

Sortilin is involved in the anterograde traffic of lysosomal enzymes and substrates. It also transports Glut4 in the opposite, retrograde, direction in a retromer-dependent fashion.
Abstract
Sortilin is a multiligand sorting receptor responsible for the anterograde transport of lysosomal enzymes and substrates. Here we demonstrate that sortilin is also involved in retrograde protein traffic. In cultured 3T3-L1 adipocytes, sortilin together with retromer rescues Glut4 from degradation in lysosomes and retrieves it to the TGN, where insulin-responsive vesicles are formed. Mechanistically, the luminal Vps10p domain of sortilin interacts with the first luminal loop of Glut4, and the cytoplasmic tail of sortilin binds to retromer. Ablation of the retromer does not affect insulin signaling but decreases the stability of sortilin and Glut4 and blocks their entry into the small vesicular carriers. As a result, Glut4 cannot reach the insulin-responsive compartment, and insulin-stimulated glucose uptake in adipocytes is suppressed. We suggest that sortilin- and retromer-mediated Glut4 retrieval from endosomes may represent a step in the Glut4 pathway vulnerable to the development of insulin resistance and diabetes.
INTRODUCTION
The regulation of blood glucose levels in mammals is achieved by insulin-dependent translocation of the glucose transporter isoform 4 (Glut4) to the plasma membrane in fat and skeletal muscle cells. Many transgenic models (reviewed in Charron et al., 1999; Minokoshi et al., 2003; Graham and Kahn, 2007), as well as nuclear magnetic resonance experiments in humans (reviewed in Shulman, 2000), show that Glut4 is responsible for the rate-limiting step in overall insulin-stimulated glucose disposal in vivo.
In insulin resistance and type 2 diabetes, insulin responsiveness of Glut4 in skeletal muscle is reduced in spite of its seemingly normal expression levels, whereas in adipose tissue, Glut4 protein is also decreased (Shepherd and Kahn, 1999; Graham and Kahn, 2007; Huang and Czech, 2007; Bogan, 2012). The latter effect is highly significant because down-regulation of the Glut4 protein specifically in adipocytes leads to systemic insulin resistance (Abel et al., 2001; Herman et al., 2012), whereas even a moderate increase in Glut4 levels improves insulin sensitivity (Atkinson et al., 2013).
Down-regulation of Glut4 has been associated with decreased expression of its mRNA (Sivitz et al., 1989). This, however, may not represent the only factor affecting Glut4 protein levels because prolonged insulin stimulation decreases the half-life of the Glut4 protein more than threefold and causes insulin resistance (Sargeant and Paquet, 1993; Ma et al., 2014). The explanation of this phenomenon is likely to lie in the cell biology of Glut4 recycling.
From the plasma membrane, Glut4 is internalized into peripheral endosomes, from where it is retrieved to a perinuclear compartment, likely the trans-Golgi network (TGN), in order to escape degradation in lysosomes (Slot et al., 1991; Foster et al., 2001; Shewan et al., 2003). Insulin-responsive vesicles (IRVs) are believed to derive from the TGN donor membranes (Bogan and Kandror, 2010). Because Glut4 has a very long half-life in spite of multiple cycles of translocation to and from the plasma membrane, its retrieval from endosomes to the TGN should be extremely efficient under normal conditions. However, in insulin resistance and diabetes, retrieval of Glut4 from endosomes may be compromised, and, correspondingly, more Glut4 molecules undergo a default trafficking pathway from endosomes to lysosomes. Indeed, a recent report shows that prolonged insulin stimulation suppresses Glut4 retrieval from endosomes and increases its lysosomal degradation (Ma et al., 2014). The mechanism of Glut4 retrieval from endosomes is not well understood and needs to be explored. To this end, it has been shown that in human skeletal muscle, retrograde trafficking of Glut4 from endosomes to the TGN involves syntaxin 10 (Esk et al., 2010) and the specific CHC22 isoform of clathrin heavy chain (Vassilopoulos et al., 2009). However, retrieval of Glut4 in adipocytes remains poorly studied.
Here we demonstrate that a sorting receptor, sortilin, plays a central role in the retrieval of Glut4 from endosomes. Specifically, the luminal Vps10 p domain of sortilin interacts with the first luminal loop of Glut4, and the cytoplasmic tail of sortilin binds to retromer, an evolutionary conserved protein complex responsible for protein selection and export from endosomes to the TGN (Bonifacino and Rojas, 2006). Thus sortilin may work as a transmembrane scaffold that links Glut4 to retromer, rerouting it from the degradative to the recycling pathway.
RESULTS
In undifferentiated 3T3-L1 preadipocytes, ectopically expressed myc7-Glut4 is localized primarily in endosomes and has a short half-life (t1/2 < 2 h; Shi and Kandror, 2005; Liu et al., 2007). On adipocyte differentiation, myc7-Glut4 becomes much more stable, and its half-life increases to >40 h (Shi and Kandror, 2005). In agreement with these results, we show here that in undifferentiated preadipocytes, a significant fraction of myc7-Glut4 is colocalized with LysoTracker, presumably in late endosomes and lysosomes, whereas in differentiated adipocytes, colocalization between myc7-Glut4 and LysoTracker is virtually undetectable (Figure 1 and Table 1, lines 1 and 2).
FIGURE 1:
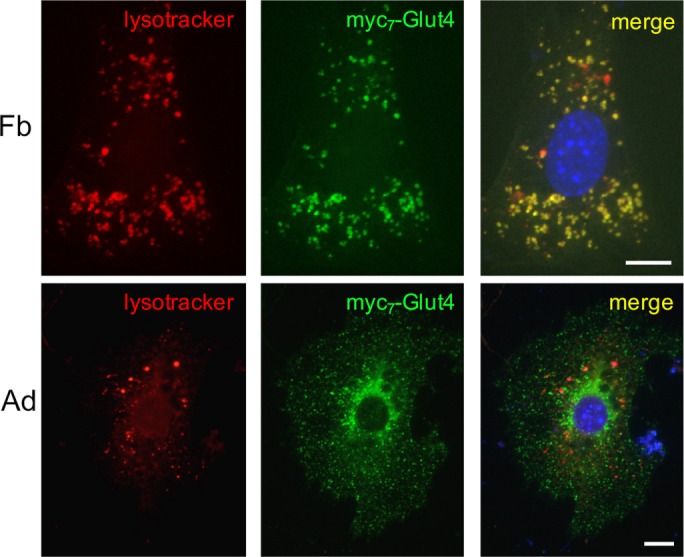
Ectopically expressed myc7-Glut4 is colocalized with LysoTracker in undifferentiated (Fb) but not in differentiated (Ad) 3T3-L1 cells. 3T3-L1 cells stably expressing myc7-Glut4 were incubated with LysoTracker, fixed, and stained with monoclonal anti-myc antibody and FITC-conjugated donkey anti-mouse IgG and analyzed by double immunofluorescence.
