

Abstract
Drug combination is an appealing strategy for combating the heterogeneity of tumors and evolution of drug resistance. However, the rationale underlying combinatorial therapy is often not well established due to lack of understandings of the specific pathways responding to the drugs, and their temporal dynamics following each treatment. Here we present several emerging trends in harnessing properties of biological systems for the optimal design of drug combinations, including the type of drugs, specific concentration, sequence of addition and the temporal schedule of treatments. We highlight recent studies showing different approaches for efficient design of drug combinations including single-cell signaling dynamics, adaption and pathway crosstalk. Finally, we discuss novel and feasible approaches that can facilitate the optimal design of combinatorial therapy.
Introduction
Tumorgenesis is an evolutionary process during which a series of mutations rise and accumulate in cells, allowing them to grow beyond physiological limitations. Depending on their history, different cancer clones can use different strategies to escape growth controls. Even in a single tumor multiple regulatory pathways can be defective including apoptosis, migration, cell cycle arrest or suppression of the immune response [1]. Such heterogeneity among individual cancer cells often limits the efficacy of a single anticancer drug to fully eliminate all cells in a tumor.
One approach for overcoming therapy resistance is by combining multiple drugs. The main rationale behind combinatorial therapy is to suppress more than one pathway and therefore to synergistically eradicate the various clones that emerge in a tumor [2]. Such approaches have been proven successful in culture cells and in the clinic [3–7]. However, despite the great interest in, and potential of combinatorial therapies, the design of drug combination (e.g. specific concentration of each drug, sequence of addition, time interval between treatments) mostly relies on the knowledge from administrating each drug alone, and in many cases on trials and errors.
The proper design of combinatorial treatments is critical for its success. Administration of one drug can lead to a dynamic response, which may increase or decrease the sensitivity of the cells to the second treatment. Such interactions may depend on the time interval between treatments, the state of the cells or the concentration of each drug. In most cases we lack the knowledge and understanding of how each drug impact cellular states that may interact epistatically with the second drug. In this review, we present recent discoveries about the dynamics of, and crosstalk between multiple signaling pathways that may have impacts on cellular states and their vulnerability, which can help rationalize the design of drug combination.
A. Signaling dynamics guide the design of combinatorial therapy
In response to external and internal inputs, signaling molecules act collectively to generate temporal changes in their level, localization or activity, here defined as “signaling dynamics”. Recently an increasing amount of evidence showed that quantitative features of signaling dynamics, such as the duration of the signal, its amplitude or accumulation rate, can carry biological information that is critical for cellular outcomes [8]. Specifically, several transcription factors have been shown to exhibit signal- and stimulus-specific dynamics that govern the transcriptional programs for differential cell fates. These include the transcription factor Msn2 in Saccharomyces cerevisiae [9], and p53 [10,11] and NF-kB [12] in human cells. The idea that signaling dynamics play an important role for cells was further strengthened by the fact that modulation of the dynamics of p53 levels and of ERK activity result in cell fate switch [11,13].
The processing of cellular information can vary dramatically between cells, and even genetically and developmentally equivalent cells may show different behaviors in response to the same stimulus. As a result, the average behavior of a population often represents a distorted version of individual patterns. For example, our studies on the p53 signaling pathway in single cells revealed a series of p53 pulses in response to DNA damage [14,15] and spontaneous p53 pulses in non-stressed conditions [16], which were masked by population averaging assays [17]. These newly identified behaviors of p53 led us to develop new models for the signaling circuits controlling p53 dynamics [10,15], and to identify a new information-transfer mechanism in this network [11]. These examples underscore the importance of tracking cellular and molecular responses at the single-cell level.
Can the dynamics of signaling molecules be used to guide the design of combinatorial therapies? In our recent work we found that when the oncogenic inhibitor of p53, MDMX, is suppressed, p53 shows two phases of dynamics in individual cells: an initial post mitotic high-amplitude pulse followed by small amplitude oscillations [18]. We further showed that these two phases of p53 dynamics are associated with activation of distinct p53 transcriptional programs: the post-mitotic pulse led to a universal p53 response activating the transcription of genes involved in multiple programs including apoptosis and other pro-death signals. The second phase of small amplitude p53 oscillations led to a specific transcriptional activation of p21, a p53 target that regulates cell cycle progression and other pro-survival signals. These results suggest that MDMX suppression results in a transcriptional program switch regulated by biphasic p53 dynamics (Fig. 1B). Importantly, these observations prompted us to examine the temporal effects in combining MDMX suppression with DNA damage.
