

Abstract
Because the majority of environmental bacteria are not easily culturable, access to many bacterially encoded secondary metabolites will be dependent on the development of improved functional metagenomic screening methods. In this study, we examined a collection of diverse Streptomyces species for the best innate ability to heterologously express biosynthetic gene clusters. We then optimized methods for constructing high quality meta-genomic cosmid libraries in the best Streptomyces host. An initial screen of a 1.5 million-membered metagenomic library constructed in Streptomyces albus, the species that exhibited the highest propensity for heterologous expression of gene clusters, led to the identification of the novel natural product metatricycloene (1). Metatricy-cloene is a tricyclic polyene encoded by a reductive, iterative polyketide-like gene cluster. Related gene clusters found in sequenced genomes appear to encode a largely unexplored collection of structurally diverse, polyene-based metabolites.
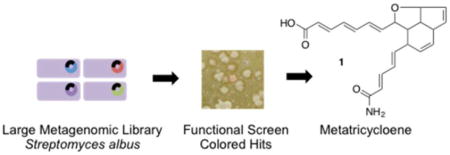
Graphical abstract
Cultured bacteria have historically been a very rich source of natural products with diverse structures and biological activities, despite making up as little as 1% of environmental microbiomes.1 Sequencing efforts aimed at cataloging the biosynthetic potential of the uncultured majority have revealed the existence of large numbers of unexplored natural product gene clusters in most environmental microbiomes.2 Functional metagenomics3 offers a means of investigating natural products encoded by uncultured bacteria. In these studies, DNA extracted directly from an environmental sample (environmental DNA, eDNA) is cloned into an easily cultured bacterium. The resulting clones are screened for phenotypes associated with the production of small molecules (e.g., color, antibacterial activity, cytotoxicity).4,5
A prerequisite for identifying metabolites using functional metagenomics is the successful heterologous expression of eDNA-derived gene clusters in a host bacterium. Despite the fact that it is known to be a poor heterologous expression host, Escherichia coli has been used for the vast majority of such studies. Only a handful of functional metagenomic studies have used hosts other the E. coli.6–8 As with E. coli, the hosts used in these studies were primarily selected for ease of growth and genetic tractability. An ideal host for functional metagenomics, on the other hand, would display high natural propensity to support the heterologous expression of diverse secondary metabolite gene clusters as well as high transformation frequency to allow for the facile construction of large metagenomic libraries.
Because the ability to heterologously express gene clusters obtained by horizontal gene transfer could provide a selective advantage to some environmental bacteria, we hypothesized that (1) naturally privileged heterologous expression strains exist in the global microbiome and (2) functional metagenomic discovery efforts would benefit from identifying these gifted strains. Here, we report on improving functional metagenomics screening through the identification of natively privileged heterologous hosts and optimization of large metagenomic library construction methods (Figure 1). Implementation of these improvements has led to the discovery of carotenoids and the biosynthetically and structurally interesting polyene, metatricycloene (1) (Figure 2). The value of these improvements is highlighted by the fact that almost 20 years after the term metagenomics was first coined,3 metatricycloene is one of the first biosynthetically complex natural products to be identified using functional metagenomic screening methods.4
Figure 1.

Improving functional metagenomics. Libraries are made in E. coli using a two-step process that removes small clones. High quality libraries are then conjugated into privileged expression hosts (S. albus) for phenotypic screening.
Figure 2.

Metatricycloene (1) structure and gene cluster.
Considering their history as prolific producers of diverse, therapeutically relevant metabolites, Streptomyces are an appealing source of heterologous expression hosts.9 Nevertheless, previous attempts at functional metagenomic screening in Streptomyces, which have been conducted using exclusively Streptomyces lividans as a host, have largely been unsuccessful at identifying secondary metabolites.10–14
Genetically engineered strains of Streptomyces have been used to improve heterologous expression of individual gene clusters.15,16 While these strains represent potential hosts for large-scale metagenomic library screening, we believe that the first step to developing the eventual best hosts is the identification of natively privileged heterologous expression strains. This would then be followed by the genetic engineering of these naturally privileged strains into even better hosts. In an effort to identify natively privileged Streptomyces species that would be more productive hosts for large metagenomic library screening efforts, we surveyed a collection of 39 Streptomyces strains for conjugation and heterologous expression efficiencies (Figure 3). These strains represent a phylogenetically diverse sampling of both commonly used and lesser-known species.
Figure 3.

