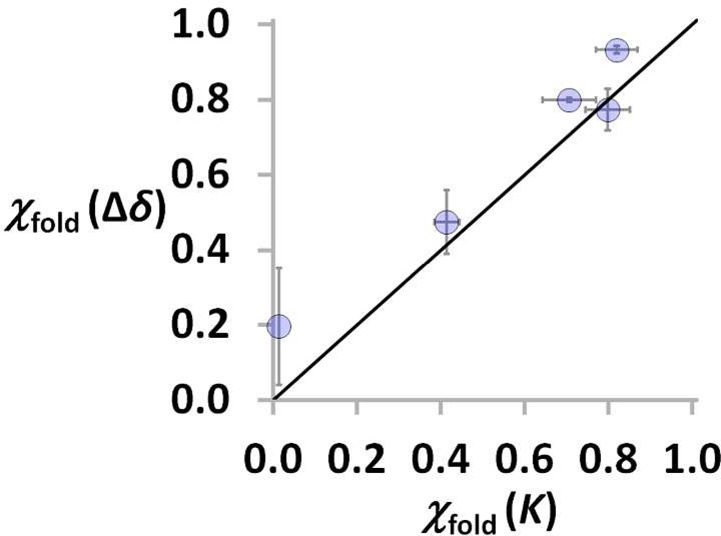
Correlation between χfold estimated using changes in chemical shift (Δδ, eq 11) and association constants (K, eq 8). The line corresponds to y = x.
Three-Dimensional Structures
Molecular mechanics calculations provide insight into the folding propensity of the AD 2-mers. A conformational search using the MMFFs force-field was performed for the monomer of each of the seven AD 2-mers (see Supporting Information for details). A folded conformation featuring an intramolecular H-bond is the lowest energy structure for all four systems that were found to fold in the NMR experiments (Figure 6).
Figure 6.
Lowest energy conformations of AD 2-mers calculated using molecular mechanics conformational searches (MMFFs force-field and CHCl3 solvation implemented in Macromodel).20
The global minimum for the C8-N system and for the C7-PO system corresponds to an open conformation, in agreement with the experimental results. However, molecular modeling suggests that the C8-NO AD 2-mer folds with an intramolecular pyridine N-oxide·phenol H-bond, which contradicts the results of the NMR experiments in Table 1. Analysis of the partial atomic charges assigned by the force-field reveals that the charge on the oxygen of the pyridine N-oxide acceptor is significantly higher than that on the oxygen of the phosphine oxide in the C8-PO AD 2-mer. The association constant for formation of a phosphine oxide·phenol H-bond in the A·D complex in Table 1 is slightly higher than the value for a pyridine N-oxide·phenol H-bond,12 so the folded conformation calculated for the C8-NO system may be an artifact of unrealistic partial atomic charges that overestimate the H-bond strength.
The structures of the complexes formed by the C8-NO and C7-PO AD 2-mers were also investigated using X-ray crystallography. The X-ray crystal structure of the methoxy analogue of the C8-NO AD 2-mer (12) shows formation of the doubly H-bonded antiparallel duplex (Figure 7). Despite the molecular modeling prediction of folding for this system, X-ray crystallography is consistent with the NMR result that the intermolecular channel leading to bimolecular duplexes is favored over the intramolecular channel leading to folded structures.
Figure 7.
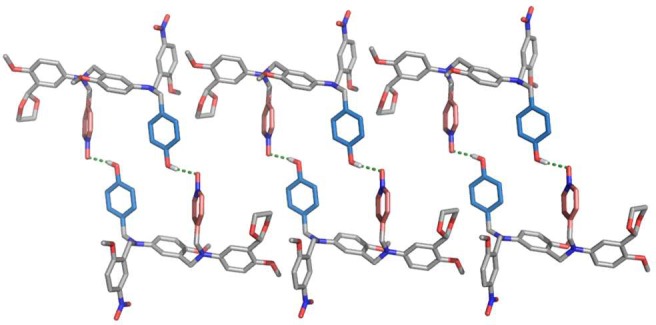
Single-crystal X-ray structure of 12, which forms a doubly H-bonded duplex in the solid state. Three adjacent unit cells with a total of six molecules of 12 are shown. Hydrogen atoms have been omitted for clarity. The backbones are shown in gray, the H-bond donor recognition units in blue, and the H-bond acceptor units in red.