Figure 1. Schedule dependent interaction between MDMX suppression and DNA damage.

(A) Mdmx suppression leads to two phases of p53 dynamics in single cells; post-mitotic pulse (blue) followed by low amplitude oscillations (yellow).
(B) The first phase of p53 activates pro-apoptotic signals, and the second phase activates pro-survival signals.
(C) The time interval between Mdmx suppression and DNA damage determines their interaction. A short interval leads to a synergistic effect and to more cell death. A long interval leads to an antagonistic effect and protect cells from death.
MDMX is overexpressed in multiple cancers including malignant melanoma, glioma and breast cancer, where p53 activity is mostly suppressed [19–21]. As a result, MDMX suppression has been suggested as a therapeutic intervention for such cancer patients [22,23]. Since most patients with MDMX overexpression bear wild type p53, we designed a combinatorial therapy composed of MDMX suppression and chemotherapy that activates p53-dependent apoptotic function. The two distinct phases of p53 dynamics (Fig. 1A) and its transcriptional program (Fig. 1B) after MDMX suppression led to a switch in the interaction between chemotherapy and MDMX inhibition depending on the time interval between the two treatments (Fig. 1C). Specifically, when DNA damage was induced during the first phase of p53 dynamics, it synergized with MDMX suppression and led to more killing of the cancer cells. However, when DNA damage was given during the second phase of p53 dynamics (p53 oscillations), MDMX suppression antagonized it and made the cells more resistance (Fig. 1C). In another recent study of p53 dynamics in response to the chemotherapy drug, Cisplatin, we revealed that the rate of p53 accumulation determined the likelihood of cell death [24]. This was caused by induction of the anti-apoptotic IAP pathway that antagonizes with p53-mediated apoptosis, leading to a combinatorial treatment where p53 is activated and the IAP pathway is inhibited, increasing the efficacy of Cisplatin. These studies showed that quantifying the dynamics of signaling molecules in single cells provide valuable temporal and spatial information on cellular status during drug treatments, which is critical for designing optimum schedule of drug combinations.
B. Drug combinations to counteract pathway adaptation
In order to maintain physiological homeostasis and to prevent signal overshoot, many signaling pathways exhibit negative feedback regulations. In many cases, negative feedbacks allow the system to reset itself back to the pre-stimulated state after the stimuli, a dynamic property also known as adaptation [25,26]. While adaptation in signaling pathways broadens the range of signal sensing and allows sensitive response to input change, it can also blunt drug response and cause drug resistance. A well-known example is the RTK-RAF-MAPK pathway adaptation to RAF inhibitors in tumors harboring BRAFV600E mutations [26,27].
RAF kinases (A, B and C) are major activating regulators of the mitogen-activated protein kinase (MAPK) cascade [28]. Growth factors active the upstream receptor tyrosine kinases (RTKs), which induce RAF kinases and the MAPK kinase ERK. ERK kinase then phosphorylates and activates transcriptional factors to promote cell cycle progression [29]. At the same time, ERK acts as a negative feedback to suppress upstream RTK and RAF kinases through direct inhibitory phosphorylations [27].
BRAF is highly mutated in various cancers including melanoma, thyroid and colorectal cancers [30]. The most common V600E mutation renders BRAFV600E constitutively active. The hyperactive BRAFV600E usually maintains strong ERK-mediated negative feedbacks thus low RTK activities. When treated with BRAFV600E specific inhibitor, there are three phases of dynamic changes in the pathway activity (Fig 2.) and [31]. Initially, the BRAFV600E and its downstream MAPK kinases are inactivated (Fig 2. Phase I). Thereafter, the ERK-mediated negative feedback is relieved which results in activation of the upstream RTK and RAF kinases (Fig 2. Phase II). Finally, the RTK and RAF kinases activate ERK and ERK-mediated feedback and the pathway reaches a new steady state that can be close to pre-treatment state (Fig 2. Phase III).
Figure 2. RTK-RAF-MAPK pathway adaptation after BRAFV600E inhibitor treatment.