Identifying Streptomyces species for use in functional metagenomics. Phylogenetically diverse Streptomyces species were tested for conjugation and heterologous expression efficiency using a set of metagenomic clones containing minimal PKS genes. Exconjugants from highly transforming strains were plated on rich media to look for color phenotypes. Broth extracts were examined by HPLC to identify species inducing the most heterologous expression events.
Target strains were interrogated for heterologous expression efficiency using a collection of 97 eDNA cosmid clones containing minimal type II polyketide synthases (PKS).17 We used type II PKS gene clusters in this study because a large number of the final metabolites and shunt products produced by type II PKS biosynthesis are colored,18 allowing for the easy visual identification of colonies heterologously expressing natural products. Colored phenotypes were observed with only four species, highlighting the inherent challenges of heterologous expression. S. lividans, S. coerulescens, and S. viridochromogenes each yielded one colored hit. S. albus yielded a total of 9 hits: 6 unique hits from phenotype screening and 3 from the subsequent examination of fermentation broths. Due to its superior heterologous expression capabilities and reasonable conjugation efficiency (Table S1), S. albus was selected for use as a metagenomic library host.
E. coli remains the most efficient host to use for constructing cosmid-based libraries from crude eDNA. For functional metagenomic screening using other hosts, libraries are first constructed in E. coli and then shuttled into a target expression host. In our experience, all E. coli-based eDNA cosmid libraries contain some empty vector and small-insert clone contamination. The appearance of small-insert clones in a large E. coli-based eDNA library is likely inevitable, because there will always be some clones that are not stable and are truncated during library propagation. Since small clones conjugate at up to 10,000-fold higher efficiencies than full length cosmids,19 even a tiny amount of small clone contamination (<0.1–0.01%) will come to dominate a library following conjugation from E. coli into the expression host. We developed a two-step library construction protocol to address this issue (Figure 1). Libraries are first constructed and propagated in E. coli to allow for the truncation of any unstable clones. Cosmid DNA is then isolated in bulk from the primary library and size selected by gel electrophoresis. Following the removal of smaller clones, the collection of full-length stable cosmids is electroporated back into E. coli, and the resulting high-quality library is mated into the desired expression host.
For this study, we constructed a 1.5 million membered DNA cosmid library from Texas desert soil, using standard eDNA cosmid cloning methods.20 This library was cleaned of small clones and conjugated into S. albus using mating conditions that we extensively optimized for shuttling large (>1 × 106 membered) metagenomic cosmid libraries into S. albus (Figure S1A–D). The quality of our final S. albus library was verified by restriction mapping of exconjugants. All clones we examined contained large insert cosmids with unique restriction maps (Figure S1E,F). This library is almost 2 orders of magnitude larger than any previously reported metagenomic library hosted in Streptomyces.12
The production of color by bacterial cultures is often an indication of small molecule biosynthesis. Color can therefore be used as a simple assay for identifying eDNA clones containing natural product biosynthetic gene clusters.6,7 Though examining clones with colored phenotypes will undoubtedly identify known classes of metabolites, since color has been the target of many previous natural product isolation studies, it also provides the opportunity to identify novel metabolites from the rarely explored biosynthetic diversity present in soil metagenomes. As an initial exploration of the natural product-encoding potential of this S. albus-based metagenomic library, we screened exconjugants directly from library mating plates for colored phenotypes. We picked 48 of the most deeply tinted colonies that appeared on these plates for further analysis. Upon retransformation of the cosmids isolated from these colored clones, 16 clones reproduced a color phenotype in S. albus. Dereplication by restriction mapping revealed 12 unique eDNA clones that displayed either yellow or orange (6), red (2), or brown (4) phenotypes (Figure S2). Each clone was fully sequenced, revealing that the eight yellow, orange or red clones contained carotenoid biosynthesis gene clusters with an assortment of tailoring genes (Figure S3). Based on LCMS-UV analysis of the acetone extracts of cell pellets and comparisons to commercial standards, the carotenoids produced by this set of clones were found to include lycopene, β-carotene, rhodopsin, dihydroxylycopene, and three more highly oxidized carotenoids (Table S3). We did not investigate these clones in more detail because carotenoids have been widely explored in culture-based studies. It is worth noting that, apart from β-carotene, none of the carotenoids have been reported in functional metagenomic screens using other hosts.6
LCMS analysis of organic extracts from cultures of one of the brown colored clones (M13) revealed a single, major, clone-specific peak (Figure 4). This metabolite was purified (1.1 mg/ L) from large-scale cultures and its structure was determined using 1 and 2D NMR as well as HRMS data. The final structure of (1) consists of a tricyclic core appended with two polyene substituents, one terminating in a carboxylic acid and the other terminating in an amide. We have named this metabolite metatricycloene. While metatricycloene did not show cytotoxicity against any bacteria, fungi, or human cells we tested, it was of interest to us because its structure does not closely resemble that of any described natural product.
Figure 4.