TABLE 1:
Quantitative analysis of protein colocalization.
| Line | Cell type | Transfected construct(s) | Staining 1 | Staining 2 | Corresponding figure | Pearson’s R |
|---|---|---|---|---|---|---|
| 1 | Preadipocyte | myc7-Glut4 | myc | LysoTracker | Figure 1 (top) | 0.88 ± 0.04 |
| 2 | Adipocyte | myc7-Glut4 | myc | LysoTracker | Figure 1 (bottom) | 0.48 ± 0.09 |
| 3 | Preadipocyte | myc7-Glut4 | myc | Vps35 | Figure 2B (top) | 0.28 ± 0.13 |
| 4 | Adipocyte | myc7-Glut4 | myc | Vps35 | Figure 2B (middle) | 0.76 ± 0.08 |
| 5 | Adipocyte | myc7-Glut4 | myc | Sortilin | Figure 4A | 0.80 ± 0.08 |
| 6 | Adipocyte | myc7-Glut4 | myc | LysoTracker | Figure 3B (bottom) | 0.86 ± 0.06 |
| Sortilin shRNA | ||||||
| 7 | Adipocyte | myc7-Glut4 | myc | Vps35 | Figure 3B (top) | 0.50 ± 0.08 |
| Sortilin shRNA | ||||||
| 8 | Preadipocyte | myc7-Glut4 | Glut4 | LysoTracker | Figure 5 (bottom) | 0.43 ± 0.08 |
| Sortilin-myc/His | ||||||
| 9 | Preadipocyte | myc7-Glut4 | Glut4 | Vps35 | Figure 5 (top) | 0.67 ± 0.10 |
| Sortilin-myc/His | ||||||
| 10 | Adipocyte | None | Glut4 | LysoTracker | Figure 8C (top) | 0.52 ± 0.11 |
| 11 | Adipocyte | None | Glut4 | Vps35 | Figure 2B (bottom) | 0.82 ± 0.09 |
| 12 | Adipocyte | Vps35 CRISPR | Glut4 | LysoTracker | Figure 8C (bottom) | 0.82 ± 0.06 |
Pearson’s R was determined by ImageJ analysis of 12–14 randomly selected cells.
Because undifferentiated 3T3-L1 cells do not express Glut4 and lack an insulin-responsive vesicular compartment, which normally represents a major “sink” for the transporter (Shi and Kandror, 2005), one possible interpretation of these results is that in undifferentiated cells, ectopically expressed Glut4 cannot be faithfully compartmentalized in the IRVs and thus undergoes rapid degradation. Alternatively, undifferentiated cells may not be able to retrieve Glut4 from endosomes to the TGN where the IRVs are formed; correspondingly, Glut4 is captured by the default pathway from endosomes to lysosomes.
Retrograde transport of proteins from endosomes to TGN depends on the sorting process performed by retromer. Mammalian retromer consists of the trimeric complex of Vps26, Vps29, and Vps35, which functions together with sorting nexins (SNX) and other auxiliary proteins (Seaman, 2012; Lucas et al., 2016) to perform this function.
Undifferentiated cells express a normal amount of retromer (Figure 2A; Yang et al., 2016). However, we have not been able to detect significant colocalization between retromer and ectopically expressed myc7-Glut4 in preadipocytes (Figure 2B, top, and Table 1, line 3); on the contrary, such colocalization becomes apparent on cell differentiation (Figure 2B, middle, and Table 1, line 4). In addition, wild-type 3T3-L1 adipocytes demonstrate significant colocalization between retromer and endogenously expressed Glut4 (Figure 2B, bottom, and Table 1, line 11). These results suggest that the retrograde traffic of Glut4 from endosomes to the TGN is inefficient in undifferentiated preadipocytes and is established on cell differentiation. To explain this phenomenon, we hypothesize that undifferentiated cells do not express a protein that couples Glut4 to retromer.
FIGURE 2:
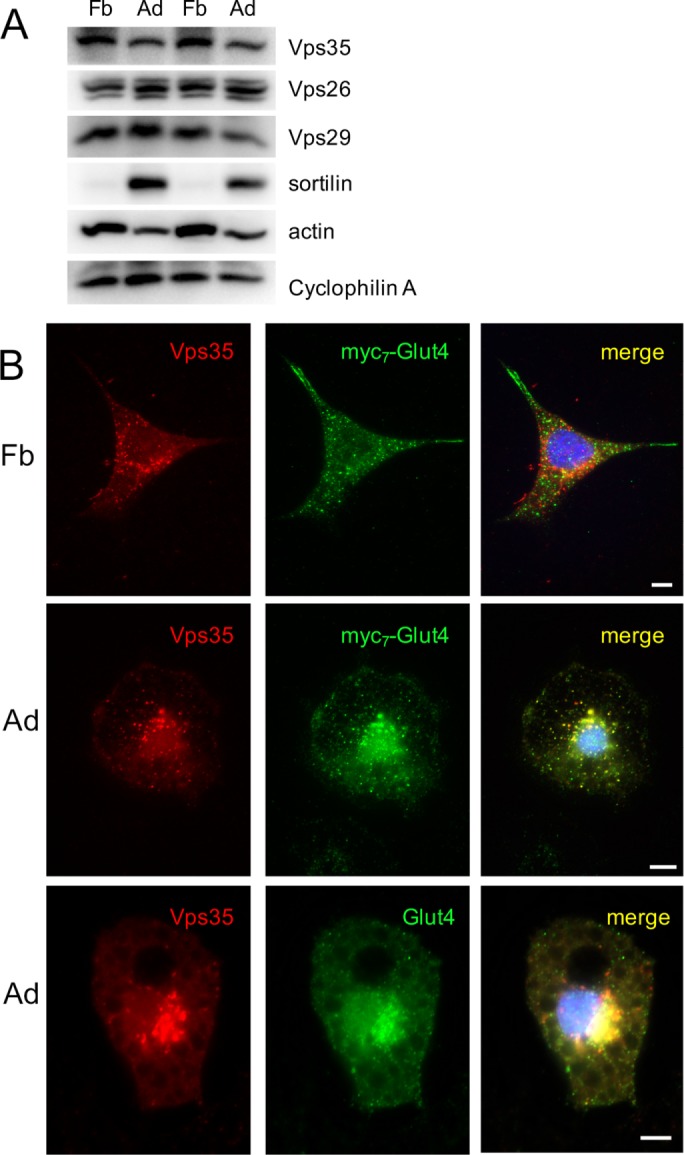
Glut4 is colocalized with Vps35 in differentiated (Ad) but not in undifferentiated (Fb) 3T3-L1 cells. (A) Total lysates (40 µg/lane) of differentiated and undifferentiated 3T3-L1 cells were analyzed by Western blotting. (B) Top and middle, 3T3-L1 cells stably expressing myc7-Glut4 were stained with goat polyclonal antibody against Vps35 and mouse monoclonal anti-myc antibody, followed by Texas red–conjugated donkey anti-goat IgG and FITC-conjugated donkey anti-mouse IgG and analyzed by double immunofluorescence. Bottom, wild-type 3T3-L1 adipocytes were stained with goat polyclonal antibody against Vps35 and rabbit polyclonal antibody against Glut4, followed by Texas red–conjugated donkey anti-goat IgG and FITC-conjugated donkey anti-rabbit IgG.
One such protein could be sortilin, which plays a pivotal role in the formation of the IRVs (Shi and Kandror, 2005; Ariga et al., 2008). In differentiating adipocytes, endogenous sortilin is expressed ∼24 h before Glut4 (Shi and Kandror, 2005), increases its stability (Shi and Kandror, 2005), and generates “static” behavior of Glut4 typical of differentiated adipocytes (Hatakeyama and Kanzaki, 2011).
To test this hypothesis, we knocked down sortilin from 3T3-L1 adipocytes that ectopically express myc7-Glut4 (Figure 3A). In line with our model, depletion of sortilin decreases colocalization of myc7-Glut4 with Vps35 (Figure 3B, top; compare line 7 to line 4 in Table 1) and, correspondingly, increases its colocalization with LysoTracker (Figure 3B, bottom; compare line 6 to line 2 in Table 1).