Administration of the BRAFV600E inhibitor leads to three phases of ERK activity. The corresponding states of the underlying molecular mechanism for each phase are shown. The dominating interactions at each phase are marked by bold arrows. High activity levels of signaling molecules are shown in bold colored circles, while low activity levels are marked by faint circles.
Due to this pathway adaptation, the strongest drug effect of BRAFV600E inhibitor is transient and only last for a few hours. In addition, it also diminishes growth dependency of tumor cells on BRAFV600E oncogene; and in the longer term may allow tumors to develop drug resistance. A potential rational drug combination design that prevents these complications is to suppress the pathway adaptation by targeting the negative feedback components. Indeed, BRAFV600E inhibitors combined with MAPK inhibitors were shown to suppress the pathway adaptation and the overall activity of the RTK-RAF-MAPK pathway [32]. This strategy is effective in increasing drug response and is currently widely used in the clinic. Similar strategies may be examined and tested in other oncogenic signaling pathways with negative feedback controls.
C. Targeting signaling crosstalk
Similar to developmental and physiological cues, drug treatments can change or “rewire” cellular states. In particular, recent evidence suggests that targeted pharmaceutical interventions of a single signaling molecule can result in enhancement of cellular functions in traditionally defined different pathways through their crosstalk (Fig. 3). For instance, pharmacological suppression of PI3K in patients bearing PIK3CA-mutant ER-positive tumors strengthens estrogen receptor (ER) function and tumor reliance on estrogen signal, which inspires a combinatorial therapy to simultaneously suppress both PI3K and ER pathways [33]. Additionally, other crosstalk in signaling pathways including PI3K/AKT/mTOR axis and PI3K/AR pathways have also been used to design combinatorial therapies [34–38].
Figure 3. Pathway crosstalk after drug treatment.
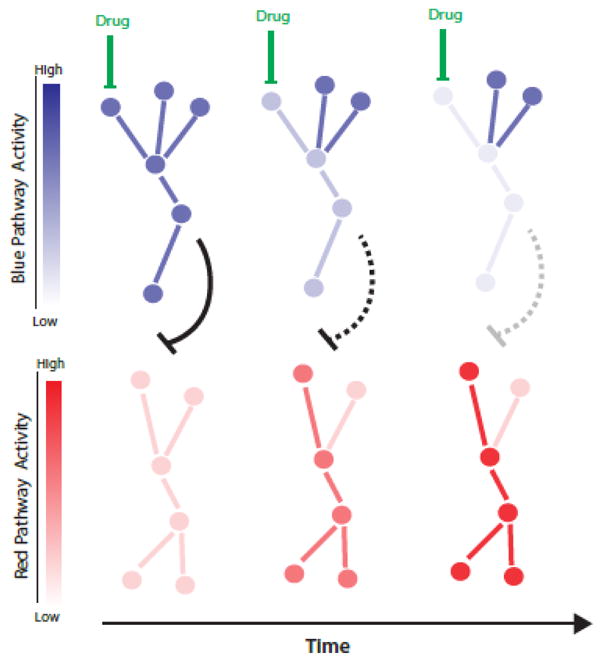
Drug administration leads to a gradual decrease in the drug-inhibited pathway (blue) followed by the activation of another signaling pathway (red) through their crosstalk reflected by the black inhibitory arrow.
Just like ripples propagating from the center of the perturbation, it takes time for a crosstalk to take place. A recent work showed that a proper temporal design of drug combination is required for targeting triple-negative breast cancers (TNBC) [39]. An inhibitor of EGFR was combined with chemotherapy drugs to synergistically kill TNBC. The synergy only existed when there was at least a minimum of four hours between the treatments. The authors then used systems profiling and a data-driven modeling (discussed below) to further identify the apoptotic pathway that changes over time after EGFR inhibition and sensitizes TNBC to the second chemotherapy. This study exemplified that the proper design of drug combinations requires understanding the temporal dynamics of crosstalk between pathways.
Despite these successful examples, we are still far from a comprehensive picture of crosstalk among signaling pathways, especially their dynamics in cell- and disease-specific contexts. For example, the time-dependent changes in apoptotic pathway in response to EGFR suppression occurs only in TNBC but not in two other tested cell types. In addition, the mechanism through which this crosstalk occur remains to be characterized [39]. Thus, there is a critical need to develop integrated approaches to identify cell- and tissue-specific regulatory mechanisms underlying pathway crosstalk in various diseases.
