

IRE1α is essential for maintaining podocyte and glomerular integrity as mice age and in glomerulonephritis. The mechanism is related, at least in part, to the maintenance of autophagy in podocytes.
Abstract
Inositol-requiring enzyme-1α (IRE1α) is an endoplasmic reticulum (ER)–transmembrane endoribonuclease kinase that plays an essential function in extraembryonic tissues during normal development and is activated during ER stress. To address the functional role of IRE1α in glomerular podocytes, we produced podocyte-specific IRE1α-deletion mice. In male mice, deletion of IRE1α in podocytes resulted in albuminuria beginning at 5 mo of age and worsening with time. Electron microscopy revealed focal podocyte foot-process effacement in 9-mo-old male IRE1α-deletion mice, as well as microvillous transformation of podocyte plasma membranes. Compared with control, glomerular cross-sectional and capillary lumenal areas were greater in deletion mice, and there was relative podocyte depletion. Levels of microtubule-associated protein 1A/1B-light chain 3 (LC3)-II expression and c-Jun N-terminal kinase-1 phosphorylation were decreased in IRE1α-deletion glomeruli, in keeping with reduced autophagy. Deletion of IRE1α exacerbated glomerular injury in anti–glomerular basement membrane nephritis. In cell culture, IRE1α dominant-negative mutants reduced the physiological (basal) accumulation of LC3B-II and the size of autophagic vacuoles but did not affect ER-associated degradation. Thus IRE1α is essential for maintaining podocyte and glomerular integrity as mice age and in glomerulonephritis. The mechanism is related, at least in part, to the maintenance of autophagy in podocytes.
INTRODUCTION
The endoplasmic reticulum (ER) is an organelle with many critical roles, including calcium homeostasis and the folding and maturation of proteins that are trafficked along the secretory pathway (Araki and Nagata, 2011). Inositol-requiring enzyme 1α (IRE1α) is an ER-transmembrane protein and a sensor of misfolded protein accumulation in the ER, which leads to ER stress (Hetz and Glimcher, 2009; Araki and Nagata, 2011; Hetz, 2012). By splicing X-box binding protein-1 (Xbp1) mRNA, IRE1α serves as a transcriptional inducer of the unfolded protein response (UPR). The importance of IRE1α in the maintenance of normal mammalian physiology and embryogenesis has been demonstrated in vivo, although its physiological mechanism of action is poorly understood (Iwawaki et al., 2009). Indeed, studies of the physiological role of IRE1α in adult mice are limited in number due to embryonic lethality of global IRE1α deletion (Iwawaki et al., 2009).
The IRE1-mediated UPR branch is the most evolutionarily conserved branch in eukaryotes. In yeast, IRE1 is the only ER sensor. In mammals, besides IRE1, two other proteins modulate distinct branches of the UPR, including activating transcription factor 6 (ATF6) and protein kinase R–like endoplasmic reticulum kinase (PERK). Mammalian IRE1 splices Xbp1 mRNA, which is homologous to yeast Hac1p. There are two isoforms of IRE1 in mammals—IRE1α, which is expressed ubiquitously, and IRE1β, which is expressed in gut epithelium (Hetz and Glimcher, 2009; Hetz, 2012).
The UPR acts to reduce the amount of misfolded protein and facilitates elimination of terminally misfolded glycoproteins. This involves up-regulation of genes encoding pro-folding ER-resident chaperones and components of ER-associated degradation (ERAD; Vembar and Brodsky, 2008). ERAD is a process that selects and retrotranslocates terminally misfolded proteins from the ER, trafficking them to the cytoplasm for degradation by the 26S proteasome (Vembar and Brodsky, 2008). IRE1α has been linked to the up-regulation of UPR and ERAD genes (Hetz and Glimcher, 2009; Hetz, 2012), but direct associations between IRE1α and substrates undergoing ERAD have not been confirmed. The general steps in ERAD have been well characterized through insights gained from yeast and mammalian studies (Vembar and Brodsky, 2008; Sun et al., 2015). Even under basal conditions, ER protein glycosylation, folding, and maturation is believed to be an imperfect process, and normally, a fraction of certain wild-type (WT) proteins is constitutively degraded via ERAD because of minor folding errors or ill-timed exposure of hydrophobic patches. Such proteins include T-cell coreceptor components (e.g., CD3δ) and the cystic fibrosis transmembrane regulator (Bonifacino et al., 1989; Varga et al., 2004). Thus constitutive, physiological ERAD is potentially required to prevent ER stress.
There is emerging evidence to support the view that autophagy is linked with the UPR and is another major mechanism to clear misfolded proteins from the secretory pathway, thereby alleviating ER stress (Cybulsky, 2013). IRE1α has been linked to autophagic degradation of misfolded proteins in studies of ER stress in vitro but not in vivo (Ogata et al., 2006; Oh and Lim, 2009; Lee et al., 2012). Autophagy is a term used to describe three interrelated pathways of protein and organellar degradation: macroautophagy (commonly referred to as autophagy), chaperone-mediated autophagy, and microautophagy (Cuervo, 2004; Huber et al., 2012). Autophagy begins with the formation of a double membrane (phagophore) that engulfs a portion of the cytoplasm. Eventually, the membrane encloses to form a vesicle (autophagosome), which then fuses with a lysosome, leading to the degradation of vesicular content by lysosomal hydrolases. Among signaling pathways known to regulate the formation of autophagosomes, inhibition of the mammalian target of rapamycin (mTOR) pathway resulting from growth factor deprivation is well established (Jung et al., 2010; Yang and Klionsky, 2010). Within this context, activation of the c-Jun amino-terminal kinase-1 (JNK1) and the death-associated protein kinase also contributes to this process by targeting beclin-1, a protein playing a critical role in the regulation of autophagy (Yang and Klionsky, 2010). Formation of autophagosomes involves conjugation of phosphatidylethanolamine to free microtubule-associated protein 1A/1B-light chain 3 (LC3), or LC3-I. Lipidated LC3 (LC3-II) redistributes from the cytosol to autophagic vacuole membranes and is therefore commonly used as a marker of autophagy. In many cells, basal autophagosome turnover and the basal quantity of autophagosomes are low. Basal/constitutive autophagy is involved in degradation of long-lived proteins and organelle turnover.
IRE1α forms a stable complex with tumor necrosis factor-α receptor–associated factor 2 (TRAF2; an E3 ubiquitin ligase and adaptor protein) and apoptosis signal–regulated kinase 1 (ASK1; Hetz and Glimcher, 2009; Hetz, 2012). The IRE1α–TRAF2 interaction has been localized to the IRE1α RNase domain (Kaneko et al., 2003). The IRE1α–TRAF2 complex has also been demonstrated to act as a positive regulator of autophagy. Double IRE1α and IRE1β–knockout (KO) mouse embryonic fibroblasts were deficient in ER stress–mediated autophagosome formation, as were WT fibroblasts expressing a dominant-negative TRAF2 mutant (Ogata et al., 2006). IRE1α knockdown in WI38 cells prevented dihydrocapsaicin-mediated autophagosome formation, and a JNK1 inhibitor prevented autophagosome formation in the presence of dihydrocapsaicin, thapsigargin, or MG132 (Oh and Lim, 2009). Knockdown of TRAF2 decreased autophagic flux induced by the autophagic enhancer tumor necrosis factor superfamily member 10 (He et al., 2012). TRAF2 was reported to colocalize with mitochondria in autophagosomes and was required for the induction of mitophagy (Yang et al., 2015). Finally, IRE1α was required for lysosomal (and proteasomal) degradation of a misfolded rhodopsin mutant (Chiang et al., 2012). Besides autophagy, IRE1α may influence proteasomal activity via ASK1; in particular, ASK1 may negatively regulate the 26S proteasome by inactivating Rpt5, an AAA ATPase of the 19S proteasome (Um et al., 2010). Given that ERAD requires 26S proteasomal activity, these studies suggest an inhibitory role of IRE1α in ERAD.
In recent years, strains of conditional IRE1α-deletion mice have been generated. Mice with IRE1α deletion in extraplacental tissues exhibited mild hypoinsulinemia, hyperglycemia, and a low-weight trend (Iwawaki et al., 2010). In T-cell–specific IRE1α-deletion mice, CD4 T-cells showed reduced interleukin-4 expression due to reduced interleukin-4 mRNA stability (Kemp et al., 2013). Deletion of IRE1α in bone marrow cells increased bone mass as the result of defective osteoclastic bone resorption (Tohmonda et al., 2015). IRE1α deletion in pro-opiomelanocortin neurons in the hypothalamus decreased fat mass by increasing energy expenditure (Xiao et al., 2016). Conditional deletion of Xbp1 has been performed in various mouse strains, and this has resulted in hyperactivation of IRE1α activity, including regulated IRE1α-dependent decay of cytosolic mRNAs (Hur et al., 2012). Moreover, the phenotypes of Xbp1 and IRE1α mice with conditional deletion in the same tissue are distinct (Hur et al., 2012), which suggests that IRE1α may perform physiological activities in a manner that does not depend on Xbp1 splicing.
Glomerular visceral epithelial cells, also known as podocytes, are highly differentiated cells in the kidney glomerulus that play an important role in the regulation of glomerular permselectivity, that is, filtration of plasma but restriction of proteins (Tryggvason and Wartiovaara, 2001; Chiang and Inagi, 2010; Greka and Mundel, 2012; Welsh and Saleem, 2012; Brinkkoetter et al., 2013). Podocytes reveal a complex morphology characterized by cell bodies and projecting foot processes that adhere to the glomerular basement membrane (GBM). Filtration slit diaphragms are localized between foot processes and help maintain the permselectivity of the glomerular filter. This intricate shape of podocytes is supported by the actin cytoskeleton, which allows podocytes to alter their shape dynamically (Tryggvason and Wartiovaara, 2001; Chiang and Inagi, 2010; Greka and Mundel, 2012; Welsh and Saleem, 2012; Brinkkoetter et al., 2013). Podocytes and glomerular endothelial cells produce the key structural components of the GBM, including collagens and laminins. Various glomerular diseases are associated with disruption of the actin cytoskeleton or surface charge of podocytes, resulting in effacement of foot processes, detachment from the GBM, and development of albuminuria and glomerulosclerosis, as well as impairment of glomerular function (Pippin et al., 2009; Chiang and Inagi, 2010).
Autophagy and ER stress have been implicated in podocyte pathophysiology (Hartleben et al., 2010; Huber et al., 2012; Cybulsky, 2013). The beclin-1–mediated pathway that regulates autophagy may be particularly important in maintaining podocyte homeostasis. Indeed, deficient JNK1 phosphorylation accompanied by deficient autophagy and elevated ER stress were reported in podocyte-specific Atg5 KO mice (Kawakami et al., 2015). Basal autophagy was suppressed in podocytes exposed to high glucose concentrations; furthermore, beclin-1 knockdown suppressed basal autophagy in podocytes and disrupted the podocyte filtration barrier integrity, as measured in vitro by albumin leakage (Fang et al., 2013). Deficient autophagy in podocytes was shown to worsen proteinuria and podocyte loss in a murine model of diabetic nephropathy (Tagawa et al., 2016). Autophagy was also implicated in the podocyte ER stress response in vitro (Cheng et al., 2015).
In the present study, we generated mice with podocyte-specific deletion of the IRE1α gene to characterize its role in podocyte pathophysiology. We show that mice with podocyte-specific deletion of IRE1α developed severe podocyte injury and albuminuria as they aged. In addition, loss of IRE1α impaired autophagy in podocytes.