The X-ray crystal structure of 47, the analogue of the C7-PO AD 2-mer with no solubilizing group, was also determined (Figure 8). There is no intramolecular folding in the solid state, in agreement with the solution-phase NMR results, but a duplex is not observed. Instead, 47 crystallizes as a linear H-bonded polymer, the state that the AD 2-mers are likely to populate at high concentrations (c > EMd).
Figure 8.
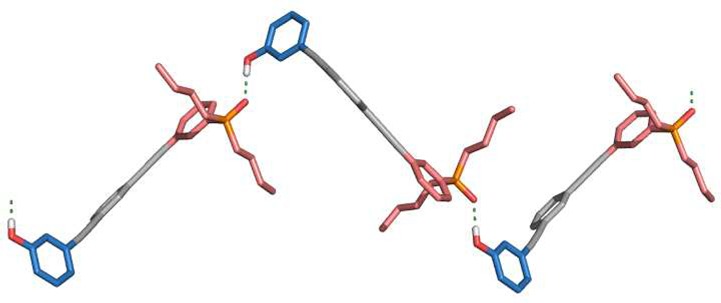
Single-crystal X-ray structure of 47, which forms a linear H-bonded polymer in the solid state. Three adjacent unit cells with a total of three molecules of 47 are shown. Hydrogen atoms have been omitted for clarity. The backbones are shown in gray, the H-bond donor recognition units in blue, and the H-bond acceptor units in red.
Conclusions
Self-complementary molecules bearing H-bond donors (D) and acceptors (A) can make either intermolecular interactions to form a duplex or intramolecular interactions to fold. A quantitative analysis of these competing equilibria is described for a family of seven different AD 2-mers, which represent the shortest mixed-sequence systems from a family of longer oligomers. The compounds were prepared using a variety of coupling chemistries (reductive amination, thiol–ene, and Sonogashira coupling) and differ in the flexibility and geometric complementarity of the backbone and in the nature of the H-bond recognition units.
The equilibrium constants for intramolecular folding, Kfold, cannot be measured directly because the folded and open conformers are in fast exchange on the NMR time scale. However, it is possible to infer the value of Kfold by comparing the equilibrium constant for self-association of an AD 2-mer with the equilibrium constant for formation of the isomeric AA·DD duplex, where the two components cannot fold. For systems that have phosphine oxide H-bond acceptors, the accuracy of this method was confirmed by comparison of the 31P NMR chemical shifts of the free and bound states. For AD 2-mers that fold to a significant extent, the phosphine oxide is involved in an intramolecular H-bond, so the free chemical shift for the monomeric state is very similar to the bound chemical shift for the AD·AD duplex. The free and bound chemical shifts for the AD·AD complex can be related to the corresponding values for the A·D and AA·DD complexes to quantify the populations of folded and open states in the monomeric form of AD, and hence Kfold. The results of the chemical shift and association constant methods show good quantitative agreement.
The three key parameters that determine the behavior of these systems are the association constant for H-bond formation between the recognition units and the effective molarities for intramolecular folding, EMf, and duplex formation, EMd. In all seven systems, EMd is larger than 1/K, which means that efficient duplex formation is observed for 1:1 complexes of two homo-oligomers, AA·DD. In four systems, EMf is comparable to EMd, which means that the AD 2-mers prefer folding over duplex formation at low concentrations. It is possible to force these systems to form AD·AD duplexes at high concentrations, but the association constants for duplex formation are 1–2 orders of magnitude lower than those observed for the isomeric AA·DD complexes. For the other three systems, EMf ≪ EMd, and no evidence for intramolecular folding was observed. This conclusion is supported by X-ray crystal structures of two of the systems that do not fold in solution: one forms the doubly H-bonded AD·AD duplex in the solid state, and the other forms a supramolecular polymer where the molecules are connected by single A·D H-bonding interactions.