D. Toward rational design of drug combinations
The preceding examples show how specific properties of signaling pathways, including their dynamics, adaptation and crosstalk can help identify companion drug combinations and the optimal temporal design of drug treatments. However, in the face of the daunting complexity of signaling pathways, what approaches would be most suitable and feasible to guide us toward an optimal design of combinatorial therapy?
One potential approach is to focus on well-characterized signaling molecules that are known to regulate critical cellular behaviors such as DNA damage response, inflammation and nutrient sensing. For example p53, NF-kB, mTOR, AKT, and ERK are hubs of signaling networks that integrate multiple upstream signals and in turn carry out multiple downstream outcomes. In this regard, their dynamics can be markers for cellular state as well as important clues for mechanistic dissection. In certain biological contexts, the dynamics themselves can be potential drug targets [40]. One illuminating example is the alternation of p53 dynamics to switch cellular decision from cell cycle arrest to senescence [11]. In this case, the construction of a kinetic model based on biochemical mechanisms of the p53 pathway provides direct links between p53 dynamics and their underlying mechanisms. Furthermore, it enables the possibility of “drugging the signaling dynamics” by designing pharmaceutical perturbations that reshape p53 dynamics decisively with precise doses and timing of drug treatments. Thus, the construction of a mechanism-based kinetic model allows precise control of signaling dynamics for pharmaceutical purposes.
With the advance of high-throughput technologies, it is now feasible to profile cancer cells with their genomes, transcriptomes and proteomes during drug responses. A recent study set an important example of using a data-driven modeling approach to identify potential combination drug targets [39]. A data set composed of transcriptional profiles, protein levels and their modification states at multiple time points after the first drug treatment was collected. By applying an unbiased, data-driven modeling analysis to this data set [41], they successfully identified signaling molecules that are mostly associated with change in cellular state, which are potential drug targets for a second treatment.
In many cases, however, there is little prior information on cellular drug response and the corresponding molecular events. In such cases the challenge is to predict the effects of drug combinations when the information is sparse and limited. A recent in silico competition of the “AstraZeneca-Sanger Drug Combination Prediction Challenge [42]” advocates the use of integrated statistical modeling and machine learning approaches to identify data features in a large but sparse data set wherein more than ten thousand drug combinations were experimentally tested measuring cell viability over a hundred drugs. While it remains to be examined how general this approach is in predicting drug synergy, it has the potential of pointing to specific new strategies for designing optimal combination therapies.
While we begin to grasp the nature of biological systems for better cancer therapy, it is evidence that a proper design of drug combinations requires multiple lines of information, including human cancer genetics, cross talk between pathways, inter-cellular signaling in the cancer microenvironment and dynamic drug responses in individual cells. In addition, quantitative and reproducible measurements of drug responses are fundamentally important. A recent study showed that the conventional drug response measurements using IC50 or Emax can be confounding due to different division rates of cells [43]. New drug-response metrics were derived to estimate drug-induced growth rate inhibition (GR), which can be critical in quantifying and predicting the effects of drug combinations. With the accumulation of such context-specific measures, our ultimate goal will be to search for general principles that can catalyze the rational design of drug combinations in multiple disease contexts.
Highlights.
Optimal scheduling of drug combinations requires understanding of the changes in cellular states after each drug.
The dynamics of signaling molecules are useful for assessing the changes in cellular states and can be potential drug targets.
Signaling adaptation due to feedback control often blunts drug responses and can be suppressed using combinatorial therapy.
Understanding the crosstalk between signaling pathways is critical for designing efficient combinatorial therapy.
Acknowledgments
We thank members of the Lahav laboratory for discussion. This research was supported by National Institute of Health grant GM083303 to G.L., F32GM105205 to S.C. and funding from the Novartis Institutes for Biomedical Research.
Footnotes
Conflict of interest statement
Nothing declared.