RESULTS
IRE1α exons 20 and 21 are deleted in podocin-Cre IRE1α flox/flox mice
In the present study, mice with loxP sites surrounding exons 20 and 21 of IRE1α gene were crossed with mice expressing a Cre recombinase under the control of the podocyte-specific podocin promoter. This breeding strategy is expected to delete exons 20 and 21 specifically in podocytes, resulting in the expression of an RNase-domain truncated (ΔR) IRE1α mutant (Iwawaki et al., 2009). In glomeruli, PCR primers were designed to amplify a 1025–base pair predeletion product of the IRE1α loxP allele and a 189–base pair postdeletion product that is expected to appear after Cre-dependent excision of IRE1α-loxP exons 20 and 21 (Supplemental Figure S1A). Mice were initially genotyped by amplifying the IRE1α and podocin-Cre genes in tail DNA. We confirmed the presence of expected IRE1α and podocin-Cre bands in IRE1αflox/flox;Cre mice (loxP-homozygous deletion) and IRE1αflox/+;Cre mice (loxP-heterozygous deletion; Supplemental Figure S1B). To confirm a functional deletion in podocytes, we conducted PCR amplification using glomerular genomic DNA from IRE1αflox/+;Cre and IRE1αflox/flox;Cre (LoxP-heterozygous and LoxP-homozygous deletion) mice. Both loxP-heterozygous and -homozygous deletion glomeruli expressed a prominent 189–base pair deletion product, which was much more intense in homozygous mice (Supplemental Figure S1C). The unspliced loxP allele (1025 base pairs) was also amplified in these mice because glomeruli contain endothelial cells, mesangial cells, and parietal epithelial cells in addition to podocytes, and Cre recombinase is not active in these nonpodocyte cells. The WT IRE1α allele, which is slightly larger than the native loxP allele (1294 base pairs), was amplified in heterozygous mice (Supplemental Figure S1C).
Male mice with podocyte-specific deletion of IRE1α develop age-dependent podocyte injury
To determine whether podocyte-specific IRE1α gene deletion perturbs podocyte health, we generated podocin-Cre IRE1αflox/flox male and female mice (IRE1αflox/flox;Cre). The podocin-Cre allele was absent in littermate controls (IRE1αflox/flox:+; WT). Monthly urine albumin/creatinine ratio was analyzed separately in male (M) and female (F) WT (+) and deletion (Cre) mice because basal albuminuria is typically greater in male mice. We did not observe significant differences in either gender between 1 and 4 mo of age (Figure 1, A and B). However, at 5 mo of age, M Cre mice developed persistent and worsening albuminuria compared with M + mice (Figure 1A). In contrast, the albumin/creatinine ratio remained relatively low in female mice at all ages, except for an increase in elderly female mice (11–13 mo), which was independent of genotype (Figure 1B). Serum creatinine in 9-mo-old male mice was not significantly different between M Cre (8.36 ± 0.35 μM; N = 5) and M + mice (9.14 ± 0.43 μM; N = 5), implying that IRE1α deletion did not affect renal function.
FIGURE 1:
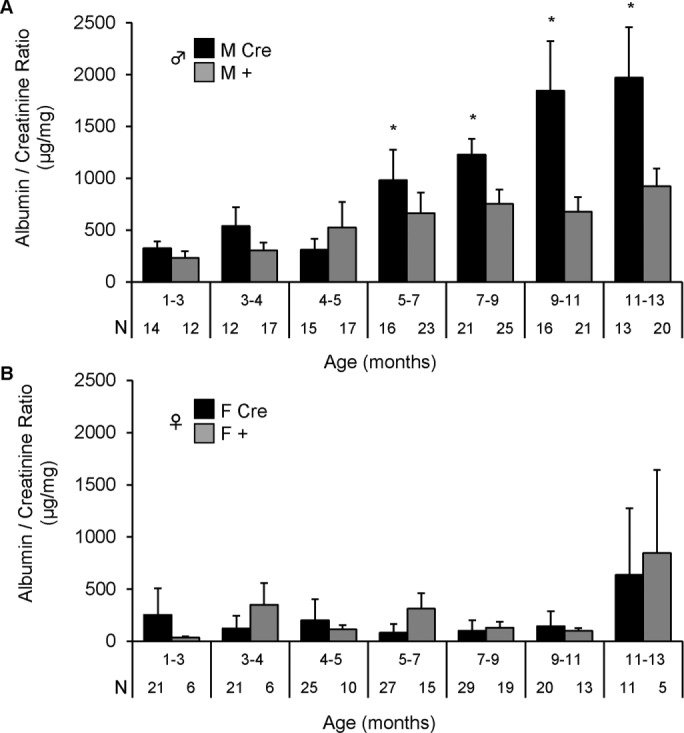
Podocyte IRE1α-deletion male mice develop albuminuria with aging. Male (A) and female (B) F3 littermates were monitored for albuminuria monthly. Podocyte IRE1α deletion in male mice (IRE1αflox/flox;Cre; M Cre) caused persistent and worsening albuminuria beginning at 5 mo of age as compared with control (IRE1αflox/flox;+; M +). Male mice: p = 4.72 × 10–4 (M Cre vs. M +; genotype); p = 2.20 × 10–6 (age); p = 0.033 (genotype × age interaction). There were no significant differences in albuminuria between female Cre (F Cre) and control (F +) mice. Number of urine samples (N) is indicated under each column.
Male mice with podocyte-specific deletion of IRE1α display enlarged glomeruli, capillary distension, and abnormal collagen deposition
Glomerular morphology and ultrastructure, as well as expression of podocyte markers, were investigated in 9-mo-old M + and M Cre mice from the same cohort used to determine age-dependent albuminuria. Based on light microscopy (periodic acid Schiff [PAS]–stained kidney sections; Figure 2A) and low-power electron micrographs (Figure 3A), M Cre glomeruli appeared to be enlarged, with particularly large glomerular capillaries, compared with M + mice. This visual impression was confirmed using an imaging-based approach demonstrating that the mean cross-sectional area of M Cre glomeruli was greater than with M + (Figure 2, A and C). This enlargement can be explained by a near-exact proportional rise in the glomerular cross-sectional area occupied by glomerular capillary lumens (Figure 2, A and D). In contrast, prealbuminuric 4- to 5-mo-old M Cre mice showed no evidence of glomerular volume expansion compared with M + mice, using PAS-stained glomeruli (unpublished data). In 9-mo-old male mice, we also used the Jones silver stain to accentuate the collagenous structures in the GBM and mesangial matrix. Using an imaging-based approach, we confirmed that 9-mo-old M Cre glomeruli contain excessive collagen in the capillary walls and mesangium compared with M + mice (Figure 2, B and E).
FIGURE 2:

Podocyte IRE1α deletion causes glomerular volume expansion and distended capillary loops, as well as collagen deposition, at 9 mo of age. (A) PAS stain. IRE1αflox/flox;Cre (M Cre) mice have enlarged glomeruli, as well as enlarged glomerular capillary lumens, compared with control (IRE1αflox/flox;+; M +). (B) There is increased capillary wall and mesangial collagen deposition in M Cre mice (Jones’ silver stain). (C) PAS-stained glomerular area is increased in M Cre mice (*p = 3.1 × 10−5). Mean of 48 M + glomeruli from three mice and 56 M Cre glomeruli from three mice. (D) Percentage of a given PAS-stained glomerulus that is occupied by capillary lumen is expanded in M Cre mice (*p = 0.0066). Average of 48 M + glomeruli from three mice, and 55 M Cre glomeruli from three mice. (E) Silver-stained glomeruli of M Cre mice have a higher fraction of glomerular tissue occupied by collagen than M + mice (*p = 1.45 × 10−5). Because the expanded capillary lumens in M Cre mice could confound this analysis, the capillary luminal area was subtracted from total glomerular cross-sectional area, so that the area of stained tissue could then be normalized to the total cross-sectional area of glomerular tissue. Imaging and quantification included 75 M + glomeruli from three mice and 74 M Cre glomeruli from three mice.
FIGURE 3:

Podocyte IRE1α deletion results in ultrastructural changes in podocytes. (A–C) Representative electron micrographs from 9-mo-old IRE1αflox/flox;+ (M +) and IRE1αflox/flox;Cre (M Cre) mice. (A) At low power, glomerular capillaries appear markedly dilated in M Cre mice (CL, capillary lumen). (B) Glomerular capillary walls from 2 M + and 2 M Cre mice reveal widened and effaced podocyte foot processes in the M Cre mice (Ψ). (C) M + and M Cre podocyte (P) cell bodies. M + mice displayed evidence of active autophagy, that is, autophagosomes or autolysosomes (AL). Lysosomes (L) were present in M Cre and M + mice. M Cre podocytes show focal foot process widening (Ψ), occludens junctions (*), and microvillous transformation of the plasma membranes (arrows). (D) Quantification of M Cre focal podocyte foot process widening (*p = 3.9 × 10−7). Foot processes were measured in 63 lengths of GBM from three M Cre mice and 20 lengths of GBM from two M + mice.
M Cre podocytes exhibit focal foot-process widening, microvillous transformation, and ectopic formation of occludens junctions
Having identified histological abnormalities in M Cre mice, we further studied glomerular ultrastructure by electron microscopy. As noted earlier, in the 9-mo-old M Cre mice, there were clear examples of capillary dilation (Figure 3A), although we did not see any morphological changes in the structure of the GBM (Figure 3, A–C). Podocyte foot processes were focally effaced, with some regions appearing relatively normal and others being severely effaced (Figure 3, B and C). Quantification of the number of foot processes per unit of length along the GBM showed that there was a significant increase in the mean foot process width (giving fewer foot processes per unit length of GBM) in M Cre mice (Figure 3D). Further, in M Cre mice, we observed podocyte microvillous transformation, a hallmark of podocyte injury, which was not apparent in M + mice (Figure 3C). Occasionally, occludens junctions had formed between some M Cre podocyte foot processes, replacing slit diaphragms, in keeping with dedifferentiation of podocytes. We noted early, mature, and late endosomes, as well as endolysosomal fusion, in both M + and M Cre podocytes (Figure 3C). Occasional phagophore engulfment, mature autophagosomes, and autolysosomes were observed in electron micrographs from M + podocytes, whereas normal or typical autophagosomes were absent in M Cre podocytes. Specifically, 11 normal autophagosomes (including a nascent phagophore and some autophagosomes fusing with lysosomes) were identified in 10 M + electron micrographs from two mice. Only five misshapen autophagosomes were identified in 20 M Cre electron micrographs from three mice, all of which contained unusual cargo or unusually shaped double membranes. We did not identify changes in the ultrastructure of podocyte organelles, including nuclei, mitochondria, rough ER, and the Golgi apparatus, between both mouse genotypes (Figure 3C).
IRE1α deletion leads to relative podocyte depletion
Next we investigated whether the albuminuria in 9-mo-old M Cre mice was associated with podocyte loss. We conducted immunofluorescence staining for Wilms tumor-1 (WT1; a protein expressed specifically in the nucleus of the podocyte) to monitor the average number of podocytes per glomerular cross section (Venkatareddy et al., 2014). Although there were no significant differences in WT1-positive nuclei per glomerulus between 9-mo-old M Cre and M + mice, WT1 counts per unit glomerular area were significantly lower in the M Cre mice, indicating relative podocyte depletion (Figure 4, A, D, and E). To further support this finding, we used immunofluorescence microscopy to compare glomerular expression of synaptopodin and podocalyxin in mice from the same cohort (Figure 4, B and C). Synaptopodin is a podocyte-specific protein, and its depletion is viewed as a marker of podocyte injury, whereas podocalyxin, which is expressed in podocytes and glomerular endothelial cells, participates in a number of important functions in podocytes (Greka and Mundel, 2012). M Cre glomeruli, which were significantly enlarged (Figure 4, F and H, and as demonstrated in Figure 2), stained positively for both synaptopodin and podocalyxin (Figure 4, B and C). Synaptopodin fluorescence normalized per unit of glomerular cross-sectional area was significantly lower in M Cre mice (Figure 4G). Podocalyxin fluorescence per unit of glomerular area tended to be reduced in M Cre mice, but the difference did not reach statistical significance (Figure 4I), perhaps due to the glomerular endothelial component, which would not be expected to change.
FIGURE 4:

Podocyte IRE1α deletion result in a reduction of WT1 and synaptopodin. (A–C) Kidney sections from 9-mo-old mice were stained with antibodies to WT1 (A), synaptopodin (B), and podocalyxin (C). Staining with nonimmune IgG is also presented (negative controls). Scale bars, 50 μm. (D, E) The number of WT1-positive nuclei per glomerulus (determined by colocalization with Hoechst nuclear stain; not shown) was assessed by visual counting. WT1 counts per glomerulus were comparable, but when expressed per 1000 µm2 of glomerular area, WT1 counts were significantly lower in M Cre mice (*p = 4.6 × 10−5); 32 M + glomeruli from three mice and 48 M Cre glomeruli from three mice. (F) Synaptopodin-stained glomeruli were enlarged in M Cre mice (*p = 0.024). (G) Quantification of synaptopodin immunofluorescence per unit glomerular area showed that intensity was lower in M Cre mice (*p = 0.046). For F and G, 74 M + glomeruli from three mice and 69 M Cre glomeruli from three mice. (H) Podocalyxin-stained glomeruli were enlarged in M Cre mice (*p = 0.0013). (I) Podocalyxin fluorescence per glomerulus and fluorescence per unit glomerular area were comparable in both groups. For H and I, 58 M + glomeruli from three mice and 54 M Cre glomeruli from three mice.