FIGURE 3:
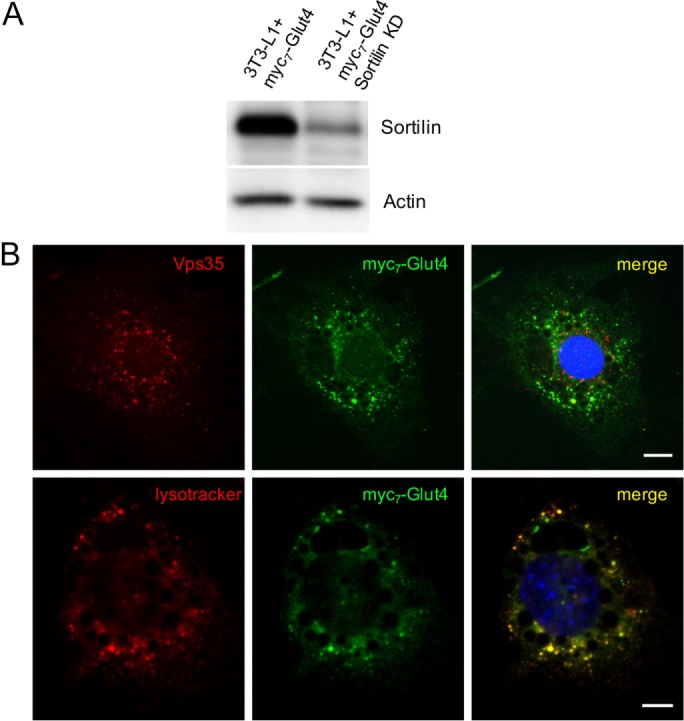
Knockdown of sortilin decreases colocalization of myc7-Glut4 with Vps35 and increases its presence in lysosomes. (A) Total lysates (40 µg/lane) prepared from 3T3-L1 adipocytes stably transfected with myc7-Glut4 and shRNA against sortilin were analyzed by Western blotting. (B) Top: 3T3-L1 adipocytes stably transfected with myc7-Glut4 and shRNA against sortilin were stained with goat polyclonal antibody against Vps35 and mouse monoclonal anti-myc antibody, followed by Texas red–conjugated donkey anti-goat IgG and FITC-conjugated donkey anti-mouse IgG. Bottom: 3T3-L1 adipocytes stably transfected with myc7-Glut4 and shRNA against sortilin were incubated with LysoTracker and stained with mouse monoclonal anti-myc antibody, followed by FITC-conjugated donkey anti-mouse IgG.
Our results are somewhat inconsistent with a recent report of significant colocalization between Vps35 and Glut4 in both differentiated and undifferentiated cells (Yang et al., 2016). We believe that this difference might be due to the level of Glut4 overexpression. In our experiments, we infected 3T3-L1 preadipocytes with myc7-Glut4–encoding lentivirus and selected for cells that ectopically express myc7-Glut4 at <10% of endogenous Glut4 levels (unpublished data), whereas the other study used electroporation, which usually results in massive overexpression of proteins, which can overflow to other compartments.
In 3T3-L1 adipocytes, sortilin shows considerable colocalization with myc7-Glut4 (Figure 4A and Table 1, line 5); however, our attempts to coimmunoprecipitate sortilin and Glut4 did not bring clear-cut results, probably because interaction of the protein-binding Vps10p domain of sortilin with its ligands is sensitive to detergents. At the same time, sortilin can be cross-linked to myc7-Glut4 using the membrane-permeable cross-linker dithiobis(succinimidyl propionate) (DSP; Shi and Kandror, 2005). In addition, yeast two-hybrid experiments suggest that the Vps10p domain interacts with the first liminal loop of Glut4 (Kim and Kandror, 2012). To confirm these results, we used the chemically synthesized peptide corresponding to the first luminal loop of Glut4 and found that it interacts with the isolated sortilin in the absence of detergents at both pH 6 (which roughly corresponds to intraendosomal pH) and pH 8 (Figure 4B). Thus three independent lines of evidence—chemical cross-linking (Shi and Kandror, 2005), the yeast two-hybrid technique (Kim and Kandror, 2012), and peptide binding (Figure 4B)—suggest that the luminal Vps10p domain of sortilin can bind to the first luminal loop of Glut4.
FIGURE 4:

Sortilin interacts with the first luminal loop of Glut4 and retromer. (A) 3T3-L1 adipocytes stably transfected with myc7-Glut4 were stained with mouse monoclonal anti-myc antibody and rabbit polyclonal sortilin antibody, followed by Texas red–conjugated donkey anti-mouse IgG and FITC-conjugated donkey anti-rabbit IgG and analyzed by double immunofluorescence. (B) Total lysates (0.5 mg) prepared from wild-type (WT) 3T3-L1 adipocytes and 3T3-L1 adipocytes stably transfected with sortilin-myc/His (S) were incubated with HisPur cobalt resin; beads were washed and additionally incubated with the peptide (100 ng) corresponding to the first luminal loop of Glut4. Eluates were analyzed by electrophoresis in Tricine gels and Western blotting. A representative result of three independent experiments. (C) The cytoplasmic tail of sortilin (CT) in the GST vector along with GST alone as a control were prebound to glutathione beads and then incubated with the cytoplasmic extract (0.5 mg) from insulin-treated and untreated 3T3-L1 adipocytes. Eluates along with input and unbound material (equal volume aliquots) were analyzed by SDS electrophoresis and Western blotting. A representative result of three independent experiments. (D) 3T3-L1 adipocytes stably expressing myc7-Glut4 were stained with mouse monoclonal anti-myc antibody, goat polyclonal antibody against Vps35, and rabbit polyclonal antibody against sortilin, followed by Alexa Fluor 647–conjugated donkey anti-mouse IgG, Alexa Fluor 546–conjugated donkey anti-goat IgG, and FITC-conjugated donkey anti-rabbit IgG and analyzed by immunofluorescence. (E) 3T3-L1 adipocytes stably expressing myc7-Glut4 or myc7-Glut4 along with sortilin-myc/His were cross-linked with DSP; sortilin-myc/His-containing complexes were isolated on HisPur cobalt resin as described in Materials and Methods and analyzed by Western blotting. A representative result of three independent experiments. (F) Sortilin may work as a transmembrane scaffold linking Glut4 to the retromer-mediated protein traffic.
In parallel, it has been shown that sortilin traffics from endosomes to the TGN in a retromer-dependent manner (Seaman, 2004; Mari et al., 2008), contains an endosome-to-TGN retrieval motif, FLV (Seaman, 2007), and coimmunoprecipitates with the retromer subunit Vps26 (Canuel et al., 2008). Because we were unable to coimmunoprecipitate sortilin and retromer with a significant yield (unpublished data), we subcloned the cytoplasmic tail of sortilin into the glutathione S-transferase (GST) vector as explained in Materials and Methods and demonstrated that in the absence of detergents, the cytoplasmic C-terminus tail of sortilin pulls down retromer, as evidenced by its subunit Vps35 (Figure 4C). Furthermore, using three-way immunofluorescence microscopy, we found that a fraction of sortilin simultaneously colocalizes with Glut4 and Vps35 (Figure 4D), suggesting that a transient complex may exist between sortilin, retromer, and Glut4. To test this idea further, we performed cross-linking of 3T3-L1 adipocytes stably expressing myc7-Glut4 and sortilin-myc/histidine (His) using the membrane-permeable, cleavable cross-linker DSP followed by the isolation of sortilin-myc/His–containing complexes on cobalt resin, which binds to His-tagged proteins. Cells that express only myc7-Glut4 served as a control for these experiments. In agreement with our previous results (Shi and Kandror, 2005), Figure 4E shows that sortilin can be cross-linked with myc7-Glut4. In addition, retromer subunits Vps26, SNX2, and, to a lesser extent, Vps35 have been found in association with sortilin-myc/His (Figure 4E). We believe that our results are consistent with those of Lucas et al. (2016), who showed that cargo proteins may interact with the interface between Vps26 and SNX and not with Vps35 as was believed previously. We thus conclude that sortilin may work as a transmembrane scaffold that links Glut4 to retromer (Figure 4F). Indeed, double expression of myc7-Glut4 and sortilin-myc/His in undifferentiated 3T3-L1 cells increases colocalization between Glut4 and Vps35 (Figure 5, top; compare line 9 to line 3 in Table 1), leads to redistribution of Glut4 from peripheral endosomes to the perinuclear compartment, and dramatically decreases colocalization of Glut4 with LysoTracker (Figure 5 bottom; compare lines 8 and 1 in Table 1).