References
- 1.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 2**.Lord CJ, Tutt ANJ, Ashworth A. Synthetic lethality and cancer therapy: lessons learned from the development of PARP inhibitors. Annu Rev Med. 2015;66:455–470. doi: 10.1146/annurev-med-050913-022545. A comprehensive review of using synthetic lethality strategy to target crosstalk in cancer cells focusing on targeting DNA damage repair pathways. [DOI] [PubMed] [Google Scholar]
- 3.Brechbiel J, Miller-Moslin K, Adjei AA. Crosstalk between hedgehog and other signaling pathways as a basis for combination therapies in cancer. Cancer Treat Rev. 2014;40:750–759. doi: 10.1016/j.ctrv.2014.02.003. [DOI] [PubMed] [Google Scholar]
- 4.Li J, Shin S, Sun Y, Yoon S-O, Li C, Zhang E, Yu J, Zhang J, Blenis J. mTORC1-driven tumor cells are highly sensitive to therapeutic targeting by antagonists of oxidative stress. Cancer Res. 2016 doi: 10.1158/0008-5472.CAN-15-2629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Andersen NJ, Boguslawski EB, Kuk CY, Chambers CM, Duesbery NS. Combined inhibition of MEK and mTOR has a synergic effect on angiosarcoma tumorgrafts. Int J Oncol. 2015;47:71–80. doi: 10.3892/ijo.2015.2989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ma X-H, Piao S-F, Dey S, McAfee Q, Karakousis G, Villanueva J, Hart LS, Levi S, Hu J, Zhang G, et al. Targeting ER stress-induced autophagy overcomes BRAF inhibitor resistance in melanoma. J Clin Invest. 2014;124:1406–1417. doi: 10.1172/JCI70454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hu-Lieskovan S, Mok S, Homet Moreno B, Tsoi J, Robert L, Goedert L, Pinheiro EM, Koya RC, Graeber TG, Comin-Anduix B, et al. Improved antitumor activity of immunotherapy with BRAF and MEK inhibitors in BRAF(V600E) melanoma. Sci Transl Med. 2015;7:279ra41. doi: 10.1126/scitranslmed.aaa4691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8**.Purvis JE, Lahav G. Encoding and decoding cellular information through signaling dynamics. Cell. 2013;152:945–956. doi: 10.1016/j.cell.2013.02.005. A review of how the dynamics of signaling molecules is shaped by the network structure and control cellular behaviors. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hao N, O’Shea EK. Signal-dependent dynamics of transcription factor translocation controls gene expression. Nat Struct Mol Biol. 2012;19:31–39. doi: 10.1038/nsmb.2192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Batchelor E, Loewer A, Mock C, Lahav G. Stimulus-dependent dynamics of p53 in single cells. Mol Syst Biol. 2011;7:488. doi: 10.1038/msb.2011.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Purvis JE, Karhohs KW, Mock C, Batchelor E, Loewer A, Lahav G. p53 dynamics control cell fate. Science. 2012;336:1440–1444. doi: 10.1126/science.1218351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Werner SL, Barken D, Hoffmann A. Stimulus specificity of gene expression programs determined by temporal control of IKK activity. Science. 2005;309:1857–1861. doi: 10.1126/science.1113319. [DOI] [PubMed] [Google Scholar]
- 13.Santos SDM, Verveer PJ, Bastiaens PIH. Growth factor-induced MAPK network topology shapes Erk response determining PC-12 cell fate. Nat Cell Biol. 2007;9:324–330. doi: 10.1038/ncb1543. [DOI] [PubMed] [Google Scholar]
- 14.Lahav G, Rosenfeld N, Sigal A, Geva-Zatorsky N, Levine AJ, Elowitz MB, Alon U. Dynamics of the p53-Mdm2 feedback loop in individual cells. Nat Genet. 2004;36:147–150. doi: 10.1038/ng1293. [DOI] [PubMed] [Google Scholar]