M Cre podocytes are deficient in autophagy
IRE1α is a mediator of the UPR and has also been linked to autophagy. To determine whether podocyte-specific deletion of IRE1α affects UPR and autophagy, we isolated glomeruli from prealbuminuric male mice (age 4–5 mo) and assessed UPR and autophagy markers by immunoblotting. M Cre mouse glomeruli exhibited elevated levels of the ER chaperones Grp78 and Grp94, suggesting activation of the ATF6 UPR pathway (Figure 5, A, C, and D). In contrast, phosphorylation of eukaryotic translation initiation factor 2α (eIF2α) on Ser-51 and the levels of C/EBP homologous protein (CHOP) were quite variable among mice, but mean values were comparable between the M + and M Cre groups, suggesting that the PERK pathway was not activated in M Cre glomeruli (Supplemental Figure S2A). In addition, M Cre glomeruli displayed elevated global ubiquitination of proteins, consistent with accumulation of misfolded proteins (Figure 5, B and H). In M Cre glomeruli, there was evidence of deficient autophagy, as determined by a lower LC3B-II/I ratio (Figure 5, A and E), and a reduced absolute amount of LC3B-II, that is, LC3B-II normalized to actin (Figure 5F). Decreased LC3B-II/I ratio or amount of LC3B-II in the M Cre glomeruli compared with M + mice implies deficient autophagic vacuole flux. Finally, in contrast to older mice (see later discussion), we did not observe reduced glomerular JNK1 (46 kDa) phosphorylation in the prealbuminuric M Cre mice; perhaps changes in JNK1 phosphorylation were below detectability (Figure 5, A and G). Phosphorylation of JNK2 (54 kDa) was poorly detected in glomerular lysates (unpublished data).
FIGURE 5:

Podocyte IRE1α deletion induces ER stress and causes deficient autophagosome turnover in 4- to 5-mo-old prealbuminuric mice. (A, B) Glomerular lysates were immunoblotted with antibodies as indicated. Representative immunoblots of four IRE1αflox/flox;+ (M +) and four IRE1αflox/flox;Cre (M Cre) mice obtained from a cohort of prealbuminuric male mice. (C, D) Densitometric quantification. M Cre mice show elevated expression of Grp78 (*p = 0.033) and Grp94 (*p = 0.0017). No change in calnexin was noted (B). (B, H) M Cre mice display elevated global polyubiquitinated proteins (*p = 0.0015). (A, E, F) M Cre mice show decreased LC3B-II, normalized either to LC3B-I (*p = 0.023) or actin (*p = 0.043; five to seven mice per group); M Cre mice show a reduced LC3B-II/I ratio (*p = 0.002). There were no differences in total LC3B-I or phospho-JNK1 (pJNK1) between groups (F, G).
The foregoing parameters were also studied in glomerular lysates from albuminuric 9-mo-old M Cre and M + mice (Figure 6A). The amount of autophagosomes was significantly lower in M Cre glomeruli, as determined by both reduced LC3B-II/I ratio and reduced LC3B-II (LC3B-II/actin ratio; Figure 6, A–C). Moreover, JNK1 phosphorylation was significantly lower in M Cre glomeruli, consistent with deficient beclin-1–associated autophagic induction (Figure 6, A and D). Grp78 and Grp94, as well as phospho-eIF2α and CHOP, did not differ between M Cre and M + mice, indicating that neither ER stress nor the UPR was substantially induced in 9-mo-old M Cre glomeruli in spite of impaired autophagy (Figure 6, A, E, and F, and Supplemental Figure S2B). We also assessed polyubiquitinated proteins in glomerular lysates from both mouse genotypes, but in contrast to our findings in prealbuminuric male mice, there was no difference between M Cre and M + mice (unpublished data). Expression levels of autophagy mediators, including beclin-1, Atg5, and Atg3, were not significantly different between 9-mo-old M Cre and M + mice, implying that autophagic activity was regulated posttranslationally and not transcriptionally in M Cre mouse glomeruli (Supplemental Figure S2B).
FIGURE 6:

Mice with podocyte IRE1α deletion are deficient in autophagy but do not show ER stress at 9 mo of age. (A) Glomerular lysates were immunoblotted with antibodies as indicated. Representative immunoblots of 5 IRE1αflox/flox;+ (M +) and 5 IRE1αflox/flox;Cre (M Cre) mice. (B–D) Densitometric quantification. M Cre mice show decreased LC3B-II, normalized either to LC3B-I (*p = 0.013) or actin (*p < 0.0001), and decreased JNK1 phosphorylation (*p = 0.048). (E–I) No significant differences between M Cre and M + mice were observed in Grp78, Grp94, podocalyxin, nephrin maturation (upper/lower band), or mature nephrin expression (upper band).
Nephrin is an important podocyte-resident protein that undergoes posttranslational modification in the ER. It is expressed as a fully glycosylated mature form (∼180 kDa), as well as an immature, ER-luminal form (∼170 kDa; Figure 6A). There were no significant differences between M + and M Cre mice in the ratio of mature to immature nephrin (Figure 6H) or total amount of mature nephrin, that is, nephrin normalized to actin (Figure 6I). For comparison, expression of podocalyxin (expressed in podocytes and glomerular endothelial cells) was also not different between M Cre and M + mice (Figure 6, A and G).
M Cre mice are more susceptible to injury in acute anti-GBM nephritis
Although deletion of IRE1α did not lead to development of overt albuminuria in mice up to 4–5 mo of age, we hypothesized that deletion of IRE1α in these mice would nonetheless predispose them to podocyte injury. To test this hypothesis, we induced acute anti-GBM nephritis in prealbuminuric, 5-mo-old M Cre and M + mice, using a single intravenous injection of anti-GBM antibody. We selected a low dose of antibody in order to allow the unmasking of any potential contribution of IRE1α to podocyte integrity. After 24 h, anti-GBM antibody tended to induce a modest increase in the albumin/creatinine ratio in the M + control mice. In contrast, M Cre mice showed a significantly greater albumin/creatinine ratio, compared with both anti-GBM injected M + mice and control M Cre mice (Figure 7, A and B). Anti-GBM antibody and complement C3 were present in glomeruli of both M Cre and M + mice (Figure 7, C–F). There was slightly greater anti-GBM antibody immunofluorescence staining in M Cre mice but no difference in C3 deposition. Given that M Cre and M + mice were injected with the same dose of anti-GBM antibody in parallel, this apparently greater antibody localization in M Cre mice may be related to minor changes in collagen composition, which was already noted in older M Cre mice (Figure 2). Because glomerular deposition of C3 was the same in M Cre and M + mice, the exaggerated increase in albuminuria in the M Cre mice was most likely due to the podocyte IRE1α deletion. The C3 deposition in Bowman’s capsule is unrelated to anti-GBM antibody injection because C3 is known to be present in Bowman’s capsule of healthy mice (Ruseva et al., 2014).
FIGURE 7:

Podocyte IRE1α deletion increases susceptibility to acute anti-GBM nephritis. (A) Male mice 5 mo of age were injected with 5 μl of sheep anti–GBM antibody (αGBM). Urine was collected preinjection and at 24 h. Kidneys were harvested at 24 h. In mice that received anti-GBM antibody, there was significantly greater albuminuria in the IRE1αflox/flox;Cre (M Cre) group than in controls (IRE1αflox/flox;+; M +). Albuminuria in anti-GBM M Cre mice was also significantly greater than the preinjection (baseline) value (*p = 0.005, ANOVA; N, number of animals). (B) The net change in albumin/creatinine ratio calculated per individual mouse for 10 anti-GBM injected M Cre mice and 6 injected M + mice was significantly greater in M Cre mice (*p = 0.049). (C–F) Anti-GBM antibody and C3 deposition in mouse glomeruli were confirmed by immunofluorescence microscopy. Quantification of sheep anti-GBM fluorescence intensity showed a slight increase in M Cre mice (*p = 4.27 × 10−7; 83 glomeruli from six GBM-injected M + mice and 110 glomeruli from seven GBM-injected M Cre mice). There were no significant differences in C3 deposition. C3 fluorescence intensity was measured in 80 glomeruli from six M + mice and 98 glomeruli from seven M Cre mice. Scale bars, 50 μm.
By electron microscopy, control M Cre mice and anti-GBM antibody-injected M + mice showed predominantly normal podocyte foot processes, with occasional focal effacement. In contrast, focal effacement was more abundant in anti–GBM-injected M Cre mice compared with injected M + mice (Figure 8, A and B). M Cre mice with anti-GBM nephritis also showed other ultrastructural signs of podocyte injury, such as podocyte basolateral plasma membranes completely fused to the GBM. In anti-GBM nephritis, M Cre mice exhibited a modest but significant podocyte loss, whereas there was no significant loss of podocytes in M + mice (Figure 8, C and D). Thus, in anti-GBM nephritis, podocyte-specific deletion of IRE1α exacerbated albuminuria, as well as podocyte loss and ultrastructural damage.
FIGURE 8:

Mice with podocyte deletion of IRE1α and anti-GBM nephritis exhibit podocyte loss and foot process effacement. (A) Representative electron micrographs showing mild foot-process widening in uninjected IRE1αflox/flox;Cre (M Cre) and injected IRE1αflox/flox;+ (M +) mice and greater widening in anti-GBM antibody–injected M Cre mice (24 h). (B) Quantification of images showing foot-process widening/effacement (N, number of animals). (C) Representative WT1 immunofluorescence images of mice with acute anti-GBM nephritis (24 h). Scale bar, 50 μm. (D) Quantification of WT1-positive cells shows that anti-GBM antibody treatment in M Cre mice resulted in a modest but significant loss of podocytes (*p = 0.027 anti-GBM vs. control in the M Cre groups). In M + mice, there was no significant decrease in podocytes after anti-GBM antibody.
IRE1α controls the physiological autophagosome formation rate
The foregoing studies in mice with podocyte-specific IRE1α RNase domain deletion showed that IRE1α contributes to maintenance of podocyte integrity in normal aging and glomerulonephritis. Moreover, IRE1α deletion appeared to disrupt autophagy in podocytes. To address the mechanism by which IRE1α regulates autophagy, we performed further studies in cultured cells. First, we overexpressed FLAG-IRE1α WT and several mutants in COS-1 cells by transient transfection and monitored IRE1α RNase activity with Xbp1 splicing in the presence of the ER stressor tunicamycin, using reverse transcriptase (RT)-PCR. Expression of IRE1α WT and the S724A phosphorylation-site mutant increased Xbp1 splicing beyond that caused by tunicamycin in control cells transfected with empty vector (Supplemental Figure S3). Expression of the K599A or ΔR mutants reduced tunicamycin-induced Xbp1 splicing, indicating that these two mutants act as dominant negative. We also tested the effect of the IRE1α RNase-directed inhibitor 4μ8C. During 6 h of cotreatment with tunicamycin, 4μ8C potently blocked Xbp1 splicing (Supplemental Figure S3).