FIGURE 5:
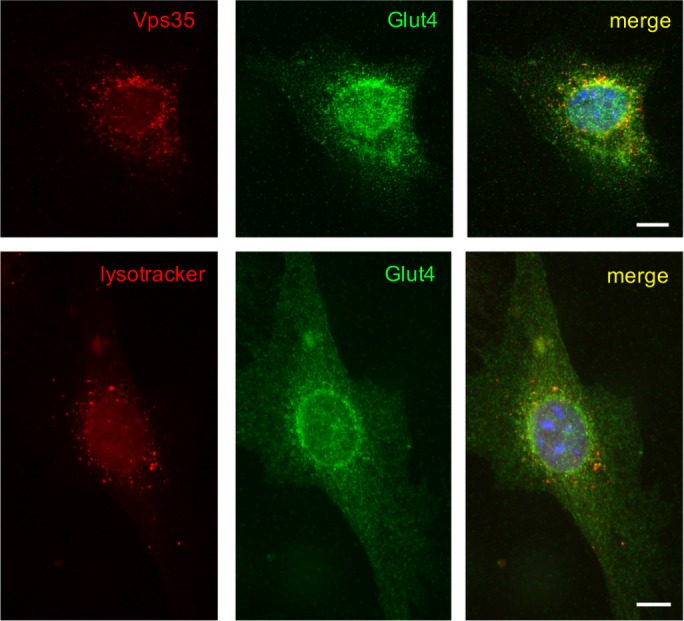
myc7-Glut4 is colocalized with Vps35 but not with LysoTracker in undifferentiated 3T3-L1 cells that express both myc7-Glut4 and sortilin-myc/His. Top: Cells were stained with goat polyclonal antibody against Vps35 and rabbit polyclonal antibody against Glut4, followed by Texas red–conjugated donkey anti-goat IgG and FITC-conjugated donkey anti-rabbit IgG. Bottom: Cells were incubated with LysoTracker, fixed, and stained with rabbit polyclonal antibody against Glut4, followed by FITC-conjugated donkey anti-rabbit IgG and analyzed by double immunofluorescence.
To estimate the functional significance of retromer in the intracellular trafficking of sortilin and Glut4, we used the clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9 system to knock out Vps35. We selected several single-cell colonies and a pooled clone of the “engineered” cells, and we used the latter for the following experiments. Figure 6A demonstrates that knockout of Vps35 decreases the expression of the two other retromer subunits, Vps29 and Vps26. Total levels of Glut4 and sortilin are also decreased. This effect can be attributed to the substantial reduction of the half-life of both proteins (Figure 6, B and C) likely due to their poor retrieval from endosomes. Because both Glut4 and sortilin are very stable proteins with t1/2 significantly >24 h, calculation of their actual half-lives in wild-type adipocytes is limited by prolonged use of inhibitors that may produce off-target effects. Knockout of Vps35 decreases t1/2 of Glut4 and sortilin to 8 ± 2 and 3 ± h, respectively, and incubation of “engineered” cells with bafilomycin A stabilizes Glut4 and sortilin and increases t1/2 of both to >20 h (Figure 6, B and C).
FIGURE 6:

Knockout of Vps35 in wild-type 3T3-L1 adipocytes decreases stability of sortilin and Glut4 in a bafilomycin A–sensitive manner. (A) total lysates (80 µg) of wild-type and CRISPR/Cas9-edited 3T3-L1 adipocytes were analyzed by Western blotting. (B) Half-life of sortilin and Glut4 was measured in wild-type and CRISPR/Cas9-edited 3T3-L1 adipocytes in the absence and presence of bafilomycin A as described in Materials and Methods. A representative result of three interdependent experiments. (C) Quantification of data shown in B. Each data point represents normalized mean value ± SE of three experiments.
Of note, it has been shown previously that knockdown of Vps26, Vps35, and SNX27 decreases intracellular levels of the Glut4 protein (Ma et al., 2014; Yang et al., 2016), and knockdown of SNX1 reduces the expression of sortilin (Mari et al., 2008). Our study suggests that these two events are functionally connected, that is, retromer may be responsible for retrieval of the sortilin–Glut4 complex.
Knockout of Vps35 does not affect insulin signaling (Figure 7A) but inhibits insulin-stimulated glucose uptake in adipocytes virtually completely (Figure 7B). We suggest that blocking the retrograde transport of Glut4 from endosomes to TGN may prevent Glut4 from entering the IRVs and hence translocation to the plasma membrane.
FIGURE 7:
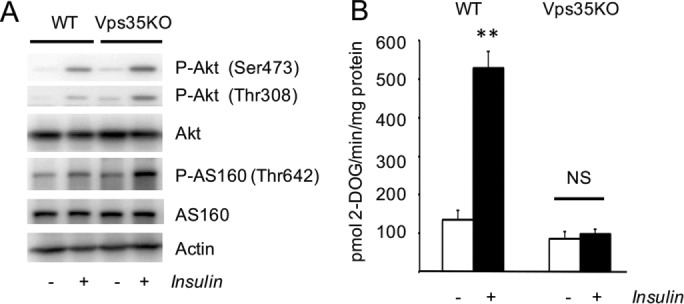
Knockout of Vps35 in 3T3-L1 adipocytes inhibits insulin-stimulated glucose uptake but not insulin signaling. (A) Total lysates (80 µg) of wild-type and CRISPR/Cas9-edited 3T3-L1 adipocytes treated and not treated with 100 nM insulin for 15 min were analyzed by Western blotting. (B) Insulin-stimulated glucose uptake was measured in wild-type and CRISPR/Cas9- edited 3T3-L1 adipocytes as described in Materials and Methods. **p < 0.01; NS, not significant. Results were analyzed by Student’s t test. A representative result of three independent experiments.
To test this idea, we separated an adipocyte extract by differential centrifugation into a heavy membrane fraction (P1), which includes endoplasmic reticulum, plasma membrane (PM), and large endosomes and TGN membranes, and a vesicular fraction (P2), which includes the IRVs and small transport vesicles (Figure 8A). In wild-type 3T3-L1 adipocytes, ∼20% of Glut4 and sortilin are localized in the vesicular fraction (Figure 8B), whereas most syntaxin 6 (TGN marker) and caveolin 1 (PM marker) are recovered in heavy membranes. Knockout of Vps35 eliminates the presence of Glut4 and sortilin in small transport vesicles presumably by blocking exit of both proteins from endosomes. In agreement with this model, the amount of Glut4 in the heavy membrane fraction is proportionally increased (Figure 8B). Immunofluorescence staining also shows that knockout of Vps35 leads to redistribution of Glut4 from the perinuclear TGN compartment in wild-type 3T3-L1 adipocytes to peripheral endosomes/lysosomes (compare Figure 8C, top and bottom) and increases colocalization of Glut4 with LysoTracker (compare lines 10 and 12 in Table 1). At the same time, neither total expression levels nor the biochemical distributions of syntaxin 6 and caveolin 1 are altered in Vps35-knockout cells (Figure 8A).
FIGURE 8:

Knockout of Vps35 in 3T3-L1 adipocytes inhibits formation of small transport vesicles containing sortilin and Glut4 and blocks their delivery from peripheral endosomes to the perinuclear compartment. (A) Total lysates (L) prepared from wild-type and CRISPR/Cas9-edited 3T3-L1 adipocytes were fractionated into heavy membrane pellet (P1) and vesicular fractions (P2), which were analyzed by Western blotting (20 µg/lane). (B) Relative distribution of Glut4 and sortilin between fractions. Presence of each protein in total lysates (L) of wild-type and CRISPR/Cas9-edited 3T3-L1 adipocytes is 100%. **p < 0.01; *p < 0.05. Results were analyzed by Student’s t test. A representative result of three independent experiments. (C) Wild- type (top) and CRISPR/Cas9-edited (bottom) 3T3-L1 adipocytes were incubated with LysoTracker, fixed, and stained with rabbit polyclonal antibody against Glut4, followed by FITC-conjugated donkey anti-rabbit IgG, and analyzed by double immunofluorescence.