- 15.Batchelor E, Mock CS, Bhan I, Loewer A, Lahav G. Recurrent initiation: a mechanism for triggering p53 pulses in response to DNA damage. Mol Cell. 2008;30:277–289. doi: 10.1016/j.molcel.2008.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Loewer A, Batchelor E, Gaglia G, Lahav G. Basal dynamics of p53 reveal transcriptionally attenuated pulses in cycling cells. Cell. 2010;142:89–100. doi: 10.1016/j.cell.2010.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lev Bar-Or R, Maya R, Segel LA, Alon U, Levine AJ, Oren M. Generation of oscillations by the p53-Mdm2 feedback loop: a theoretical and experimental study. Proc Natl Acad Sci US A. 2000;97:11250–11255. doi: 10.1073/pnas.210171597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18**.Chen S-H, Forrester W, Lahav G. Schedule-dependent interaction between anticancer treatments. Science. 2016;351:1204–1208. doi: 10.1126/science.aac5610. The first study using live imaging and single-cell dynamics to reveal a switch in cellular states that guided the design for drug combination. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gembarska A, Luciani F, Fedele C, Russell EA, Dewaele M, Villar S, Zwolinska A, Haupt S, de Lange J, Yip D, et al. MDM4 is a key therapeutic target in cutaneous melanoma. Nat Med. 2012;18:1239–1247. doi: 10.1038/nm.2863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Danovi D, Meulmeester E, Pasini D, Migliorini D, Capra M, Frenk R, de Graaf P, Francoz S, Gasparini P, Gobbi A, et al. Amplification of Mdmx (or Mdm4) directly contributes to tumor formation by inhibiting p53 tumor suppressor activity. Mol Cell Biol. 2004;24:5835–5843. doi: 10.1128/MCB.24.13.5835-5843.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Riemenschneider MJ, Büschges R, Wolter M, Reifenberger J, Boström J, Kraus JA, Schlegel U, Reifenberger G. Amplification and overexpression of the MDM4 (MDMX) gene from 1q32 in a subset of malignant gliomas without TP53 mutation or MDM2 amplification. Cancer Res. 1999;59:6091–6096. [PubMed] [Google Scholar]
- 22.Wade M, Li Y-C, Wahl GM. MDM2, MDMX and p53 in oncogenesis and cancer therapy. Nat Rev Cancer. 2013;13:83–96. doi: 10.1038/nrc3430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Karni-Schmidt O, Lokshin M, Prives C. The Roles of MDM2 and MDMX in Cancer. Annu Rev Pathol. 2016;11:617–644. doi: 10.1146/annurev-pathol-012414-040349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Paek AL, Liu JC, Loewer A, Forrester WC, Lahav G. Cell-to-Cell Variation in p53 Dynamics Leads to Fractional Killing. Cell. 2016;165:631–642. doi: 10.1016/j.cell.2016.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ma W, Trusina A, El-Samad H, Lim WA, Tang C. Defining network topologies that can achieve biochemical adaptation. Cell. 2009;138:760–773. doi: 10.1016/j.cell.2009.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26*.Fey D, Matallanas D, Rauch J, Rukhlenko OS, Kholodenko BN. The complexities and versatility of the RAS-to-ERK signalling system in normal and cancer cells. Semin Cell Dev Biol. 2016 doi: 10.1016/j.semcdb.2016.06.011. A review of the system properties of the RTK-RAS-ERK pathway including its dynamics and adaptation and their relationship to drug resistance. [DOI] [PubMed] [Google Scholar]
- 27.Lito P, Rosen N, Solit DB. Tumor adaptation and resistance to RAF inhibitors. Nat Med. 2013;19:1401–1409. doi: 10.1038/nm.3392. [DOI] [PubMed] [Google Scholar]
- 28.Desideri E, Cavallo AL, Baccarini M. Alike but Different: RAF Paralogs and Their Signaling Outputs. Cell. 2015;161:967–970. doi: 10.1016/j.cell.2015.04.045. [DOI] [PubMed] [Google Scholar]
- 29.Lavoie H, Therrien M. Regulation of RAF protein kinases in ERK signalling. Nat Rev Mol Cell Biol. 2015;16:281–298. doi: 10.1038/nrm3979. [DOI] [PubMed] [Google Scholar]
- 30.Rahman MA, Salajegheh A, Smith RA, Lam AK-Y. BRAF inhibitor therapy for melanoma, thyroid and colorectal cancers: development of resistance and future prospects. Curr Cancer Drug Targets. 2014;14:128–143. doi: 10.2174/1568009614666140121150930. [DOI] [PubMed] [Google Scholar]