To confirm the role of IRE1α in regulating autophagy, we transiently transfected COS-1 cells with FLAG-IRE1α (WT, S724A, K599A, or ΔR) or empty vector. We changed culture medium after 22 h to remove residual transfection reagent, as well as to replenish amino acids and nutrients to ensure that experimental differences were not caused by starvation-induced autophagy. Then we treated cells with chloroquine to block degradation of autophagosomes and allow surface components (e.g., LC3B-II) and contents to accumulate in the cytoplasm (Tanida et al., 2008; Ni et al., 2011). Expression of FLAG-IRE1α WT and mutants is presented in Figure 9A. Densitometric quantification showed that expression of FLAG-IRE1α WT and K599A was comparable, whereas S724A and ΔR expression was 1.5-fold greater than WT (p < 0.0001; 24 experiments). The rate of autophagic vacuole formation/accumulation was determined by monitoring levels of LC3B-II, as LC3B-I levels were comparable in all cells. Basal LC3B-II accumulation was modest, as determined in cells transfected with empty vector, in the absence of chloroquine treatment, whereas chloroquine induced LC3B-II accumulation at 2 and 6 h (Figure 9, A and C). Overexpression of IRE1α WT and S724A slightly enhanced the rate of chloroquine-induced LC3B-II accumulation compared with chloroquine-treated vector-transfected cells, but this was not statistically significant (Figure 9, A and C). Conversely, overexpression of both dominant-negative mutants IRE1α K599A and ΔR significantly reduced the chloroquine-induced LC3B-II accumulation rate at 2 and 6 h compared with cells overexpressing IRE1α WT (Figure 9, A and C). In addition, IRE1α ΔR overexpression significantly reduced the accumulation rate compared with the empty vector transfection (chloroquine treated), and K599A tended to do so as well.
FIGURE 9:
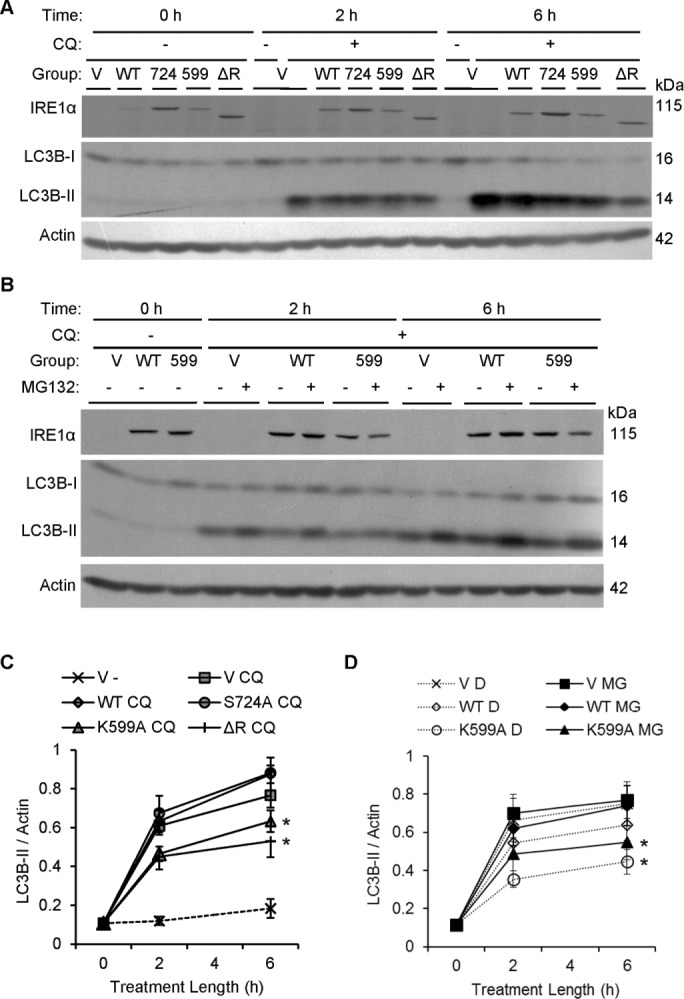
IRE1α facilitates physiological formation of autophagic vacuoles. (A) COS-1 cells transiently transfected with vector (V), FLAG-IRE1α WT, or mutants were treated with or without chloroquine (CQ, 50 μM). Levels of LC3B were measured by immunoblotting after 2 or 6 h. FLAG-IRE1α expression was confirmed using anti-FLAG antibody. (C) Quantification of the rate of LC3B-II accumulation shows that overexpression of IRE1α K599A and ΔR significantly reduces the formation of LC3B-II vs. IRE1α WT. IRE1α ΔR also significantly lowers the rate of LC3B-II formation vs. control (vector), whereas IRE1α K599A tends to lower the rate of formation vs. control (*p = 5.40 × 10−7 [IRE1α]; p = 1.09 × 10−9 [CQ]; p < 2.2 × 10−16 [time]; p = 0.0018 [IRE1α × time interaction]; p = 1.07 × 10−4 [CQ × time interaction]; seven experiments). (B) COS-1 cells transfected with vector (V), IRE1α WT, or K599A were treated with chloroquine (CQ, 50 μM) plus vehicle (DMSO [D]) or MG132 (MG, 25 μM). Overexpression of IRE1α K599A reduced the rate of LC3B-II accumulation independently of MG132 treatment. (D) Quantification of the rate of LC3B-II accumulation over the 6-h chloroquine treatment period shows that overexpression of IRE1α K599A significantly reduced the rate of LC3B-II accumulation vs. control (empty vector) and vs. IRE1α WT. Differences were significant at 2 h and increased further at 6 h (*p = 0.0047 [IRE1α]; p = 1.1 × 10−14 [time]). These effects were independent of MG132. Six experiments.
It has been reported that inhibition of the proteasome can enhance accumulation of autophagosomes (Ding et al., 2007; Wang et al., 2013). In the next study, we examined whether proteasomal inhibition can stimulate autophagy during the same experimental time course as the one that revealed involvement of IRE1α. Cells expressing vector, FLAG-IRE1α WT, or K599A were cotreated with chloroquine and either vehicle (dimethyl sulfoxide [DMSO]) or 25 μM MG132. LC3B-II accumulation increased at 2 and 6 h in all groups, but the increase in LC3B-II was significantly lower at both time points in the IRE1α K599A group (Figure 9, B and D). MG132 tended to increase LC3B-II at 2 and 6 h in each of the three groups, but differences between MG132- and vehicle-treated cells were not statistically significant (Figure 9, B and D). We also conducted separate experiments to verify that MG132 cannot itself modulate the autophagosome formation rate in COS-1 cells. After 6 h of incubation, neither 25 μM MG132 treatment alone nor MG132 cotreatment with chloroquine could potentiate the autophagosome formation rate beyond that caused by chloroquine alone (unpublished data).
IRE1α determines the volume rather than the number of autophagosomes
To extend our investigation of the effects of IRE1α on LC3-II levels, we studied formation of LC3B-II–positive autophagosomes (autophagic “puncta”) in COS-1 cells by monitoring mCherry–enhanced green fluorescent protein (eGFP)–LC3B fluorescence (N’Diaye et al., 2009; Mizushima et al., 2010). mCherry-eGFP-LC3B-I is expressed diffusely in the cytoplasm; however, after induction of autophagy, mCherry-eGFP-LC3B-II binds to the surface of the autophagosomes, giving bright puncta that are dually mCherry and eGFP fluorescent. eGFP is quenched in the acidic lysosomes, whereas mCherry is acid stable; therefore eGFP-mCherry–positive puncta are autophagosomes or autolysosomes deacidified by chloroquine, whereas mCherry-positive puncta are active lysosomes or autolysosomes.
COS-1 cells grown on collagen-coated coverslips were cotransfected with FLAG-IRE1α (WT, S724A, K599A, or ΔR) or empty vector, along with mCherry-eGFP-LC3B (Figure 10, A–C). Cells were either untreated or treated with chloroquine. Quantifying only the eGFP fluorescence of puncta that colocalized with mCherry allowed us to monitor autophagic puncta independent of lysosomes, autolysosomes, diffuse LC3B-I and green-channel autofluorescence. Puncta area increased in the presence of chloroquine in vector-, IRE1α WT–, and S724A-transfected cells. Compared with these groups, both the K599A and ΔR mutants reduced the cell area occupied by autophagic puncta (Figure 10, A and B). Increased puncta area was largely explained by increased puncta size (Figure 10C) and not number of puncta. Immunofluorescence using FLAG antibody revealed FLAG-IRE1α WT and mutants in respective transfected cells, with robust perinuclear expression of IRE1α and, to a lesser extent, filamentous-cytoplasmic expression, as expected for an ER transmembrane protein (Figure 12D).
FIGURE 10:

IRE1α modulates autophagosome volume. (A) COS-1 cells were transiently cotransfected with FLAG-IRE1α WT or mutants and mCherry-eGFP-LC3B. Cells were untreated or treated with chloroquine (CQ, 50 μM) and examined after 6 h. The mCherry-eGFP-LC3B reporter was not transfected in the cells shown in the first row (negative control). (B) Quantification revealed that the punctal area (the cross-sectional area occupied by autophagic puncta per 1000 μm2 of total cell area) was lower in chloroquine-treated K599A- and ΔR-overexpressing cells compared with treated WT or vector/control (*p = 1.11 × 10−10 [IRE1α]; p < 2.2 × 10−16 [CQ]; p = 4.19 × 10−8 [IRE1α × CQ interaction]). (C) Cross-sectional area per autophagosome (punctum size) paralleled punctal area. IRE1α WT or S724A increased punctum size compared with control, K599A, or ΔR (*p = 4.20 × 10−5 [IRE1α]; p < 2.2 × 10−16 [CQ]; p = 7.82 × 10−3 [IRE1α × CQ interaction]). (D) Cells expressing empty vector or FLAG-IRE1α WT or mutants were immunostained with anti-FLAG antibody (nonimmune IgG in controls). IRE1α WT. and mutants show cytoplasmic staining with perinuclear accentuation. For B and C, the number of trials and the number of quantified cells are indicated beneath columns.
FIGURE 12:
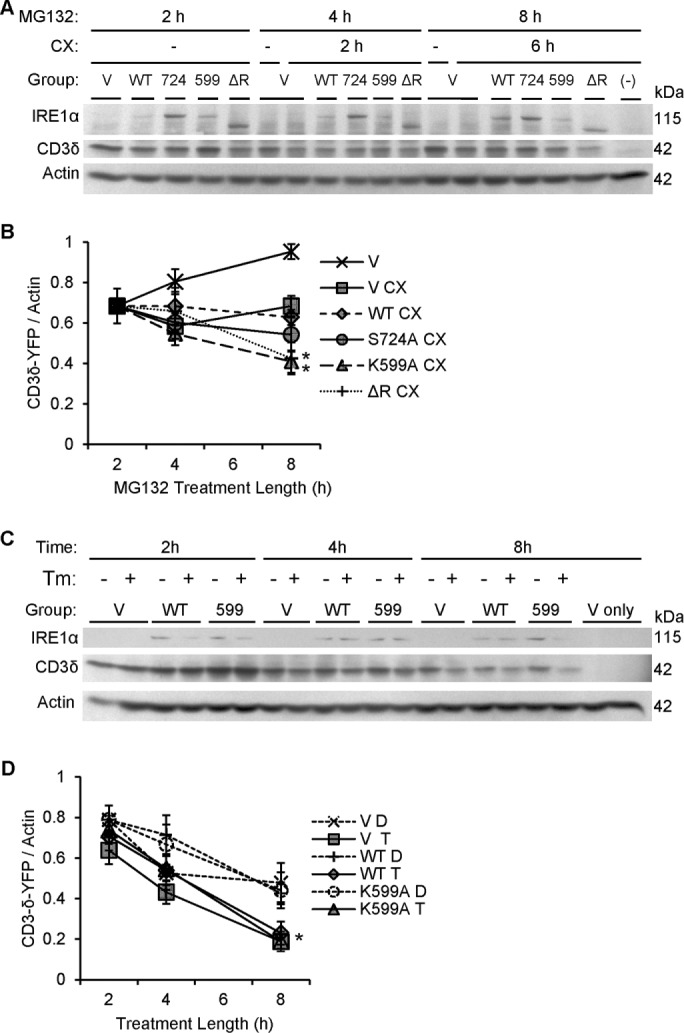
Dominant-negative IRE1α mutants (K599A and ΔR) accelerate degradation of CD3δ-YFP. (A) COS-1 cells cotransfected with FLAG-IRE1α (WT or mutants) and the ERAD reporter CD3δ-YFP were pretreated with MG132 (MG, 10 μM) to partially block the proteasome. At 2 h, vehicle (DMSO) or cycloheximide (CX, 25 μg/ml) was added. Lysates were immunoblotted after another 2 or 6 h, as indicated. Expression of IRE1α and CD3δ-YFP was confirmed using anti-FLAG and anti-GFP antibodies, respectively. Overexpression of IRE1α K599A or ΔR enhanced CD3δ-YFP degradation. The lane labeled “–” is a mock-transfected, cycloheximide-treated control sample, which confirms the CD3δ-YFP signal specificity. (B) Densitometric quantification showed that in the presence of cycloheximide, overexpression of IRE1α K599A or ΔR enhanced CD3δ-YFP degradation compared with vector. In addition, IRE1α K599A enhanced CD3δ-YFP degradation compared with WT (*p = 0.003 [IRE1α]; p = 0.0065 [CX]; p = 0.003 [IRE1α × time interaction; five experiments]. (C) COS-1 cells coexpressing IRE1α WT and CD3δ-YFP were cotreated with cycloheximide together with either vehicle (DMSO [D]) or tunicamycin (Tm or T, 5 μg/ml). Lysates were immunoblotted 2, 4, and 8 h posttreatment. The lanes labeled “V only” are without transfection of CD3δ-YFP. (D) Densitometric quantification shows that tunicamycin stimulated degradation of CD3δ-YFP in vector- and IRE1α-transfected cells. IRE1α did not modulate the tunicamycin-induced degradation of CD3δ-YFP, that is, overexpression of IRE1α WT did not potentiate tunicamycin-induced degradation of CD3δ-YFP, nor did IRE1α K599A exert an inhibitory effect (*p = 4.12 × 10−5 [Tm]; p = 3.70 × 10−15 [CX + time]; six experiments).