DISCUSSION
Sortilin and MPR represent multiligand sorting receptors that are known to be involved in anterograde transport of lysosomal enzymes and substrates (Bonifacino and Rojas, 2006; Burd, 2011; Coutinho et al., 2012). It is usually believed, within the framework of this model, that both sortilin and MPR carry their ligands from the TGN to endosomes and are retrieved back to TGN “empty handed”—that is, not carrying any ligand. The central finding of our study is that sortilin transports Glut4 in the opposite, retrograde, direction, thus rerouting the transporter from the degradative to the recycling pathway. In other words, sortilin may target one set of ligands from the TGN to endosomes and a different set of ligands (in our experiments, Glut4) back from endosomes to the TGN. Another substantial finding is that interaction of sortilin with retromer plays a central role in the retrograde traffic of Glut4.
In undifferentiated 3T3-L1 preadipocytes, which express retromer but not sortilin, ectopically expressed Glut4 has a short half-life (Shi and Kandror, 2005; Liu et al., 2007) and is found largely in endosomes/lysosomes (Figure 1). Overexpression of sortilin together with Glut4 in undifferentiated cells stabilizes Glut4 and reroutes it to the recycling pathway (Figure 5; Shi and Kandror, 2005). On the contrary, knockdown of sortilin or retromer decreases the half-life of Glut4 and blocks its delivery to the insulin-responsive vesicular compartment. Mechanistically, sortilin binds to the first luminal loop of Glut4 via its luminal Vps10p domain, thus recruiting Glut4 to the retromer-dependent retrograde trafficking machinery.
In a recent report, Yang et al. (2016) suggested that retromer in conjunction with SNX27 mediates insulin-dependent translocation of Glut4 to the plasma membrane. Although we cannot exclude this possibility, we did not detect significant translocation of retromer to the plasma membrane in response to insulin stimulation (unpublished data). Thus we favor a model according to which retromer delivers the sortilin/Glut4 complex from endosomes to the TGN, where the IRVs are formed.
Previously we showed that sortilin plays an important role not only in Glut4 retrieval from endosomes, as demonstrated here, but also in the process of IRV formation on donor TGN membranes (Huang et al., 2013). Of interest, the adaptor proteins that mediate formation of the IRVs are likely to be Golgi-localized, γ-ear-containing, Arf-binding proteins (GGAs; Li and Kandror, 2005). In other words, retromer is responsible for the formation of Glut4/sortilin-containing endosome-to-TGN transport carriers (see also Mari et al., 2008), whereas GGAs may drive the formation of the IRVs on the TGN.
We believe that our findings are physiologically significant because a strong reverse correlation between levels of sortilin expression and insulin resistance has been revealed by several research groups. Thus exposure of cultured cells to saturated fatty acids (Tsuchiya et al., 2010; Bi et al., 2013) or inhibitors of phosphoinositide 3-kinase and Akt (Li et al., 2015) decreases sortilin expression in vitro. In vivo, expression of sortilin is attenuated in ob/ob, db/db, and high fat diet–fed mice (Keller et al., 2008; Kaddai et al., 2009; Ai et al., 2012) and obese humans (Kaddai et al., 2009; Bi et al., 2013). By the same token, expression of Vps35 is decreased in mouse models of diabetes (Morabito et al., 2014), and a recent genome-wide association study identified a type 2 diabetes–related single-nucleotide polymorphism at the Vps26 locus (Kooner et al., 2011). Thus sortilin- and retromer-mediated Glut4 retrieval from endosomes may represent a “weak” step in the Glut4 pathway susceptible to the development of insulin resistance and diabetes.
MATERIALS AND METHODS
Reagents and antibodies
Insulin and other chemicals were obtained from Sigma-Aldrich (St, Louis, MO). Bovine serum and fetal bovine serum (FBS) were from Atlanta Biologicals (Lawrenceville, GA). DMEM, Opti-MEM, and Dulbecco’s phosphate-buffered saline (PBS) were purchased from Invitrogen (Carlsbad, CA). [3H]2-Deoxyglucose was purchased from PerkinElmer (Waltham, MA). Mouse monoclonal antibodies against the myc epitope and β-actin, rabbit polyclonal antibodies against cyclophilin A, Akt, phospho-Akt (Ser-473), phospho-Akt (Thr-308), and phospho-AS160 (Thr-642) were from Cell Signaling Technology (Danvers, MA). Rabbit polyclonal antibodies against the myc epitope, sortilin, Vps26, and Glut4 were from Abcam (Cambridge, MA), against Vps29 from Novus Biologicals (Littleton, CO), and against GST from Millipore (Billerica, MA). Mouse monoclonal antibody against sortilin, syntaxin 6, and SNX2 and rabbit polyclonal antibody against caveolin 1 were from BD Bioscience Pharmingen (San Diego, CA). Goat polyclonal antibody against Vps35 was from Imgenex (San Diego, CA). Chicken polyclonal antibody against the C-terminus of AS160/TBC1D4 (HPTNDKAKAGNKP), generated by Quality Controlled Biochemicals (Hopkinton, MA), was a kind gift of Michael Czech (University of Massachusetts Medical School). Horseradish peroxidase (HRP)–conjugated anti-mouse and anti-rabbit immunoglobulin G (IgG) were from Cell Signaling Technology. HRP-conjugated anti-goat IgG, fluorescein isothiocyanate (FITC)–conjugated donkey anti-rabbit antibody, Texas red–conjugated donkey anti-mouse antibody, Alexa Fluor 647–conjugated donkey anti-mouse antibody, Alexa Fluor 546–conjugated donkey anti-goat antibody, LysoTracker Red DND-99, ProLong Gold Antifade Mountant with DAPI, and Halt Protease and Phosphatase Inhibitor Cocktail were from Thermo Fisher Scientific (Waltham, MA).
Stable cell lines
Preparation, culturing, and differentiation of 3T3-L1 cells stably transfected with pBabe-myc7-Glut4, mLNCX2-sortilin-myc/His, and double transfected with pBabe-myc7-Glut4 and mLNCX2-sortilin-myc/His, as well as cells depleted of sortilin with the help of pBabe–short hairpin RNA, were described previously (Shi and Kandror, 2005). Cells were grown in DMEM containing 10% calf bovine serum. Two days after confluence, cells were transferred to the differentiation medium (DMEM with 10% FBS, 0.174 μM insulin, 1 μM dexamethasone, and 0.5 mM 3-isobutyl-1-methylxanthine). After 48 h, differentiation medium was replaced with DMEM containing 10% FBS.
Immunofluorescence
Undifferentiated and differentiated 3T3-L1 cells were grown on coverslips coated with collagen IV (Sigma-Aldrich). Serum-starved cells were fixed with 4% paraformaldehyde in PBS (pH 7.4) for 10 min and permeabilized with 0.2% Triton X-100 for 3 min at room temperature. After blocking with 5% donkey serum for 1 h, cells were stained overnight at 4°C with primary antibodies, followed by incubation with secondary antibody for 1 h at room temperature. In several experiments, cells before fixing were incubated with LysoTracker (0.4 μM) for 40 min at 37°C. Antifade solution was used for mounting cells on slides. Slides were examined with the help of the Axio Observer Z1 fluorescence microscope equipped with the Hamamatsu digital camera C10600/ORCA-R2 and AxioVision 4.8.1 program (Carl Zeiss, Thornwood, NY). Each scale bar is 5 μm. Each image shows a representative result of at least three independent experiments.