- 31.Lito P, Pratilas CA, Joseph EW, Tadi M, Halilovic E, Zubrowski M, Huang A, Wong WL, Callahan MK, Merghoub T, et al. Relief of profound feedback inhibition of mitogenic signaling by RAF inhibitors attenuates their activity in BRAFV600E melanomas. Cancer Cell. 2012;22:668–682. doi: 10.1016/j.ccr.2012.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Corcoran RB, Atreya CE, Falchook GS, Kwak EL, Ryan DP, Bendell JC, Hamid O, Messersmith WA, Daud A, Kurzrock R, et al. Combined BRAF and MEK Inhibition With Dabrafenib and Trametinib in BRAF V600-Mutant Colorectal Cancer. J Clin Oncol. 2015;33:4023–4031. doi: 10.1200/JCO.2015.63.2471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bosch A, Li Z, Bergamaschi A, Ellis H, Toska E, Prat A, Tao JJ, Spratt DE, Viola-Villegas NT, Castel P, et al. PI3K inhibition results in enhanced estrogen receptor function and dependence in hormone receptor-positive breast cancer. Sci Transl Med. 2015;7:283ra51. doi: 10.1126/scitranslmed.aaa4442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Carracedo A, Ma L, Teruya-Feldstein J, Rojo F, Salmena L, Alimonti A, Egia A, Sasaki AT, Thomas G, Kozma SC, et al. Inhibition of mTORC1 leads to MAPK pathway activation through a PI3K-dependent feedback loop in human cancer. J Clin Invest. 2008;118:3065–3074. doi: 10.1172/JCI34739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chandarlapaty S, Sawai A, Scaltriti M, Rodrik-Outmezguine V, Grbovic-Huezo O, Serra V, Majumder PK, Baselga J, Rosen N. AKT inhibition relieves feedback suppression of receptor tyrosine kinase expression and activity. Cancer Cell. 2011;19:58–71. doi: 10.1016/j.ccr.2010.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mendoza MC, Er EE, Blenis J. The Ras-ERK and PI3K-mTOR pathways: cross-talk and compensation. Trends Biochem Sci. 2011;36:320–328. doi: 10.1016/j.tibs.2011.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Carver BS, Chapinski C, Wongvipat J, Hieronymus H, Chen Y, Chandarlapaty S, Arora VK, Le C, Koutcher J, Scher H, et al. Reciprocal feedback regulation of PI3K and androgen receptor signaling in PTEN-deficient prostate cancer. Cancer Cell. 2011;19:575–586. doi: 10.1016/j.ccr.2011.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38*.Ni J, Ramkissoon SH, Xie S, Goel S, Stover DG, Guo H, Luu V, Marco E, Ramkissoon LA, Kang YJ, et al. Combination inhibition of PI3K and mTORC1 yields durable remissions in mice bearing orthotopic patient-derived xenografts of HER2-positive breast cancer brain metastases. Nat Med. 2016 doi: 10.1038/nm.4120. A study of using patient-derived xenografts to understand mechanism of efficacy and responsiveness of targeted drug combinations. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lee MJ, Ye AS, Gardino AK, Heijink AM, Sorger PK, MacBeath G, Yaffe MB. Sequential application of anticancer drugs enhances cell death by rewiring apoptotic signaling networks. Cell. 2012;149:780–794. doi: 10.1016/j.cell.2012.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40**.Behar M, Barken D, Werner SL, Hoffmann A. The dynamics of signaling as a pharmacological target. Cell. 2013;155:448–461. doi: 10.1016/j.cell.2013.09.018. The first theoretical study describing how signaling dynamics emerged from molecular mechanisms can be used as drug targets. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Janes KA, Yaffe MB. Data-driven modelling of signal-transduction networks. Nat Rev Mol Cell Biol. 2006;7:820–828. doi: 10.1038/nrm2041. [DOI] [PubMed] [Google Scholar]
- 42.https://www.synapse.org/#!Synapse:syn4231880 * The AstraZeneca-Sanger Drug Combination Prediction DREAM Challenge website that describes the goals, raw data and competition results.
- 43**.Hafner M, Niepel M, Chung M, Sorger PK. Growth rate inhibition metrics correct for confounders in measuring sensitivity to cancer drugs. Nat Methods. 2016;13:521–527. doi: 10.1038/nmeth.3853. A novel metrics for quantitatively assessing drug response. [DOI] [PMC free article] [PubMed] [Google Scholar]