Induction of autophagy via IRE1α does not require IRE1α RNase activity
Given that IRE1α K599A (a kinase- and RNase-inactive mutant) and ΔR (RNase-domain deletion mutant that can bind ATP) both reduced the autophagosome formation rate (Figure 9), we then determined whether this process requires the physical presence of the RNase domain or RNase activity. COS-1 cells were cotreated with chloroquine plus vehicle or the IRE1α RNase inhibitor 4μ8C. Over a 24-h period, 4μ8C had no significant effect on the LC3B-II accumulation rate (Figure 11A). In addition, there were no differences in either the LC3B-II/I ratio or the LC3B-II/actin ratio (Figure 11, C and D). To validate that 4μ8C inhibited IRE1α RNase activity effectively, we coincubated cells with 4μ8C and tunicamycin and demonstrated that 4μ8C abolished tunicamycin-induced Xbp1 splicing (Figure 11B). Therefore these data strongly suggest that autophagosome formation requires IRE1α kinase activity and the presence of the C-terminal RNase domain (perhaps a protein-binding site) but not IRE1α RNase activity. Note that IRE1α WT can potentially induce IRE1α clusterization, resulting in Xbp1 splicing under unstressed conditions. Such an effect cannot be entirely excluded under these experimental conditions.
FIGURE 11:

IRE1α-mediated autophagosome formation does not require RNase activity. (A) COS-1 cells were treated with chloroquine (CQ, 50 μM), the IRE1α RNase inhibitor 4μ8c (10 μM), or vehicle (DMSO). Quantification of the rate of autophagosome formation for up to 24 h as reported by LC3B-II normalized to actin (C) or the LC3B-II to I ratio (D) revealed no significant differences in the presence or absence of 4μ8c, although LC3B-II increased over time (C: p = 2.21 × 10−6; D: p = 9.34 × 10−5; three experiments). (B) Tunicamycin (T, 10 μg/ml) induced Xbp1 splicing, and this effect was blocked by 4μ8c (4μ, 10 μM) at 6 and 24 h. NR, no–reverse transcriptase negative control; NT, no-template negative control; Tx, treatment.
Inhibition of IRE1α permits substrate shunting into ERAD
Our findings implicated a role for IRE1α in mediating basal autophagic flux. However, besides autophagy, IRE1α has been linked to ERAD. Thus we examined whether degradation of ER misfolded proteins also involves IRE1α-dependent ERAD or autophagy-ERAD cross-talk. Degradation of ectopically expressed CD3δ–yellow fluorescent protein (YFP) in the presence of cycloheximide (to block protein synthesis) was used to monitor ERAD (Menendez-Benito et al., 2005). CD3δ-YFP is retrotranslocated from the ER into the cytoplasm and then degraded by the proteasome. COS-1 cells were cotransfected with CD3δ-YFP and IRE1α, and, after 24 h, they were treated with cycloheximide for 6 h. Over the 6-h period, the amount of CD3δ-YFP decreased by ∼40% (similar to the results shown in Figure 12D, vector/DMSO group). Neither IRE1α WT nor K599A overexpression affected the rate of CD3δ-YFP degradation compared with control (similar to Figure 12D). We next examined whether IRE1α can modulate ERAD in the presence of reduced proteasomal capacity (partial inhibition of the proteasome with MG132). COS-1 cells were cotransfected with CD3δ-YFP and IRE1α WT, S724A, K599A, ΔR, or empty vector (Figure 12A). After 24 h, cells were treated for 2 h with a low dose of MG132 (10 μM) to partially inhibit the proteasome and allow for CD3δ-YFP accumulation. Then cycloheximide was added to inhibit protein synthesis. In the presence of low-dose MG132, the level of CD3δ-YFP was near constant over time in cycloheximide-treated cells that had been cotransfected with empty vector or IRE1α WT (Figure 12, A and B). This result was predicted for the empty vector control because MG132 would partially inhibit proteasomal degradation of CD3δ-YFP. Overexpression of IRE1α WT did not overcome partial proteasomal blockade by promoting degradation of CD3δ-YFP, indicating that IRE1α was unable to enhance ERAD. However, overexpression of both K599A and ΔR mutants significantly enhanced degradation of CD3δ-YFP (Figure 12, A and B) while blocking autophagic flux at comparable time points (Figure 9C). IRE1α S724A overexpression mildly enhanced degradation of CD3δ-YFP, although this effect was not statistically significant (Figure 12B). Note that the level of CD3δ-YFP tended to increase in the absence of cycloheximide in control cells (Figure 12B), suggesting that there was ongoing synthesis of the reporter; consequently, all of the effects of IRE1α WT and mutants on CD3δ-YFP were studied in the presence of cycloheximide, where changes in CD3δ-YFP reflect degradation. Together, these results are in keeping with the view that under basal conditions, IRE1α is a positive regulator of autophagy but not ERAD. Also note that partial inhibition of proteasomal activity with a low dose of MG132 was required to observe enhancement of ERAD by the K599A and ΔR mutants.
Tunicamycin enhances ERAD independently of IRE1α
In this experiment, we investigated the regulatory role of IRE1α in cells subjected to ER stress induced by tunicamycin. In a preliminary study, we found that 6-h incubation with 5 μg/ml tunicamycin potently blocked CD3δ-YFP glycosylation (unpublished data), consistent with induction of protein misfolding and ER stress. COS-1 cells were cotransfected with IRE1α (WT or K599A) or empty vector together with CD3δ-YFP to monitor ERAD. After 24 h, cells were treated for 2 h with either vehicle (DMSO) or 5 μg/ml tunicamycin plus cycloheximide to simultaneously induce ER protein misfolding while inhibiting protein synthesis. Tunicamycin treatment in all cells significantly enhanced the degradation of CD3δ-YFP, but this was not affected by IRE1α WT or K599A (Figure 12, C and D). Thus ERAD can be up-regulated secondary to misfolded protein accumulation; however, this occurs independently of ectopic IRE1α overexpression or inhibition of endogenous IRE1α.
DISCUSSION
Overall our study demonstrates the importance of physiological IRE1α activity in podocyte homeostasis and establishes an important link between IRE1α and autophagy. Deletion of the IRE1α RNase domain in podocytes in vivo induced podocyte injury and perturbed glomerular capillary wall integrity during the aging process (Figures 1 and 3) and in acute glomerulonephritis (Figures 7 and 8). Moreover, deletion of IRE1α in podocytes resulted in glomerular volume expansion (in particular, capillary volume), relative podocyte depletion, reduction of synaptopodin, and enhanced collagen deposition (Figures 2 and 4). In keeping with an earlier study on podocyte-specific Atg5 deletion in mice (Hartleben et al., 2010), the deletion of IRE1α in podocytes, together with our findings in cell culture, links podocyte autophagy flux with global glomerular health as mice age.
In 5- to 13-mo podocyte-specific IRE1α-deficient mice, albuminuria was observed in males but not females (Figure 1). This gender-specific difference may result from genetic determinants in mouse glomerular physiology. In general, male mice have a higher basal albumin/creatinine ratio than females. This might be due to gender-specific differences in basal intraglomerular capillary pressure, including differences in sensitivity to angiotensin II and/or norepinephrine at the glomerular afferent and efferent arterioles. Alternatively, gender-specific differences may determine the nature of cross-talk between podocytes and glomerular endothelial cells, perhaps involving vascular endothelial growth factor and/or nitric oxide, which are factors known to mediate vascular dilation (Tryggvason and Wartiovaara, 2001; Chiang and Inagi, 2010; Greka and Mundel, 2012; Welsh and Saleem, 2012; Brinkkoetter et al., 2013).
IRE1α in podocytes may be involved in the regulation of intraglomerular hemodynamic forces that exert outward hydrostatic pressure from within the glomerular capillaries, as well as glomerular capillary structure and volume. In podocyte-specific IRE1α-deficient males, intraglomerular structural changes were observed, such as dilated glomerular capillaries, glomerular volume expansion, and enhanced collagen deposition in the glomerulus. Such perturbations could lead to age-dependent capillary wall weakening and distension of podocytes, eventually leading to podocyte detachment. Deposition of collagen in the glomerulus could represent a compensatory response for structural deficiency, eventually leading to glomerulosclerosis. Indeed, deposition of collagen in the mesangial regions of glomeruli most likely reflects early glomerulosclerosis. Within this context, the precise mechanism underlying podocyte-mediated capillary dilation in the absence of IRE1α remains to be established.
In association with albuminuria, podocyte-specific IRE1α deletion resulted in ultrastructural changes of injury and dedifferentiation, including microvillous transformation of plasma membranes, formation of occludens junctions, and focal effacement of foot processes (Figure 3). When the WT1 counts are taken together with evidence of glomerular volume expansion (Figure 2) and the relative depletion of the podocyte marker synaptopodin (Figure 4), the same number of podocytes would reside upon volume-expanded glomerular capillaries, that is, fewer podocytes per unit glomerular volume, implying relative podocyte depletion (Puelles and Bertram, 2015). Remaining podocytes would stretch and hypertrophy. Over time, the relative podocyte depletion would lead to injury, as reflected by the ultrastructural changes.
Before the onset of albuminuria, podocyte-specific, IRE1α-deficient young male mice showed increased susceptibility to develop acute anti-GBM nephritis. At the low dose of anti-GBM antibody we used, IRE1α-sufficient mice did not develop a significant increase in the urine albumin/creatinine ratio. In contrast, the podocyte-specific, IRE1α-deficient mice exhibited a fourfold rise in this ratio above the preinjection (baseline) ratio and approximately threefold increase versus the control group (Figure 7). Podocyte-specific, IRE1α-deficient mice with anti-GBM nephritis also displayed evidence of severe foot process effacement and loss of podocytes compared with the control group (Figure 8). This result indicates that IRE1α is important in the maintenance of podocyte structure and glomerular permselectivity not only during aging, but also in response to acute glomerular injury.
Before the onset of albuminuria, male podocyte-specific, IRE1α-deficient mice showed glomerular UPR activation (increases in Grp78 and Grp94), accumulation of polyubiquitinated proteins, and deficient autophagosome turnover (Figure 5). After the onset of podocyte injury and albuminuria (at 9 mo of age), evidence of UPR activation and accumulation of polyubiquitinated proteins was absent in podocyte-specific, IRE1α-deficient mice compared with control mice, whereas the defect in autophagy persisted (Figure 6). The ER stress response in the prealbuminuric, podocyte-specific, IRE1α-deficient mice may have been induced by another branch of the UPR, perhaps the ATF6 pathway. By 9 mo of age, enhancement of ERAD in the podocyte-specific, IRE1α-deficient mice may have attenuated the UPR response by eliminating the accumulated polyubiquitinated proteins (despite deficient autophagy). Our finding in cell culture that inhibition of IRE1α by the IRE1α ΔR mutant permits substrate shunting into the ERAD pathway supports this possibility (Figure 12).