GST pull-down experiments with the C-terminus of sortilin
The 252–base pair oligonucleotide corresponding to the cytoplasmic C-terminus of human sortilin was amplified by PCR from the full-length sortilin cDNA (purchased from OriGene, Rockville, MD) using the forward primer 5′-CGGGGTACCAAACAGAATTCCAAGTCA-3′ and the reverse primer 5′-CGCGGATCCTATTCCAAGAGGTCCTCATC-3′ (underlined sequences represent the KpnI and BamHI restriction sites). Amplified sequence was cloned into the pGEM-T vector (Promega, Madison, WI). For bacterial expression, pET-42a vector (with the GST-His tag; Millipore) and pGEM-T vector with the sortilin C-terminus were digested with KpnI and BamHI. A digested fragment corresponding to the sortilin tail, as well as pET-42a vector, was gel purified, ligated and transformed into Escherichia coli BL21-CodonPlus (DE3)-RIPL strain (Agilent Technologies, Santa Clara, CA) according to manufacturer’s instructions. Individual colonies were grown overnight in 10 ml of Luria–Bertani broth (LB; 10 g/l tryptone, 5 g/l yeast extract, 10 g/l NaCl, pH 7.5). The overnight culture was added to fresh LB (1:100 vol) containing 1 mM of isopropyl β-d-1 thiogalactopyranoside, incubated at 37°C for 5 h at 250 rpm, and pelleted. The cell pellet was resuspended in PBS with protease inhibitor cocktail, sonicated three times for 45 s on ice, and centrifuged at 12,000 rpm for 10 min. The supernatant was then incubated with Glutathione Sepharose 4B beads (GE Healthcare LifeSciences, Pittsburgh, PA) at 4°C overnight on an orbital shaker. Beads were then washed three times with PBS. 3T3L1 adipocytes were serum starved for 4 h and treated with either insulin (100 nM) or carrier (5 µM HCl) for 15 min. Cells were washed and collected in PBS with protease inhibitor cocktail and homogenized with 10 strokes through the ball-bearing cell cracker (Isobiotec, Heidelberg, Germany). Homogenates were then centrifuged at 200,000 × g for 2 h. The supernatant was then incubated with GST-sortilin C-terminus- as well as control GST-glutathione beads overnight at 4°C on an orbital shaker. On the next day, beads were washed with PBS three times and eluted with Laemmli sample buffer, and eluted proteins were analyzed by Western blotting.
Binding of sortilin to the first luminal loop of Glut4
A myc-tagged (underlined) peptide corresponding to the first luminal loop of Glut4 EQKLISEEDLNAPQKVIEQSYNATWLGRQGPGGPSSIPPGTLTTLWA was synthesized by GeneScript (Piscataway, NJ). Wild-type and S 3T3-L1 preadipocytes were lysed in 30 mM NaCl, 10 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), 5% glycerol, 0.5% Triton X-100, and 10 mM imidazole with protease inhibitors at pH 7.4, and cell lysates were incubated with HisPur cobalt resin (Thermo Scientific, Grand Island, NY) for 90 min on an orbital shaker at 4°C. Beads were washed four times with washing buffer (50 mM Na2HPO4/NaH2PO4, 300 mM NaCl, and 20 mM imidazole) with pH 6.5 or 8 and incubated with the peptide (100 ng in 100 µl) in the washing buffer with the corresponding pH for 30 min at 4°C. After that, the beads were washed four times with the washing buffer at pH 6.5 or 8. The proteins were eluted with tricine sample buffer (Bio-Rad, Hercules, CA). Eluates were analyzed by electrophoresis in tricine gradient gels (10–20%), followed by Western blotting with anti-myc antibody.
Cross-linking and isolation of His-tagged proteins
Cross-linking was performed as previously described (Shi and Kandror, 2005) with minor modifications. Briefly, cells were washed twice with PBS and once with KRP buffer (12.5 mM HEPES, 120 mM NaCl, 6 mM KCl, 1.2 mM MgSO4, 1.0 mM CaCl2, 0.6 mM Na2HPO4, 0.4 mM NaH2PO4, 2.5 mM d-glucose, pH 7.4), and DSP was added to final concentration 2 mM for 30 min at room temperature. Then quenching buffer (50 mM Tris, 150 mM NaCl, pH 7.4) was added for 15 min at 4°C, followed by two washes with the same buffer. Cells were lysed in lysis buffer (10 mM HEPES, 30 mM NaCl, 5% glycerol, 10 mM imidazole, 0.5% Triton X-100, pH 7.4) with the protease inhibitor cocktail specialized for the isolation of His-tagged proteins (Thermo Fisher Scientific), and cell lysates were cleared by centrifugation at 16,000 × g for 20 min. The lysate was incubated with the HisPur cobalt resin at 4°C for 4 h with rotation and then washed four times with wash buffer (50 mM Na2HPO4/NaH2PO4, 300 mM NaCl, 20 mM imidazole, pH 7.4). Elution was carried out with elution buffer (50 mM Na2HPO4/NaH2PO4, 300 mM NaCl, 300 mM imidazole, pH 7.4) with 50 mM dithiothreitol at 37°C for 30 min, and eluted proteins were analyzed by Western blotting.
CRISPR/Cas9 genome editing
Genomic editing was performed in 3T3-L1 preadipocytes using the CRISPR/Cas9 system as previously described (Sanjana et al., 2014). Briefly, four single guide RNAs (sgRNAs) targeting exons 2–5 of the Vps35 gene (5′-ATGACTGAACCTTCACAGCC -3′, 5′-GAGCTCTCCAAGCATATTGG-3′, 5′-TGATGAACTGCACTACTTGG-3′, and 5′-GATTTGGTAGAAATGTGCCG-3′, respectively) were designed using online software (http://crispr.mit.edu; http://chopchop.cbu.uib.no). The lentiCRISPR v2 plasmid (52961; Addgene) was used to express sgRNAs and the Cas9 protein along with the envelope plasmid pCMV-VSV-G and the packaging plasmid psPAX2. Cells infected with lentiviruses were treated with puromycin (3 μg/ml) for the selection of stable transfectants, and single-cell colonies were isolated and grown in the medium with 3 μg/ml puromycin. The genomic region surrounding the sgRNA target site was amplified by PCR and sequenced. The primers used for the PCR were as follows: 2F, TCCTACCACAGGTTTCTTTGATGCC; 2R, TCAGGGCTCTTTGTTCCTGAGTCTC; 3F, TATAGCCCTGGTGAGGAGTTAGGC; 3R, ATCCATTTCAGAGGAGGCAGGGTG; 4F, CCATGGACCTCTCTCTGTGCCAGG; 4R, ATGTCTCTGCAGCTAAGTGCTGGGA; 5F, GGGACTGACCACTGAAATCAGTCA; and 5R, CAGTGAGTACCAGCCACTCAAAGCC.
Measurements of protein stability
Aqueous solutions of cycloheximide (50 μg/ml) or emetine (10 μM) with or without bafilomycin A (100 nM) were added to plates at time zero. At the indicated time intervals, the cells were rinsed three times with PBS and harvested in ice-cold RIPA buffer (Millipore) with the protease inhibitor cocktail. Cell lysates were vortexed, rotated at 4°C for 30 min, and spun for 20 min at 16,000 × g in a microcentrifuge at 4°C. The presence of the individual proteins in supernatants was analyzed by Western blotting.
[3H]2-deoxyglucose uptake
This assay was performed in six-well plates. Cells were washed three times with serum-free DMEM, starved for 4 h, washed twice with warm Krebs-Ringer-HEPES (KRH) buffer without glucose (121 mM NaCl, 4.9 mM KCl, 1.2 mM MgSO4, 0.33 mM CaCl2, 12 mM HEPES, pH 7.4), and treated with either 100 nM insulin or carrier (5 µM HCl) at 37°C for 15 min. Radioactive 2-deoxyglucose (0.1 mM, 0.625 μCi/ml) was added to cells for 5 min. The assay was terminated by aspirating the radioactive medium and the cells were washed three times with 2 ml of ice-cold KRH containing 25 mM d-glucose. Each well was then extracted with 400 μl of 0.1% SDS in KRH buffer without glucose, and 300-μl aliquots were removed for determination of radioactivity by liquid scintillation counting. Measurements were made in triplicate and corrected for specific activity and nonspecific diffusion (as determined in the presence of 5 μM cytochalasin B), which was <5% of the total uptake. The protein concentration was determined using the BCA Protein Assay Kit (Thermo Fisher Scientific) and used to normalize counts.