We previously found that the value of EMd for duplex formation does not depend greatly on the conformational flexibility of the backbone. This work shows that folding equilibria are more sensitive to the nature of the backbone. The three AD 2-mers that do not fold have recognition units connected by fewer than five rotatable bonds, but when the backbone is more flexible, folding is observed. This observation provides a convenient rule of thumb for the design of systems where duplex formation is favored over intramolecular folding of neighboring recognition units. In longer oligomers, the probability of folding between recognition units that are remote in sequence will increase, even for relatively rigid backbones. These more complex folding patterns are something that we hope to exploit in the development of synthetic information molecules that exhibit both sequence-selective duplex formation and sequence-directed folding properties. Work on the synthesis of longer mixed-sequence oligomers using the systems identified here that do not fold is in progress (Figure 9).
Figure 9.
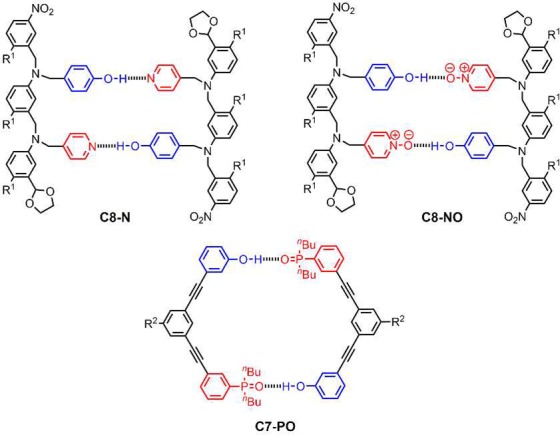
Structures of duplexes formed by AD 2-mers that do not fold (R1 = (2-ethylhexyl)oxy; R2 = S-β-citronelloxy).
Acknowledgments
We thank the Engineering and Physical Sciences Research Council (EP/J008044/2) and European Research Council (ERC-2012-AdG 320539-duplex) for funding. We thank Mr. Harry Adams (University of Sheffield) and Dr. Andrew Bond (University of Cambridge) for the help with X-ray crystallographic data collection and analysis.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.7b01357.
The authors declare no competing financial interest.
Supplementary Material
References
- a Watson J. D.; Crick F. H. Nature 1953, 171, 964. 10.1038/171964b0. [DOI] [PubMed] [Google Scholar]; b Sinden R. R.DNA Structure and Function; Academic Press: San Diego, CA, 1994. [Google Scholar]
- a Anfinsen C. B. Science 1973, 181, 223. 10.1126/science.181.4096.223. [DOI] [PubMed] [Google Scholar]; b Branden C.; Tooze J.. Introduction to Protein Structure, 2nd ed.; Garland Science: New York, 1999. [Google Scholar]; c Bikard D.; Loot C.; Baharoglu Z.; Mazel D. Microbiol. Mol. Biol. Rev. 2010, 74, 570. 10.1128/MMBR.00026-10. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Wells R. D. Trends Biochem. Sci. 2007, 32, 271. 10.1016/j.tibs.2007.04.003. [DOI] [PubMed] [Google Scholar]; e Burge S.; Parkinson G. N.; Hazel P.; Todd A. K.; Neidle S. Nucleic Acids Res. 2006, 34, 5402. 10.1093/nar/gkl655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Ellington A. D.; Szostak J. W. Nature 1990, 346, 818. 10.1038/346818a0. [DOI] [PubMed] [Google Scholar]; b Stoltenburg R.; Reinemann C.; Strehlitz B. Biomol. Eng. 2007, 24, 381. 10.1016/j.bioeng.2007.06.001. [DOI] [PubMed] [Google Scholar]; c Tan W.; Wang K.; Drake T. J. Curr. Opin. Chem. Biol. 2004, 8, 547. 10.1016/j.cbpa.2004.08.010. [DOI] [PubMed] [Google Scholar]