The mechanism by which IRE1α maintains podocyte homeostasis and glomerular integrity as mice age, involves autophagy, at least in part (Figures 5 and 6). IRE1α can bind TRAF2 and activate ASK1, which in turn can activate JNK1 and beclin-1 to stimulate autophagy (Nishitoh et al., 2002). The reduced activation-specific phosphorylation of JNK1 in glomeruli of mice with podocyte-specific IRE1α deletion supports the role of JNK1 as a mediator of autophagy (Figure 6); however, the pathway will require further elucidation, as JNK1 phosphorylation was not reduced significantly in prealbuminuric mice, despite reduced autophagy (Figure 5). Podocytes display a relatively higher basal rate of autophagy compared with other cells, and based on studies in podocyte-specific, Atg5 KO mice, autophagy appears to be a homeostatic mechanism to maintain podocyte integrity and a protective mechanism against aging and glomerular injury (Hartleben et al., 2010; Kawakami et al., 2015). Thus the Atg5 KO mice developed focal and global glomerulosclerosis by 22 mo of age (Hartleben et al., 2010). Impaired autophagy in podocyte-specific, IRE1α-deficient mice may perturb the synthesis and delivery of key structural proteins required for slit diaphragm assembly or podocyte–GBM interactions. Alternatively, impaired autophagy could affect the production of secreted factors necessary for maintenance of glomerular structure, such as vascular endothelial growth factor.
There are some limitations to quantifying enhanced autophagic flux in vivo. Thus, to confirm the role of IRE1α in autophagy, we measured the conversion of LC3-I to LC3-II in cultured cells expressing IRE1α WT and various mutants. Our results indicate that IRE1α constitutively regulates formation of autophagosomes—specifically, their volume (Figures 9 and 10). Overexpression of two dominant-negative IRE1α mutants (K599A and ΔR) reduced the mean cross-sectional area of autophagosomes by ∼30%, compared with IRE1α WT. In terms of autophagosome volume, this translates into a ∼40% reduction. Of interest, the effect of IRE1α on formation of autophagosomes required both the IRE1α kinase and RNase domains but not RNase activity (Figures 9– 11). Therefore IRE1α activation through the RNase-independent activation of the ASK1-TRAF2-JNK signaling pathway (Kaneko et al., 2003) could promote an ER framework (“cradle”) for autophagosome formation (Hayashi-Nishino et al., 2009; Yla-Anttila et al., 2009).
Alternatively, IRE1α is believed to up-regulate the expression of factors that mediate ERAD (Hetz and Soto, 2003; Hetz and Glimcher, 2009), although a link between IRE1α and ERAD has not been established conclusively (Chiang et al., 2012; Sun et al., 2015). In the present study, overexpression of both IRE1α K599A and ΔR mutants in cultured cells significantly enhanced degradation of the ERAD reporter CD3δ-YFP while blocking autophagic flux (Figures 9 and 12). These results are in keeping with the view that IRE1α is a positive regulator of autophagy but not of ERAD. Indeed, IRE1α might constitutively inhibit ERAD via ASK1-mediated Rpt5 phosphorylation in the 19S regulatory cap of the 26S proteasome (Um et al., 2010). Given the lack of an ERAD-specific endogenous reporter in the mouse glomerulus, we cannot make definite conclusions on the potential role of IRE1α in ERAD in vivo.
Recently mice with podocyte-specific deletion of Xbp1 were characterized (Hassan et al., 2016). In contrast to the podocyte-specific deletion of IRE1α, podocyte Xbp1-deficient mice displayed no proteinuria or histological evidence of glomerular injury up to 1 yr of age. However, when deletion of Xbp1 was combined with podocyte-specific deletion of Sec 63 (a chaperone required for protein folding in the ER), the double-deletion mice developed albuminuria by 8–10 wk of age, which continued to progress over a 1-yr period. There were associated ultrastructural changes in the podocytes, glomerulosclerosis, and apoptosis of podocytes, which correlated with activation of JNK and caspases. These results suggest that deletion of Xbp1 and Sec 63 resulted in overactivation of IRE1α and consequently a cytotoxic UPR.
In summary, the present study provides important functional information on the contribution of IRE1α to podocyte homeostasis and injury. We expect that our findings will enhance interest in testing compounds that modulate IRE1α for their therapeutic protective effects on podocyte injury and glomerulonephritis.
MATERIALS AND METHODS
Reagents and antibodies
Cell culture reagents were purchased from Wisent (Saint-Jean-Baptiste, Canada). Electrophoresis and immunoblotting reagents were from Bio-Rad Laboratories (Mississauga, Canada), Sigma-Aldrich (St. Louis, MO), Thermo Fisher Scientific (Burlington, Canada), and Amersham GE Healthcare (Baie d’Urfé, Canada). Lipofectamine 2000 and OptiMEM were from Invitrogen Life Technologies (Burlington, Canada). Rabbit monoclonal anti–phospho-JNK (4668S), rabbit anti-JNK (9252S), rabbit anti-LC3B (2775S), rabbit anti–beclin-1 (3495), rabbit anti-Atg3 (3415), and rabbit anti-Atg5 (12994) were from Cell Signaling Technology (Danvers, MA). Mouse anti-GFP B-2 (sc-9996), rat anti-Grp94 9G10 (sc-32249), goat anti–synaptopodin P-19 (sc-21537), rabbit anti-WT1 C-19 (sc-192), and mouse anti-CHOP (sc-7357) antibodies were from Santa Cruz Biotechnology (Santa Cruz, CA). Goat anti-podocalyxin (AF1556) was purchased from R & D systems (Minneapolis, MN). Mouse M2 anti-FLAG (F3165 or F1804), rabbit anti-ubiquitin (U5379), and rabbit anti-actin (A2066) antibodies were purchased from Sigma-Aldrich. Rabbit anti-Grp78 antibody (SPA-826) was purchased from Enzo Life Sciences (Ann Arbor, MI). Rabbit anti–phospho-eIF2α Ser51 (44728G) was from Invitrogen. Rabbit anti-nephrin antiserum was kindly provided by Tomoko Takano (McGill University, Montreal, Canada) and was described previously (Elimam et al., 2016). Fluorescein isothiocyanate (FITC)-conjugated goat anti-mouse complement C3 antibody (Cappel 55500) and rabbit anti-sheep immunoglobulin G (IgG; 0865205) were purchased from MP Biomedicals (Santa Ana, CA).
Plasmids
Plasmids encoding full-length IRE1α WT or S724A, K599A or RNase-deletion (ΔR) mutants in-frame with a C-terminal FLAG epitope tag were kindly provided by Yong Liu (Shanghai Institutes for Biological Sciences, Shanghai, China; Qiu et al., 2010). The plasmid encoding CD3δ-YFP was described previously (Menendez-Benito et al., 2005). pBABE-puro-mCherry-eGFP-LC3B was purchased from Addgene (plasmid 22418). pRC/RSV empty vector (Invitrogen) was used as a mock-transfection control. Plasmid identities were confirmed by restriction digestion analyses and DNA sequencing.
Cell culture and transfection
Kidney fibroblastic COS-1 cells were cultured in DMEM with 10% fetal bovine serum. Cells were used between passages 4 and 50. For immunoblot studies, 2 × 105 cells were plated into 35-mm plates in 2 ml of medium. To prepare cells on coverslips for microscopy studies of autophagic puncta, 5 × 104 cells were plated in 22-mm wells on bovine collagen–coated coverslips in 1 ml of culture medium. Cells were grown at 37°C in 5% CO2 and constant humidity until ∼70% confluency. Cells were transiently transfected with plasmid DNAs using Lipofectamine 2000 according to the manufacturer’s instructions. For experiments, culture medium was replaced 22 h posttransfection with fresh medium (37°C) to prevent starvation-mediated autophagy and remove residual transfection reagents. Experiments were initiated after 2 h. For immunoblotting studies, transfections involved 1 μg of IRE1α-FLAG or pRC/RSV, which was combined with 1 μg CD3δ-YFP in studies of ERAD. For fluorescence microscopy, transfections involved 0.3 μg of pBABE-puro-mCherry-eGFP-LC3B plasmid, 0.5 μg of pRC/RSV, or 0.5 μg of IRE1α.
Mouse breeding and genotyping
IRE1α flox/flox mice (loxP sites surrounding IRE1α exons 20 and 21) maintained on a C57BL/6 and 129 mixed background were generated as described previously (Iwawaki et al., 2009). The IRE1α flox/flox mice were crossed with podocin-Cre/+ mice of a mixed ICR/129/B6 background in the F0 generation to give F1 podocin-Cre/+; IRE1α flox/+ mice. F1 mice were backcrossed with F0 IRE1α flox/flox mice to give 25% podocin-Cre/+; IRE1α flox/flox mice, 25% podocin-Cre/+; IRE1α flox/+ mice, 25% +/+; IRE1α flox/flox mice, and 25% +/+; IRE1α flox/+ mice in the F2 generation. Thereafter F2 podocin-Cre/+; IRE1α flox/flox males were crossed with F2 +/+; IRE1α flox/flox females to give F3 littermates that were 50% podocin-Cre/+; IRE1α flox/flox and 50% +/+; IRE1α flox/flox. Animal protocols were reviewed and approved by the McGill University Animal Care Committee.
For genotyping, PCR was conducted on tail DNA, which was extracted after a 1 h digestion with proteinase K. The podocin-Cre allele was amplified with forward primer 5′-CGTACTGACGGTGGGAGAAT-3′ and reverse primer 5′-TGCATGATCTCCGGTATTGA-3′, resulting in a 374–base pair product. The IRE1α WT and flox alleles were amplified with forward primer 5′-CCGAGCCATGAGAAACAAGG-3′ and reverse primer 5′-CCCTGCCAGGATGGTCATGG-3′. The WT allele appeared as a 254–base pair band, and the floxed allele appeared as a 229–base pair band.
To confirm podocyte IRE1α deletion, glomeruli were isolated from mouse kidneys by differential sieving as described previously (Salant and Cybulsky, 1988), and genomic DNA was purified. PCR primers were designed to amplify a 1025–base pair unspliced loxP allele and a 189–base pair deletion product that results from Cre-dependent excision of IRE1α exons 20 and 21, with both primers flanking the loxP sites that surround IRE1α exons 20 and 21: forward 5′-CCAGTGCTCTTGAAAAGAGG-3′ and reverse 5′-CCCTGCCAGGATGGTCATGG-3′. In parallel, this PCR program was used to amplify the 374–base pair product of the glomerular podocin-Cre gene, using the same primers as described.
Anti-GBM nephritis
Sheep anti-rat GBM antiserum production was described previously (Salant and Cybulsky, 1988). Mice were injected intravenously with 5 μl of antiserum (Salant and Cybulsky, 1988). In preliminary studies, we established that the 5-μl dose of antibody induced modest albuminuria.
Measurement of urinary albumin, creatinine, and serum creatinine
Spot urine samples were collected from mice in the morning, immediately placed on ice, and subsequently frozen. Urinary albumin concentration was measured using the Mouse Albumin ELISA Quantification Kit (Bethyl Laboratories, Montgomery, TX) according to the manufacturer’s instructions. Urinary creatinine was measured using a creatinine assay kit from Cayman Chemical Company (Ann Arbor, MI). The kit uses a picric acid–based method. All samples were analyzed at least in duplicate. Albumin and creatinine concentrations were obtained using the standard curve equation of best fit, and the albumin/creatinine ratio was calculated in micrograms/milligram for each animal. For serum creatinine, mice were exsanguinated by cardiac puncture. Serum creatinine was measured using an enzymatic assay on the Olympus AU5822 chemistry system (Beckman Coulter) in the Biochemistry Laboratory at the McGill University Health Centre.
Immunoblotting
COS-1 cells and isolated mouse glomeruli were lysed in buffer containing 1% Triton X-100 (lysis buffer) supplemented with 10 μl/ml protease inhibitor cocktail (BioShop) as described previously (Elimam et al., 2016). Cytosolic protein concentrations were measured with the Bradford assay. Samples were subjected to SDS–PAGE, and proteins were electrophoretically transferred to a polyvinylidene fluoride membrane of 0.2 μm pore size. Membranes were blocked in a solution of 5% bovine serum albumin (BSA) in 1× Tris-buffered saline supplemented with 0.1% Tween-20 (1× TTBS). Membranes were then incubated in primary antibody in 5% BSA in 1× TTBS, overnight, followed by incubation in horseradish peroxidase–conjugated secondary antibody for 1 h at 22°C.