Subcellular fractionation of 3T3-L1 cells
3T3-Ll adipocytes or preadipocytes were washed three times with serum-free DMEM, warmed to 37°C, and starved in the same medium for 2 h. Cells were treated with 100 nM insulin or carrier (5 mM HCl at 1000 × dilution) in DMEM for 15 min at 37°C. Cells were then washed three times with cold HES buffer (250 mM sucrose, 20 mM HEPES, 1 mM EDTA, pH 7.4) with protease inhibitor cocktail and harvested in the same buffer (0.3–1 ml/10-cm dish). Homogenization was performed in a ball-bearing homogenizer (Isobiotec) with a 12-µm clearance by 10 strokes for adipocytes and 15 strokes for preadipocytes. Homogenates were centrifuged at 14,000 × g for 20 min (P1). Membrane vesicles in supernatants (P2) were concentrated by pelleting at 200,000 × g for 90 min. Pellets were resuspended in 0.1–0.25 ml of HES buffer.
Gel electrophoresis and Western blotting
Proteins were separated in SDS–polyacrylamide gels according to Laemmli (1970) and transferred to Immobilon-P membranes (Millipore). After transfer, the membrane was blocked with 5% BSA in PBS with 0.5% Tween 20 for 1 h. Blots were probed overnight with specific primary antibodies at 4°C, followed by 1-h incubation at room temperature with HRP-conjugated secondary antibodies. Protein bands were detected with the enhanced chemiluminescence substrate kit (PerkinElmer Life Sciences, Boston, MA) using a Bio-Rad ChemiDoc XRS+ System (Hercules, CA).
Acknowledgments
We thank Richard E. Fine and John Wells (Edith Nourse Rogers Memorial Veterans Hospital, Bedford, MA) for reagents and helpful discussions and Hira Chaudhry for help with several experiments. This work was supported by Research Grants DK52057 and DK107498 from the National Institutes of Health to K.V.K. X.P. and M.S. were supported by Institutional Training Grant 2T32DK007201 from the National Institutes of Health.
Abbreviations used:
- AS160
Akt substrate of 160 kDa
- Cas9
CRISPR associated protein 9
- CRISPR
clustered regularly interspaced short palindromic repeats
- DSP
dithiobis (succinimidyl propionate)
- FITC
fluorescein isothiocyanate
- GGA
Golgi-localized, gamma-ear-containing, ARF-binding protein
- Glut4
glucose transporter isoform 4
- GST
glutathione S-transferase
- HEPES
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- HRP
horseradish peroxidase
- KRH
Krebs-Ringer-HEPES
- KRP
Krebs-Ringer-phosphate
- PBS
Dulbecco’s phosphate-buffered saline
- PM
plasma membrane
- SNX
sorting nexin
- TGN
trans-Golgi network
- Vps10p
vacuolar protein sorting 10 protein
- Vps35
vacuolar protein sorting–associated protein 35.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E16-11-0777) on April 27, 2017.
REFERENCES
- Abel ED, Peroni O, Kim JK, Kim Y-B, Boss O, Hadro E, Minnemann T, Shulman GI, Kahn BB. Adipose-selective targeting of the GLUT4 gene impairs insulin action in muscle and liver. Nature. 2001;409:729–733. doi: 10.1038/35055575. [DOI] [PubMed] [Google Scholar]
- Ai D, Baez JM, Jiang H, Conlon DM, Hernandez-Ono A, Frank-Kamenetsky M, Milstein S, Fitzgerald K, Murphy AJ, Woo CW, et al. Activation of ER stress and mTORC1 suppresses hepatic sortilin-1 levels in obese mice. J Clin Invest. 2012;122:1677–1687. doi: 10.1172/JCI61248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ariga M, Nedachi T, Katagiri H, Kanzaki M. Functional role of sortilin in myogenesis and development of insulin-responsive glucose transport system in C2C12 myocytes. J Biol Chem. 2008;283:10208–10220. doi: 10.1074/jbc.M710604200. [DOI] [PubMed] [Google Scholar]
- Atkinson BJ, Griesel BA, King CD, Josey MA, Olson AL. Moderate GLUT4 overexpression improves insulin sensitivity and fasting triglyceridemia in high-fat diet-fed transgenic mice. Diabetes. 2013;62:2249–2258. doi: 10.2337/db12-1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bi L, Chiang JY, Ding WX, Dunn W, Roberts B, Li T. Saturated fatty acids activate ERK signaling to downregulate hepatic sortilin 1 in obese and diabetic mice. J Lipid Res. 2013;54:2754–2762. doi: 10.1194/jlr.M039347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogan JS. Regulation of glucose transporter translocation in health and diabetes. Annu Rev Biochem. 2012;81:507–532. doi: 10.1146/annurev-biochem-060109-094246. [DOI] [PubMed] [Google Scholar]
- Bogan JS, Kandror KV. Biogenesis and regulation of insulin-responsive vesicles containing GLUT4. Curr Opin Cell Biol. 2010;22:506–512. doi: 10.1016/j.ceb.2010.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonifacino JS, Rojas R. Retrograde transport from endosomes to the trans-Golgi network. Nat Rev Mol Cell Biol. 2006;7:568–579. doi: 10.1038/nrm1985. [DOI] [PubMed] [Google Scholar]
- Burd CG. Physiology and pathology of endosome-to-Golgi retrograde sorting. Traffic. 2011;12:948–955. doi: 10.1111/j.1600-0854.2011.01188.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canuel M, Lefrancois S, Zeng J, Morales CR. AP-1 and retromer play opposite roles in the trafficking of sortilin between the Golgi apparatus and the lysosomes. Biochem Biophys Res Commun. 2008;366:724–730. doi: 10.1016/j.bbrc.2007.12.015. [DOI] [PubMed] [Google Scholar]
- Charron MJ, Katz EB, Olson AL. GLUT4 gene regulation and manipulation. J Biol Chem. 1999;274:3253–3256. doi: 10.1074/jbc.274.6.3253. [DOI] [PubMed] [Google Scholar]
- Coutinho MF, Prata MJ, Alves S. A shortcut to the lysosome: the mannose-6-phosphate-independent pathway. Mol Genet Metab. 2012;107:257–266. doi: 10.1016/j.ymgme.2012.07.012. [DOI] [PubMed] [Google Scholar]
- Esk C, Chen CY, Johannes L, Brodsky FM. The clathrin heavy chain isoform CHC22 functions in a novel endosomal sorting step. J Cell Biol. 2010;188:131–144. doi: 10.1083/jcb.200908057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster LJ, Li D, Randhawa VK, Klip A. Insulin accelerates inter-endosomal glut4 traffic via phosphatidylinositol 3-kinase and protein kinase B. J Biol Chem. 2001;276:44212–44221. doi: 10.1074/jbc.M102964200. [DOI] [PubMed] [Google Scholar]
- Graham TE, Kahn BB. Tissue-specific alterations of glucose transport and molecular mechanisms of intertissue communication in obesity and type 2 diabetes. Horm Metab Res. 2007;39:717–721. doi: 10.1055/s-2007-985879. [DOI] [PubMed] [Google Scholar]
- Hatakeyama H, Kanzaki M. Molecular basis of insulin-responsive GLUT4 trafficking systems revealed by single molecule imaging. Traffic. 2011;12:1805–1820. doi: 10.1111/j.1600-0854.2011.01279.x. [DOI] [PubMed] [Google Scholar]
- Herman MA, Peroni OD, Villoria J, Schon MR, Abumrad NA, Bluher M, Klein S, Kahn BB. A novel ChREBP isoform in adipose tissue regulates systemic glucose metabolism. Nature. 2012;484:333–338. doi: 10.1038/nature10986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang G, Buckler-Pena D, Nauta T, Singh M, Asmar A, Shi J, Kim JY, Kandror KV. Insulin responsiveness of glucose transporter 4 in 3T3-L1 cells depends on the presence of sortilin. Mol Biol Cell. 2013;24:3115–3122. doi: 10.1091/mbc.E12-10-0765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang S, Czech MP. The GLUT4 glucose transporter. Cell Metab. 2007;5:237–252. doi: 10.1016/j.cmet.2007.03.006. [DOI] [PubMed] [Google Scholar]