- a Blackburn G. M.; Gait M. J.; Loakes D.; Williams D. M.. Nucleic Acids in Chemistry and Biology, 3rd ed.; RSC Publishing: London, 2006. [Google Scholar]; b Conn G. L.; Draper D. E. Curr. Opin. Struct. Biol. 1998, 8, 278. 10.1016/S0959-440X(98)80059-6. [DOI] [PubMed] [Google Scholar]; c Bevilacqua P. C.; Blose J. M. Annu. Rev. Phys. Chem. 2008, 59, 79. 10.1146/annurev.physchem.59.032607.093743. [DOI] [PubMed] [Google Scholar]; d Rana T. M. Nat. Rev. Mol. Cell Biol. 2007, 8, 23. 10.1038/nrm2085. [DOI] [PubMed] [Google Scholar]
- a Kramer R.; Lehn J.-M.; Marquis-Rigault A. Proc. Natl. Acad. Sci. U. S. A. 1993, 90, 5394. 10.1073/pnas.90.12.5394. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Marquis A.; Smith V.; Harrowfield J.; Lehn J.-M.; Herschbach H.; Sanvito R.; Leize-Wagner E.; van Dorsselaer A. Chem. - Eur. J. 2006, 12, 5632. 10.1002/chem.200600143. [DOI] [PubMed] [Google Scholar]; c Anderson H. L. Inorg. Chem. 1994, 33, 972. 10.1021/ic00083a022. [DOI] [Google Scholar]; d Taylor P. N.; Anderson H. L. J. Am. Chem. Soc. 1999, 121, 11538. 10.1021/ja992821d. [DOI] [Google Scholar]; e Berl V.; Huc I.; Khoury R. G.; Krische M. J.; Lehn J.-M. Nature 2000, 407, 720. 10.1038/35037545. [DOI] [PubMed] [Google Scholar]; f Berl V.; Huc I.; Khoury R. G.; Lehn J.-M. Chem. - Eur. J. 2001, 7, 2810.. [DOI] [PubMed] [Google Scholar]
- a Tanaka Y.; Katagiri H.; Furusho Y.; Yashima E. Angew. Chem. 2005, 117, 3935. 10.1002/ange.200501028. [DOI] [PubMed] [Google Scholar]; b Ito H.; Furusho Y.; Hasegawa T.; Yashima E. J. Am. Chem. Soc. 2008, 130, 14008. 10.1021/ja806194e. [DOI] [PubMed] [Google Scholar]; c Yamada H.; Furusho Y.; Ito H.; Yashima E. Chem. Commun. 2010, 46, 3487. 10.1039/c002170a. [DOI] [PubMed] [Google Scholar]; d Yashima E.; Ousaka N.; Taura D.; Shimomura K.; Ikai T.; Maeda K. Chem. Rev. 2016, 116, 13752. 10.1021/acs.chemrev.6b00354. [DOI] [PubMed] [Google Scholar]
- a Sánchez-Quesada J.; Seel C.; Prados P.; de Mendoza J.; Dalcol I.; Giralt E. J. Am. Chem. Soc. 1996, 118, 277. 10.1021/ja953243d. [DOI] [Google Scholar]; b Bisson A. P.; Carver F. J.; Eggleston D. S.; Haltiwanger R. C.; Hunter C. A.; Livingstone D. L.; McCabe J. F.; Rotger C.; Rowan A. E. J. Am. Chem. Soc. 2000, 122, 8856. 10.1021/ja0012671. [DOI] [Google Scholar]; c Bisson A. P.; Hunter C. A. Chem. Commun. 1996, 1723. 10.1039/cc9960001723. [DOI] [Google Scholar]; d Gong B.; Yan Y.; Zeng H.; Skrzypczak-Jankunn E.; Kim Y. W.; Zhu J.; Ickes H. J. Am. Chem. Soc. 1999, 121, 5607. 10.1021/ja990904o. [DOI] [Google Scholar]; e Gong B. Synlett 2001, 582. 10.1055/s-2001-13350. [DOI] [Google Scholar]; f Zeng H.; Miller R. S.; Flowers R. A.; Gong B. J. Am. Chem. Soc. 2000, 122, 2635. 10.1021/ja9942742. [DOI] [Google Scholar]; g Gong B. Acc. Chem. Res. 2012, 45, 2077. 10.1021/ar300007k. [DOI] [PubMed] [Google Scholar]; h Yang Y.; Yang Z.-Y.; Yi Y.-P.; Xiang J.