Reverse transcriptase-PCR for Xbp1
Cells were lysed, and mRNA was purified using the RNeasy Mini Kit (Qiagen) according to the manufacturer’s instructions. Purified mRNA integrity was verified using electrophoresis. Bands were assessed for the integrity of rRNA under ultraviolet light. Xbp1 forward and reverse primers flanked the 26-nucleotide region that is normally spliced and excised by IRE1α. Xbp1 forward primer was 5′-CCAAAAACTTTTGCTAGAAAATCA-3′, and the reverse primer was 5′-CAGAATCCATGGGGAGATGT-3′.
Histology and light microscopy
At the time of kidney harvesting, segments of renal cortex were collected on ice, cut to 1.5 mm × 2 mm rectangular blocks with the cortical-to-medullary (apical-to-distal) axis lengthwise, and fixed in 4% paraformaldehyde in phosphate-buffered saline (PBS). Tissues were paraffin embedded, sectioned at 3–4 μm thickness, and stained with PAS or Jones’ silver stain. Kidney sections were examined using a Zeiss AXIO Observer.Z1 microscope. Images were collected with a Zeiss AxioCam MRm monochrome camera (Carl Zeiss, Oberkochen, Germany) at an exposure length set to avoid camera saturation.
For PAS- and Jones’ silver–stained glomeruli, cross-sectional area was measured in ImageJ as follows. Using the “freehand shape” tool, a glomerulus was outlined, and cross-sectional area in micrometers squared was obtained using the “measure” tool. To determine the fraction of a glomerular cross-section occupied by glomerular capillary lumens, the image was converted to 8-bit grayscale with the same freehand outline surrounding the glomerulus. Using the “threshold” tool set to “percentile” mode, a glomerulus was thresholded such that the capillary lumens were selected/isolated. Using the “measure” tool, the thresholded area was obtained in micrometers squared. The ratio [thresholded area (μm2)]/[total glomerular cross-sectional area (μm2)] gave the fraction of glomerular cross sections occupied by capillary lumens. For Jones’ silver–stained glomeruli, the “threshold” tool set to “percentile” mode was again used as follows: each glomerulus was thresholded so that darkly stained glomerular tissue was selected/isolated. Using the “measure” tool, the area occupied by silver-stained tissue was obtained in micrometers squared. The equation [stained area (μm2)]/[(glomerular cross-sectional area − capillary lumen area) (μm2)] gave the proportion of glomerular tissue that was stained.
Immunofluorescence staining
Isolation, embedding, and cryosectioning (5-μm sections) of renal cortex were described previously (Elimam et al., 2016). WT1 immunofluorescent stain was described previously (Elimam et al., 2016). For synaptopodin and podocalyxin immunostaining, kidney sections were air dried and fixed in 4% paraformaldehyde. Slides were washed in PBS and incubated in blocking solutions (containing BSA or nonimmune serum) for 1 h at 22°C in a humidified chamber. Slides were incubated with primary antibody or nonimmune control IgG overnight at 4°C, in a humidified chamber. Slides were washed with PBS and incubated with secondary antibody for 1 h at 22°C in a humidified chamber. In general, as a final step, stained slides were washed with PBS and incubated with 1 μg/ml Hoechst H33342 nuclear stain for 10 min at 37°C in a humidified chamber.
For sheep anti-GBM and C3 immunofluorescence staining, cryosections were air dried and fixed in acetone at −20°C. Slides were blocked in 5% normal rabbit serum (sheep IgG) or 5% normal goat serum (C3) and 5% BSA in PBS for 15 min at 24°C. Slides were incubated with FITC-conjugated rabbit anti-sheep IgG or FITC-conjugated goat anti-mouse C3 for 2 h at 22°C. Sections were stained with Hoechst H33342 as described.
Fluorescence imaging was performed on a Zeiss AXIO Observer.Z1 microscope. Images were collected with a Zeiss AxioCam MRm monochrome camera at an exposure length set to avoid saturation. For WT1 images, the number of fluorescent red nuclei that colocalized with Hoechst nuclear stain were counted (per glomerulus). For podocalyxin, synaptopodin, sheep anti-GBM antibody, and C3, glomeruli were encircled with a freehand region of interest (ImageJ). Cross-sectional area and fluorescence intensity were obtained within the freehand region of interest using the “measure” tool in ImageJ.
Electron microscopy and quantification of podocyte foot-process effacement
Kidney cortex was separated on ice, cut into 1.5 × 2 μm tissue blocks, and fixed in a solution of 2.5% glutaraldehyde in sodium cacodylate buffer (0.2 M C2H7AsO2, pH 7.4, with 0.1% CaCl and 0.2 M sucrose) at 4°C. Tissue preparation for electron microscopy and electron microscopic imaging was carried out at the McGill University Facility for Electron Microscopy Research as described previously (Elimam et al., 2016). ImageJ software was calibrated according to the image magnification, and the number of podocyte foot processes was counted along a given length of GBM. Absolute foot process width (FPW) was calculated in micrometers as follows: FPW = π/4 × ∑GBM length/∑slits (Gundersen et al., 1980; Elimam et al., 2016).
Fluorescence quantification of autophagic puncta and FLAG immunostaining
Cells transfected with mCherry-eGFP-LC3B and FLAG-hIRE1α adherent to coverslips were washed with PBS at 22°C and fixed in ice-cold 4% paraformaldehyde in PBS for 15 min. Coverslips were washed three times with PBS at room temperature and incubated in 1 μg/ml Hoechst H33342 nuclear stain in PBS for 10 min at 37°C. Coverslips were then washed with PBS at 22°C and mounted on slides. Fluorescence imaging was conducted as described. Images were analyzed for puncta using ImageJ software as described previously (Elimam et al., 2016), except that particles <0.1 μm2 and >100 μm2 were excluded.
For immunofluorescence staining of the IRE1α FLAG epitope, coverslips were fixed, blocked, and incubated with monoclonal anti-FLAG primary antibody. Next coverslips were incubated with Alexa 546–conjugated donkey anti-mouse IgG in 3% BSA in PBS for 1 h at 37°C. Coverslips were then washed with PBS and incubated with 1 μg/ml Hoechst H33342 nuclear stain and mounted as described.
Statistics
Data are presented as mean ± SEM. Unless otherwise indicated, data were compared as arbitrary units (percentage of maximum value in the data set). All statistical tests were conducted with the program R (The R Foundation). One-tail t tests were used to assess differences between two groups. For more than two groups, analysis of variance (ANOVA) was used (Rao, 2007) as specified in the figure legends. For two-way ANOVAs conducted on data sets for which groups had unequal sample numbers, type 2 sum-of-squares ANOVA tests were conducted for data sets with no significant interaction terms. For data sets with a substantial interaction between factors, the type 1 ANOVA was conducted twice, with one factor leading the other and then vice versa. Once both ANOVA tests were completed, the test that gave the least significant p values for all comparisons was used. All post hoc comparisons and adjustments of the critical p value were conducted according to the Bonferroni method.
Supplementary Material
Acknowledgments
We thank Julie Guillemette for expert technical assistance and Jeffrey Brodsky for helpful suggestions. This work was supported by Research Grants MOP-125988 and MOP-133492 from the Canadian Institutes of Health Research, studentships from the Fonds de recherche du Québec–Santé and McGill University Health Centre Research Institute (D.R.K.), and the Catherine McLaughlin Hakim Chair (A.V.C.).
Abbreviations used:
- ASK1
apoptosis signal-regulated kinase 1
- CHOP
C/EBP homologous protein
- eIF2α
eukaryotic translation initiation factor 2α
- ER
endoplasmic reticulum
- ERAD
ER-associated degradation
- GBM
glomerular basement membrane
- GFP
green fluorescent protein
- IRE1α
inositol-requiring enzyme-1α
- JNK
c-Jun amino-terminal kinase
- KO
knockout
- LC3
microtubule-associated protein 1A/1B-light chain 3
- TRAF2
tumor necrosis factor-α receptor receptor–associated factor 2
- UPR
unfolded protein response
- WT
wild type
- WT1
Wilms tumor-1
- Xbp1
X-box binding protein-1
- YFP
yellow fluorescent protein.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E16-12-0828) on April 20, 2017.
REFERENCES
- Araki K, Nagata K. Protein folding and quality control in the ER. Cold Spring Harb Perspect Biol. 2011;3:a007526. doi: 10.1101/cshperspect.a007526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonifacino JS, Suzuki CK, Lippincott-Schwartz J, Weissman AM, Klausner RD. Pre-Golgi degradation of newly synthesized T-cell antigen receptor chains: intrinsic sensitivity and the role of subunit assembly. J Cell Biol. 1989;109:73–83. doi: 10.1083/jcb.109.1.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinkkoetter PT, Ising C, Benzing T. The role of the podocyte in albumin filtration. Nat Rev Nephrol. 2013;9:328–336. doi: 10.1038/nrneph.2013.78. [DOI] [PubMed] [Google Scholar]
- Cheng YC, Chang JM, Chen CA, Chen HC. Autophagy modulates endoplasmic reticulum stress-induced cell death in podocytes: a protective role. Exp Biol Med (Maywood) 2015;240:467–476. doi: 10.1177/1535370214553772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang CK, Inagi R. Glomerular diseases: genetic causes and future therapeutics. Nat Rev Nephrol. 2010;6:539–554. doi: 10.1038/nrneph.2010.103. [DOI] [PubMed] [Google Scholar]
- Chiang WC, Messah C, Lin JH. IRE1 directs proteasomal and lysosomal degradation of misfolded rhodopsin. Mol Biol Cell. 2012;23:758–770. doi: 10.1091/mbc.E11-08-0663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuervo AM. Autophagy: many paths to the same end. Mol Cell Biochem. 2004;263:55–72. doi: 10.1023/B:MCBI.0000041848.57020.57. [DOI] [PubMed] [Google Scholar]
- Cybulsky AV. The intersecting roles of endoplasmic reticulum stress, ubiquitin-proteasome system, and autophagy in the pathogenesis of proteinuric kidney disease. Kidney Int. 2013;84:25–33. doi: 10.1038/ki.2012.390. [DOI] [PubMed] [Google Scholar]
- Ding WX, Ni HM, Gao W, Yoshimori T, Stolz DB, Ron D, Yin XM. Linking of autophagy to ubiquitin-proteasome system is important for the regulation of endoplasmic reticulum stress and cell viability. Am J Pathol. 2007;171:513–524. doi: 10.2353/ajpath.2007.070188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elimam H, Papillon J, Kaufman DR, Guillemette J, Aoudjit L, Gross RW, Takano T, Cybulsky AV. Genetic ablation of calcium-independent phospholipase A2gamma induces glomerular injury in mice. J Biol Chem. 2016;291:14468–14482. doi: 10.1074/jbc.M115.696781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang L, Zhou Y, Cao H, Wen P, Jiang L, He W, Dai C, Yang J. Autophagy attenuates diabetic glomerular damage through protection of hyperglycemia-induced podocyte injury. PLoS One. 2013;8:e60546. doi: 10.1371/journal.pone.0060546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greka A, Mundel P. Cell biology and pathology of podocytes. Annu Rev Physiol. 2012;74:299–323. doi: 10.1146/annurev-physiol-020911-153238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gundersen HJ, Seefeldt T, Osterby R. Glomerular epithelial foot processes in normal man and rats. Distribution of true width and its intra- and inter-individual variation. Cell Tissue Res. 1980;205:147–155. doi: 10.1007/BF00234450. [DOI] [PubMed] [Google Scholar]