HPLC UV (270 nm) traces of culture broth extracts from M13 and M13 oxidase gene knockout mutants.
Approximately one-third of the open reading frames present on M13 have their closest relative in the genome of a Kibdelosporangium species (Table S2).21 The NCBI database contains only a small number of unique 16S rRNA sequences from cultured Kibdelosporangium strains. While a number of natural products have been identified from bacteria in this genus, it remains a rarely cultured Actinobacterial genera.21
Bioinformatic analysis of the open reading frames on M13 revealed a collection of polyketide/fatty acid synthesis-like genes (Figure 2, Table S2). This includes a gene predicted to encode a rarely described didomain ketosynthase (KS:KS*) comprised of two FabF-like domains (MtcI), an acyl carrier protein (ACP) (MtcF), malonyl-CoA-ACP transacylase (AT) (MtcD), a 3-ketoacyl-ACP ketoreductase (KR) (MtcE), and an acyl-ACP dehydratase (DH) (MtcG). Based on the genes present on M13 and the structure of metatricycloene, we proposed the biosynthetic scheme shown in Figure 5.
Figure 5.

Proposed biosynthetic scheme for (1). Dashed bonds indicate potential locations for MtcA oxidation.
In our proposal, the terminal amide arises from a malonyl starter unit that is modified by the predicted amidotransferase MtcK. MtcK is 66% identical to OxyD, which carries out the same reaction in oxytetracycline biosynthesis. The ACP and KS:KS* proteins are predicted to catalyze 10 rounds of chain elongation using malonyl CoA. Only the first KS domain in MtcI contains a complete complement of catalytic residues. This KS domain is therefore predicted to be responsible for chain elongation. The second KS domain (KS*) is missing a conserved active site histidine (H303, Figure S6), and we therefore believe it functions as a chain length factor (CLF). Contrary to what is seen in type II aromatic polyketide biosynthesis where three distinct proteins (ACP, KS, and CLF) are used to generate a nonreduced polyacetate, the polyketide precursor in metatricycloene biosynthesis is reduced to a polyene by predicted ketoreductase (MtcE) and dehydratase (MtcG) enzymes (Figure 5). In our scheme, the polyene is then oxidized and undergoes one C–O and two C–C cyclizations to give the tricyclic ring system.
To better understand the biosynthesis of metatricycloene, we saturated the M13 cosmid with transposons. Cosmids containing transposons in genes predicted to encode three different oxidation enzymes were individually transformed back into S. albus and assayed for the ability to encode the production of metatricyloene. Only in the case of the FAD-linked oxidase knockout (MtcA) was 1 no longer observed in culture broth extracts (Figure 4). It remains to be seen whether this oxidase is solely involved in the oxidation of metatricycloene or whether it also “guides” the cyclization of 1, as metatricycloene is the only major clone specific metabolite seen in M13 cultures.
Metatricycloene biosynthesis is related to that of the aryl polyenes.22 Aryl polyenes, however, arise from the use of distinct KS and CLF proteins, similar to what is seen in aromatic PKS biosynthesis. Reductive biosynthetic systems using KS:KS*-like didomain proteins have been characterized from culture-based bacterial screening efforts in only a few instances. Two examples are granadaene (2) and andrimid (3) (Figure 6B).23,24 In granadaene biosynthesis, the polyene is decorated with ornithine and the deoxy sugar rhamnose. In andrimid biosynthesis, the polyene is incorporated into a hybrid nonribosomal peptide/polyketide.
Figure 6.