-F.; Chen C.-F.; Wan L.-J.; Shuai Z.-G. J. Org. Chem. 2007, 72, 4936. 10.1021/jo070525a. [DOI] [PubMed] [Google Scholar]; i Chu W.-J.; Yang Y.; Chen C.-F. Org. Lett. 2010, 12, 3156. 10.1021/ol101068n. [DOI] [PubMed] [Google Scholar]; j Chu W.-J.; Chen J.; Chen C.-F.; Yang Y.; Shuai Z. J. Org. Chem. 2012, 77, 7815. 10.1021/jo301434a. [DOI] [PubMed] [Google Scholar]; k Archer E. A.; Krische M. J. J. Am. Chem. Soc. 2002, 124, 5074. 10.1021/ja012696h. [DOI] [PubMed] [Google Scholar]; l Gong H.; Krische M. J. J. Am. Chem. Soc. 2005, 127, 1719. 10.1021/ja044566p. [DOI] [PubMed] [Google Scholar]
- a Dervan P. B.; Burli R. W. Curr. Opin. Chem. Biol. 1999, 3, 688. 10.1016/S1367-5931(99)00027-7. [DOI] [PubMed] [Google Scholar]; b Renneberg D.; Dervan P. B. J. Am. Chem. Soc. 2003, 125, 5707. 10.1021/ja0300158. [DOI] [PubMed] [Google Scholar]; c Dervan P. B.; Doss R. M.; Marques M. A. Curr. Med. Chem.: Anti-Cancer Agents 2005, 5, 373. 10.2174/1568011054222346. [DOI] [PubMed] [Google Scholar]; d Doss R. M.; Marques M. A.; Foister S.; Chenoweth D. M.; Dervan P. B. J. Am. Chem. Soc. 2006, 128, 9074. 10.1021/ja0621795. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Meier J. L.; Yu A. S.; Korf I.; Segal D. J.; Dervan P. B. J. Am. Chem. Soc. 2012, 134, 17814. 10.1021/ja308888c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Gellman S. H. Acc. Chem. Res. 1998, 31, 173. 10.1021/ar960298r. [DOI] [Google Scholar]; b Hill D. J.; Mio M. J.; Prince R. B.; Hughes T. S.; Moore J. S. Chem. Rev. 2001, 101, 3893. 10.1021/cr990120t. [DOI] [PubMed] [Google Scholar]; c Goodman C. M.; Choi S.; Shandler S.; DeGrado W. F. Nat. Chem. Biol. 2007, 3, 252. 10.1038/nchembio876. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Guichard G.; Huc Chem. Commun. 2011, 47, 5933. 10.1039/c1cc11137j. [DOI] [PubMed] [Google Scholar]; e Juwarker H.; Suk J.-m.; Jeong K.-S. Chem. Soc. Rev. 2009, 38, 3316. 10.1039/b909034g. [DOI] [PubMed] [Google Scholar]; f Gan Q.; Ferrand Y.; Bao C.; Kauffmann B.; Grélard A.; Jiang H.; Huc I. Science 2011, 331, 1172. 10.1126/science.1200143. [DOI] [PubMed] [Google Scholar]; g Gopalakrishnan R.; Frolov A. I.; Knerr L.; Drury W. J. III; Valeur E. J. Med. Chem. 2016, 59, 9599. 10.1021/acs.jmedchem.6b00376. [DOI] [PubMed] [Google Scholar]
- a Chandramouli N.; Ferrand Y.; Lautrette G.; Kauffmann B.; Mackereth C. D.; Laguerre M.; Dubreuil D.; Huc I. Nat. Chem. 2015, 7, 334. 10.1038/nchem.2195. [DOI] [PubMed] [Google Scholar]; b Lautrette G.; Wicher B.; Kauffmann B.; Ferrand Y.; Huc I. J. Am. Chem. Soc. 2016, 138, 10314. 10.1021/jacs.6b05913. [DOI] [PubMed] [Google Scholar]