- Hartleben B, Godel M, Meyer-Schwesinger C, Liu S, Ulrich T, Kobler S, Wiech T, Grahammer F, Arnold SJ, Lindenmeyer MT, et al. Autophagy influences glomerular disease susceptibility and maintains podocyte homeostasis in aging mice. J Clin Invest. 2010;120:1084–1096. doi: 10.1172/JCI39492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassan H, Tian X, Inoue K, Chai N, Liu C, Soda K, Moeckel G, Tufro A, Lee AH, Somlo S, et al. Essential role of X-box binding protein-1 during endoplasmic reticulum stress in podocytes. J Am Soc Nephrol. 2016;27:1055–1065. doi: 10.1681/ASN.2015020191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi-Nishino M, Fujita N, Noda T, Yamaguchi A, Yoshimori T, Yamamoto A. A subdomain of the endoplasmic reticulum forms a cradle for autophagosome formation. Nat Cell Biol. 2009;11:1433–1437. doi: 10.1038/ncb1991. [DOI] [PubMed] [Google Scholar]
- He W, Wang Q, Xu J, Xu X, Padilla MT, Ren G, Gou X, Lin Y. Attenuation of TNFSF10/TRAIL-induced apoptosis by an autophagic survival pathway involving TRAF2- and RIPK1/RIP1-mediated MAPK8/JNK activation. Autophagy. 2012;8:1811–1821. doi: 10.4161/auto.22145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hetz C. The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat Rev Mol Cell Biol. 2012;13:89–102. doi: 10.1038/nrm3270. [DOI] [PubMed] [Google Scholar]
- Hetz C, Glimcher LH. Fine-tuning of the unfolded protein response: assembling the IRE1alpha interactome. Mol Cell. 2009;35:551–561. doi: 10.1016/j.molcel.2009.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hetz C, Soto C. Protein misfolding and disease: the case of prion disorders. Cell Mol Life Sci. 2003;60:133–143. doi: 10.1007/s000180300009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber TB, Edelstein CL, Hartleben B, Inoki K, Jiang M, Koya D, Kume S, Lieberthal W, Pallet N, Quiroga A, et al. Emerging role of autophagy in kidney function, diseases and aging. Autophagy. 2012;8:1009–1031. doi: 10.4161/auto.19821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hur KY, So JS, Ruda V, Frank-Kamenetsky M, Fitzgerald K, Koteliansky V, Iwawaki T, Glimcher LH, Lee AH. IRE1alpha activation protects mice against acetaminophen-induced hepatotoxicity. J Exp Med. 2012;209:307–318. doi: 10.1084/jem.20111298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwawaki T, Akai R, Kohno K. IRE1alpha disruption causes histological abnormality of exocrine tissues, increase of blood glucose level, and decrease of serum immunoglobulin level. PLoS One. 2010;5:e13052. doi: 10.1371/journal.pone.0013052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwawaki T, Akai R, Yamanaka S, Kohno K. Function of IRE1 alpha in the placenta is essential for placental development and embryonic viability. Proc Natl Acad Sci USA. 2009;106:16657–16662. doi: 10.1073/pnas.0903775106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung CH, Ro SH, Cao J, Otto NM, Kim DH. mTOR regulation of autophagy. FEBS Lett. 2010;584:1287–1295. doi: 10.1016/j.febslet.2010.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneko M, Niinuma Y, Nomura Y. Activation signal of nuclear factor-kappa B in response to endoplasmic reticulum stress is transduced via IRE1 and tumor necrosis factor receptor-associated factor 2. Biol Pharm Bull. 2003;26:931–935. doi: 10.1248/bpb.26.931. [DOI] [PubMed] [Google Scholar]
- Kawakami T, Gomez IG, Ren S, Hudkins K, Roach A, Alpers CE, Shankland SJ, D’Agati VD, Duffield JS. Deficient autophagy results in mitochondrial dysfunction and FSGS. J Am Soc Nephrol. 2015;26:1040–1052. doi: 10.1681/ASN.2013111202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemp KL, Lin Z, Zhao F, Gao B, Song J, Zhang K, Fang D. The serine-threonine kinase inositol-requiring enzyme 1alpha (IRE1alpha) promotes IL-4 production in T helper cells. J Biol Chem. 2013;288:33272–33282. doi: 10.1074/jbc.M113.493171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H, Noh JY, Oh Y, Kim Y, Chang JW, Chung CW, Lee ST, Kim M, Ryu H, Jung YK. IRE1 plays an essential role in ER stress-mediated aggregation of mutant huntingtin via the inhibition of autophagy flux. Hum Mol Genet. 2012;21:101–114. doi: 10.1093/hmg/ddr445. [DOI] [PubMed] [Google Scholar]
- Menendez-Benito V, Verhoef LG, Masucci MG, Dantuma NP. Endoplasmic reticulum stress compromises the ubiquitin-proteasome system. Hum Mol Genet. 2005;14:2787–2799. doi: 10.1093/hmg/ddi312. [DOI] [PubMed] [Google Scholar]
- Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell. 2010;140:313–326. doi: 10.1016/j.cell.2010.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- N’Diaye EN, Kajihara KK, Hsieh I, Morisaki H, Debnath J, Brown EJ. PLIC proteins or ubiquilins regulate autophagy-dependent cell survival during nutrient starvation. EMBO Rep. 2009;10:173–179. doi: 10.1038/embor.2008.238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni HM, Bockus A, Wozniak AL, Jones K, Weinman S, Yin XM, Ding WX. Dissecting the dynamic turnover of GFP-LC3 in the autolysosome. Autophagy. 2011;7:188–204. doi: 10.4161/auto.7.2.14181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishitoh H, Matsuzawa A, Tobiume K, Saegusa K, Takeda K, Inoue K, Hori S, Kakizuka A, Ichijo H. ASK1 is essential for endoplasmic reticulum stress-induced neuronal cell death triggered by expanded polyglutamine repeats. Genes Dev. 2002;16:1345–1355. doi: 10.1101/gad.992302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogata M, Hino S, Saito A, Morikawa K, Kondo S, Kanemoto S, Murakami T, Taniguchi M, Tanii I, Yoshinaga K, et al. Autophagy is activated for cell survival after endoplasmic reticulum stress. Mol Cell Biol. 2006;26:9220–9231. doi: 10.1128/MCB.01453-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh SH, Lim SC. Endoplasmic reticulum stress-mediated autophagy/apoptosis induced by capsaicin (8-methyl-N-vanillyl-6-nonenamide) and dihydrocapsaicin is regulated by the extent of c-Jun NH2-terminal kinase/extracellular signal-regulated kinase activation in WI38 lung epithelial fibroblast cells. J Pharmacol Exp Ther. 2009;329:112–122. doi: 10.1124/jpet.108.144113. [DOI] [PubMed] [Google Scholar]
- Pippin JW, Brinkkoetter PT, Cormack-Aboud FC, Durvasula RV, Hauser PV, Kowalewska J, Krofft RD, Logar CM, Marshall CB, Ohse T, Shankland SJ. Inducible rodent models of acquired podocyte diseases. Am J Physiol Renal Physiol. 2009;296:F213–F229. doi: 10.1152/ajprenal.90421.2008. [DOI] [PubMed] [Google Scholar]
- Puelles VG, Bertram JF. Counting glomeruli and podocytes: rationale and methodologies. Curr Opin Nephrol Hypertens. 2015;24:224–230. doi: 10.1097/MNH.0000000000000121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu Y, Mao T, Zhang Y, Shao M, You J, Ding Q, Chen Y, Wu D, Xie D, Lin X, Gao X, et al. A crucial role for RACK1 in the regulation of glucose-stimulated IRE1alpha activation in pancreatic beta cells. Sci Signal. 2010;3:ra7. doi: 10.1126/scisignal.2000514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao KV. Biostatistics: A Manual of Statistical Methods for Use in Health, Nutrition and Anthropology. New Delhi, India: Jaypee Brothers Medical Publishers; 2007. [Google Scholar]
- Ruseva MM, Takahashi M, Fujita T, Pickering MC. C3 dysregulation due to factor H deficiency is mannan-binding lectin-associated serine proteases (MASP)-1 and MASP-3 independent in vivo. Clin Exp Immunol. 2014;176:84–92. doi: 10.1111/cei.12244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salant DJ, Cybulsky AV. Experimental glomerulonephritis. Methods Enzymol. 1988;162:421–461. doi: 10.1016/0076-6879(88)62096-9. [DOI] [PubMed] [Google Scholar]
- Sun S, Shi G, Sha H, Ji Y, Han X, Shu X, Ma H, Inoue T, Gao B, Kim H, et al. IRE1alpha is an endogenous substrate of endoplasmic-reticulum-associated degradation. Nat Cell Biol. 2015;17:1546–1555. doi: 10.1038/ncb3266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tagawa A, Yasuda M, Kume S, Yamahara K, Nakazawa J, Chin-Kanasaki M, Araki H, Araki S, Koya D, Asanuma K, et al. Impaired podocyte autophagy exacerbates proteinuria in diabetic nephropathy. Diabetes. 2016;65:755–767. doi: 10.2337/db15-0473. [DOI] [PubMed] [Google Scholar]
- Tanida I, Ueno T, Kominami E. LC3 and autophagy. Methods Mol Biol. 2008;445:77–88. doi: 10.1007/978-1-59745-157-4_4. [DOI] [PubMed] [Google Scholar]
- Tohmonda T, Yoda M, Iwawaki T, Matsumoto M, Nakamura M, Mikoshiba K, Toyama Y, Horiuchi K. IRE1alpha/XBP1-mediated branch of the unfolded protein response regulates osteoclastogenesis. J Clin Invest. 2015;125:3269–3279. doi: 10.1172/JCI76765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tryggvason K, Wartiovaara J. Molecular basis of glomerular permselectivity. Curr Opin Nephrol Hypertens. 2001;10:543–549. doi: 10.1097/00041552-200107000-00009. [DOI] [PubMed] [Google Scholar]
- Um JW, Im E, Park J, Oh Y, Min B, Lee HJ, Yoon JB, Chung KC. ASK1 negatively regulates the 26 S proteasome. J Biol Chem. 2010;285:36434–36446. doi: 10.1074/jbc.M110.133777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varga K, Jurkuvenaite A, Wakefield J, Hong JS, Guimbellot JS, Venglarik CJ, Niraj A, Mazur M, Sorscher EJ, Collawn JF, Bebok Z. Efficient intracellular processing of the endogenous cystic fibrosis transmembrane conductance regulator in epithelial cell lines. J Biol Chem. 2004;279:22578–22584. doi: 10.1074/jbc.M401522200. [DOI] [PubMed] [Google Scholar]
- Vembar SS, Brodsky JL. One step at a time: endoplasmic reticulum-associated degradation. Nat Rev Mol Cell Biol. 2008;9:944–957. doi: 10.1038/nrm2546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatareddy M, Wang S, Yang Y, Patel S, Wickman L, Nishizono R, Chowdhury M, Hodgin J, Wiggins PA, Wiggins RC. Estimating podocyte number and density using a single histologic section. J Am Soc Nephrol. 2014;25:1118–1129. doi: 10.1681/ASN.2013080859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang XJ, Yu J, Wong SH, Cheng AS, Chan FK, Ng SS, Cho CH, Sung JJ, Wu WK. A novel crosstalk between two major protein degradation systems: regulation of proteasomal activity by autophagy. Autophagy. 2013;9:1500–1508. doi: 10.4161/auto.25573. [DOI] [PubMed] [Google Scholar]
- Welsh GI, Saleem MA. The podocyte cytoskeleton—key to a functioning glomerulus in health and disease. Nat Rev Nephrol. 2012;8:14–21. doi: 10.1038/nrneph.2011.151. [DOI] [PubMed] [Google Scholar]
- Xiao Y, Xia T, Yu J, Deng Y, Liu H, Liu B, Chen S, Liu Y, Guo F. Knockout of inositol-requiring enzyme 1alpha in pro-opiomelanocortin neurons decreases fat mass via increasing energy expenditure. Open Biol. 2016;6:160131. doi: 10.1098/rsob.160131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang KC, Ma X, Liu H, Murphy J, Barger PM, Mann DL, Diwan A. Tumor necrosis factor receptor-associated factor 2 mediates mitochondrial autophagy. Circ Heart Fail. 2015;8:175–187. doi: 10.1161/CIRCHEARTFAILURE.114.001635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Klionsky DJ. Eaten alive: a history of macroautophagy. Nat Cell Biol. 2010;12:814–822. doi: 10.1038/ncb0910-814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yla-Anttila P, Vihinen H, Jokitalo E, Eskelinen EL. 3D tomography reveals connections between the phagophore and endoplasmic reticulum. Autophagy. 2009;5:1180–1185. doi: 10.4161/auto.5.8.10274. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.