- Kaddai V, Jager J, Gonzalez T, Najem-Lendom R, Bonnafous S, Tran A, Le Marchand-Brustel Y, Gual P, Tanti JF, Cormont M. Involvement of TNF-alpha in abnormal adipocyte and muscle sortilin expression in obese mice and humans. Diabetologia. 2009;52:932–940. doi: 10.1007/s00125-009-1273-3. [DOI] [PubMed] [Google Scholar]
- Keller MP, Choi Y, Wang P, Davis DB, Rabaglia ME, Oler AT, Stapleton DS, Argmann C, Schueler KL, Edwards S, et al. A gene expression network model of type 2 diabetes links cell cycle regulation in islets with diabetes susceptibility. Genome Res. 2008;18:706–716. doi: 10.1101/gr.074914.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Kandror KV. The first luminal loop confers insulin responsiveness to the glucose transporter 4. Mol Biol Cell. 2012;23:910–917. doi: 10.1091/mbc.E11-10-0839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kooner JS, Saleheen D, Sim X, Sehmi J, Zhang W, Frossard P, Been LF, Chia KS, Dimas AS, Hassanali N, et al. Genome-wide association study in individuals of South Asian ancestry identifies six new type 2 diabetes susceptibility loci. Nat Genet. 2011;43:984–989. doi: 10.1038/ng.921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- Li J, Matye DJ, Li T. Insulin resistance induces posttranslational hepatic sortilin 1 degradation in mice. J Biol Chem. 2015;290:11526–11536. doi: 10.1074/jbc.M115.641225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li LV, Kandror KV. Golgi-localized, gamma-ear-containing, Arf-binding protein adaptors mediate insulin-responsive trafficking of glucose transporter 4 in 3T3-L1 adipocytes. Mol Endocrinol. 2005;19:2145–2153. doi: 10.1210/me.2005-0032. [DOI] [PubMed] [Google Scholar]
- Liu LB, Omata W, Kojima I, Shibata H. The SUMO conjugating enzyme Ubc9 is a regulator of GLUT4 turnover and targeting to the insulin-responsive storage compartment in 3T3-L1 adipocytes. Diabetes. 2007;56:1977–1985. doi: 10.2337/db06-1100. [DOI] [PubMed] [Google Scholar]
- Lucas M, Gershlick DC, Vidaurrazaga A, Rojas AL, Bonifacino JS, Hierro A. Structural mechanism for cargo recognition by the retromer complex. Cell. 2016;167:1623–1635 e14. doi: 10.1016/j.cell.2016.10.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma J, Nakagawa Y, Kojima I, Shibata H. Prolonged insulin stimulation down-regulates GLUT4 through oxidative stress-mediated retromer inhibition by a protein kinase CK2-dependent mechanism in 3T3-L1 adipocytes. J Biol Chem. 2014;289:133–142. doi: 10.1074/jbc.M113.533240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mari M, Bujny MV, Zeuschner D, Geerts WJ, Griffith J, Petersen CM, Cullen PJ, Klumperman J, Geuze HJ. SNX1 defines an early endosomal recycling exit for sortilin and mannose 6-phosphate receptors. Traffic. 2008;9:380–393. doi: 10.1111/j.1600-0854.2007.00686.x. [DOI] [PubMed] [Google Scholar]
- Minokoshi Y, Kahn CR, Kahn BB. Tissue-specific ablation of the GLUT4 glucose transporter or the insulin receptor challenges assumptions about insulin action and glucose homeostasis. J Biol Chem. 2003;278:33609–33612. doi: 10.1074/jbc.R300019200. [DOI] [PubMed] [Google Scholar]
- Morabito MV, Berman DE, Schneider RT, Zhang Y, Leibel RL, Small SA. Hyperleucinemia causes hippocampal retromer deficiency linking diabetes to Alzheimer’s disease. Neurobiol Dis. 2014;65:188–192. doi: 10.1016/j.nbd.2013.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanjana NE, Shalem O, Zhang F. Improved vectors and genome-wide libraries for CRISPR screening. Nat Methods. 2014;11:783–784. doi: 10.1038/nmeth.3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sargeant RJ, Paquet MR. Effect of insulin on the rates of synthesis and degradation of GLUT1 and GLUT4 glucose transporters in 3T3-L1 adipocytes. Biochem J. 1993;290:913–919. doi: 10.1042/bj2900913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seaman MN. Cargo-selective endosomal sorting for retrieval to the Golgi requires retromer. J Cell Biol. 2004;165:111–122. doi: 10.1083/jcb.200312034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seaman MN. Identification of a novel conserved sorting motif required for retromer-mediated endosome-to-TGN retrieval. J Cell Sci. 2007;120:2378–2389. doi: 10.1242/jcs.009654. [DOI] [PubMed] [Google Scholar]
- Seaman MN. The retromer complex—endosomal protein recycling and beyond. J Cell Sci. 2012;125:4693–4702. doi: 10.1242/jcs.103440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepherd PR, Kahn BB. Glucose transporters and insulin action. Implications for insulin resistance and diabetes mellitus. N Engl J Med. 1999;341:248–257. doi: 10.1056/NEJM199907223410406. [DOI] [PubMed] [Google Scholar]
- Shewan AM, van Dam EM, Martin S, Luen TB, Hong W, Bryant NJ, James DE. GLUT4 recycles via a trans-Golgi network (TGN) subdomain enriched in Syntaxins 6 and 16 but not TGN38: involvement of an acidic targeting motif. Mol Biol Cell. 2003;14:973–986. doi: 10.1091/mbc.E02-06-0315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J, Kandror KV. Sortilin is essential and sufficient for the formation of Glut4-storage vesicles in 3T3-L1 adipocytes. Dev Cell. 2005;9:99–108. doi: 10.1016/j.devcel.2005.04.004. [DOI] [PubMed] [Google Scholar]
- Shulman GI. Cellular mechanisms of insulin resistance. J Clin Invest. 2000;106:171–176. doi: 10.1172/JCI10583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sivitz WI, DeSautel SL, Kayano T, Bell GI, Pessin JE. Regulation of glucose transporter messenger RNA in insulin-deficient states. Nature. 1989;340:72–74. doi: 10.1038/340072a0. [DOI] [PubMed] [Google Scholar]
- Slot SW, Geuze HJ, Gigengack S, Lienhard GE, James DE. Immuno-localization of the insulin-regulatable glucose transporter in brown adipose tissue of the rat. J Cell Biol. 1991;113:123–135. doi: 10.1083/jcb.113.1.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuchiya Y, Hatakeyama H, Emoto N, Wagatsuma F, Matsushita S, Kanzaki M. Palmitate-induced down-regulation of sortilin and impaired GLUT4 trafficking in C2C12 myotubes. J Biol Chem. 2010;285:34371–34381. doi: 10.1074/jbc.M110.128520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassilopoulos S, Esk C, Hoshino S, Funke BH, Chen CY, Plocik AM, Wright WE, Kucherlapati R, Brodsky FM. A role for the CHC22 clathrin heavy-chain isoform in human glucose metabolism. Science. 2009;324:1192–1196. doi: 10.1126/science.1171529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Hong LK, Follett J, Wabitsch M, Hamilton NA, Collins BM, Bugarcic A, Teasdale RD. Functional characterization of retromer in GLUT4 storage vesicle formation and adipocyte differentiation. FASEB J. 2016;30:1037–1050. doi: 10.1096/fj.15-274704. [DOI] [PubMed] [Google Scholar]