- a Stross A. E.; Iadevaia G.; Hunter C. A. Chem. Sci. 2016, 7, 94. 10.1039/C5SC03414K. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Iadevaia G.; Stross A. E.; Neumann A.; Hunter C. A. Chem. Sci. 2016, 7, 1760. 10.1039/C5SC04467G. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Stross A. E.; Iadevaia G.; Hunter C. A. Chem. Sci. 2016, 7, 5686. 10.1039/C6SC01884J. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Núñez-Villanueva D.; Hunter C. A. Chem. Sci. 2017, 8, 206. 10.1039/C6SC02995G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Hunter C. A. Angew. Chem., Int. Ed. 2004, 43, 5310. 10.1002/anie.200301739. [DOI] [PubMed] [Google Scholar]; b Calero C. S.; Farwer J.; Gardiner E. J.; Hunter C. A.; Mackey M.; Scuderi S.; Thompson S.; Vinter J. G. Phys. Chem. Chem. Phys. 2013, 15, 18262. 10.1039/c3cp53158a. [DOI] [PubMed] [Google Scholar]
- a Adrian J. C.; Wilcox C. S. J. Am. Chem. Soc. 1991, 113, 678. 10.1021/ja00002a048. [DOI] [Google Scholar]; b Adrian J. C.; Wilcox C. S. J. Am. Chem. Soc. 1992, 114, 1398. 10.1021/ja00030a040. [DOI] [Google Scholar]
- Krishnamurthy V. M.; Semetey V.; Bracher P. J.; Shen N.; Whitesides G. M. J. Am. Chem. Soc. 2007, 129, 1312. 10.1021/ja066780e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Paffen T. F. E.; Ercolani G.; de Greef T. F. A.; Meijer E. W. J. Am. Chem. Soc. 2015, 137, 1501. 10.1021/ja5110377. [DOI] [PubMed] [Google Scholar]; b Teunissen A. J. P.; Paffen T. F. E.; Ercolani G.; de Greef T. F. A.; Meijer E. W. J. Am. Chem. Soc. 2016, 138, 6852. 10.1021/jacs.6b03421. [DOI] [PubMed] [Google Scholar]
- Hubbard T. A.; Brown A. J.; Bell I. A. W.; Cockroft S. L. J. Am. Chem. Soc. 2016, 138, 15114. 10.1021/jacs.6b09130. [DOI] [PubMed] [Google Scholar]
- a Misuraca M. C.; Grecu T.; Freixa Z.; Garavini V.; Hunter C. A.; van Leeuwen P. W. N. M.; Segarra-Maset M. D.; Turega S. M. J. Org. Chem. 2011, 76, 2723. 10.1021/jo2000397. [DOI] [PubMed] [Google Scholar]; b Hunter C. A.; Misuraca M. C.; Turega S. M. J. Am. Chem. Soc. 2011, 133, 582. 10.1021/ja1084783. [DOI] [PubMed] [Google Scholar]; c Hunter C. A.; Misuraca M. C.; Turega S. M. J. Am. Chem. Soc. 2011, 133, 20416. 10.1021/ja208330y. [DOI] [PubMed] [Google Scholar]; d Hunter C. A.; Misuraca M. C.; Turega S. M. Chem. Sci. 2012, 3, 2462. 10.1039/c2sc20358h. [DOI] [Google Scholar]; e Hunter C. A.; Misuraca M. C.; Turega S. M. Chem. Sci. 2012, 3, 589. 10.1039/C1SC00635E. [DOI] [Google Scholar]; f Sun H.; Hunter C. A.; Navarro C.; Turega S. J. Am. Chem. Soc. 2013, 135, 13129. 10.1021/ja406235d. [DOI] [PubMed] [Google Scholar]; g Adams H.; Chekmeneva E.; Hunter C. A.; Misuraca M. C.; Navarro C.; Turega S. M. J. Am. Chem. Soc. 2013, 135, 1853. 10.1021/ja310221t. [DOI] [PubMed] [Google Scholar]
- a Murray T. J.; Zimmerman S. C. J. Am. Chem. Soc. 1992, 114, 4010. 10.1021/ja00036a079. [DOI] [Google Scholar]; b Zimmerman S. C.; Murray T. J. Tetrahedron Lett. 1994, 35, 4077. 10.1016/S0040-4039(00)73116-9. [DOI] [Google Scholar]
- Hunter C. A.; Anderson H. L. Angew. Chem., Int. Ed. 2009, 48, 7488. 10.1002/anie.200902490. [DOI] [PubMed] [Google Scholar]
- MacroModel, version 9.8; Schrödinger, LLC, New York, 2014.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.