(A) Phylogenetic tree of KS:KS* proteins. Representative gene clusters associated with KS:KS* genes from different clades. Striped clades contain clusters that do not have complete sets (KS:KS*, DH, and KR) of poylene encoding genes. (B) Characterized molecules that use didomain ketosynthase proteins.
A search of sequenced genomes for KS:KS* didomain homologues revealed a number of KS:KS* genes that are adjacent to DH, KR, and ACP genes, as well as unique sets of genes potentially encoding tailoring enzymes. KS:KS* genes group by phylogenetic origin and are often found in species that have rarely been explored for the production of bioactive secondary metabolites, such as rare Actinobacteria and Firmicutes. The diverse collection of potential tailoring genes found around KS:KS* homologues suggests that they may be involved in the biosynthesis of overlooked polyene-based natural products (Figure 6A). For example, within the KS:KS* clade in which MtcI falls, there are multiple gene clusters that appear to encode different tailoring enzymes including diverse oxidases. Due to the underexplored nature of this reductive class of polyketides, the KS:KS* gene should be a productive probe for identifying gene clusters that encode novel natural products in sequenced genomes and metagenomes.
Although the limitations of S. albus as a metagenomic screening host remain to be seen, our discovery of both isoprenoid (carotenoid) and polyketide (metatricycloene) encoding gene clusters suggests that it will prove useful for identifying gene clusters that are more biosynthetically diverse than the type II PKS clusters that we used for strain selection. The identification of metatricycloene and multiple carotenoid biosynthetic gene clusters are rare examples of identifying complex biosynthetic gene clusters using functional metage-nomics.4 Based on the work presented here, a larger-scale exploration of bacteria for gifted heterologous expressers, coupled with genome engineering of naturally privileged heterologous expression hosts and larger-insert eDNA cloning methods should help to make functional metagenomics a more routine tool for identifying novel natural products.
Supplementary Material
Acknowledgments
We thank Carolina Adura Alcaino and HTSRC for their help. This work was supported by Grant U01 GM110714.
Footnotes
The authors declare no competing financial interest.
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.6b02921.
Additional experimental details, NMR data, gene table, TE knockout data, and colony images (PDF)
References
- 1.Torsvik V, Goksoyr J, Daae FL. Appl Environ Microbiol. 1990;56:782. doi: 10.1128/aem.56.3.782-787.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Charlop-Powers Z, Owen JG, Reddy BV, Ternei MA, Brady SF. Proc Natl Acad Sci U S A. 2014;111:3757. doi: 10.1073/pnas.1318021111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Handelsman J, Rondon MR, Brady SF, Clardy J, Goodman RM. Chem Biol. 1998;5:R245. doi: 10.1016/s1074-5521(98)90108-9. [DOI] [PubMed] [Google Scholar]
- 4.Iqbal HA, Feng Z, Brady SF. Curr Opin Chem Biol. 2012;16:109. doi: 10.1016/j.cbpa.2012.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Banik JJ, Brady SF. Curr Opin Microbiol. 2010;13:603. doi: 10.1016/j.mib.2010.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Craig JW, Chang FY, Brady SF. ACS Chem Biol. 2009;4:23. doi: 10.1021/cb8002754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Craig JW, Chang FY, Kim JH, Obiajulu SC, Brady SF. Appl Environ Microbiol. 2010;76:1633. doi: 10.1128/AEM.02169-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Martinez A, Kolvek SJ, Yip CL, Hopke J, Brown KA, MacNeil IA, Osburne MS. Appl Environ Microbiol. 2004;70:2452. doi: 10.1128/AEM.70.4.2452-2463.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.de Lima Procopio RE, da Silva IR, Martins MK, de Azevedo JL, de Araujo JM. Braz J Infect Dis. 2012;16:466. doi: 10.1016/j.bjid.2012.08.014. [DOI] [PubMed] [Google Scholar]
- 10.Lussier FX, Chambenoit O, Cote A, Hupe JF, Denis F, Juteau P, Beaudet R, Shareck F. J Ind Microbiol Biotechnol. 2011;38:1321. doi: 10.1007/s10295-010-0915-2. [DOI] [PubMed] [Google Scholar]
- 11.Martinez A, Kolvek SJ, Hopke J, Tiong Yip CL, Osburne MS. Appl Environ Microbiol. 2005;71:1638. doi: 10.1128/AEM.71.3.1638-1641.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McMahon MD, Guan C, Handelsman J, Thomas MG. Appl Environ Microbiol. 2012;78:3622. doi: 10.1128/AEM.00044-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang GY, Graziani E, Waters B, Pan W, Li X, McDermott J, Meurer G, Saxena G, Andersen RJ, Davies J. Org Lett. 2000;2:2401. doi: 10.1021/ol005860z. [DOI] [PubMed] [Google Scholar]
- 14.Courtois S, Cappellano CM, Ball M, Francou FX, Normand P, Helynck G, Martinez A, Kolvek SJ, Hopke J, Osburne MS, et al. Appl Environ Microbiol. 2003;69:49. doi: 10.1128/AEM.69.1.49-55.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gomez-Escribano JP, Bibb MJ. Microb Biotechnol. 2011;4:207. doi: 10.1111/j.1751-7915.2010.00219.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Komatsu M, Uchiyama T, Omura S, Cane DE, Ikeda H. Proc Natl Acad Sci U S A. 2010;107:2646. doi: 10.1073/pnas.0914833107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Feng Z, Kallifidas D, Brady SF. Proc Natl Acad Sci U S A. 2011;108:12629. doi: 10.1073/pnas.1103921108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Staunton J, Weissman KJ. Nat Prod Rep. 2001;18:380. doi: 10.1039/a909079g. [DOI] [PubMed] [Google Scholar]
- 19.Hanahan D. J Mol Biol. 1983;166:557. doi: 10.1016/s0022-2836(83)80284-8. [DOI] [PubMed] [Google Scholar]
- 20.Brady SF. Nat Protoc. 2007;2:1297. doi: 10.1038/nprot.2007.195. [DOI] [PubMed] [Google Scholar]
- 21.Ogasawara Y, Torrez-Martinez N, Aragon AD, Yackley BJ, Weber JA, Sundararajan A, Ramaraj T, Edwards JS, Melancon CE., 3rd Genome Announce. 2015;3:e00114–15. doi: 10.1128/genomeA.00114-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schoner TA, Gassel S, Osawa A, Tobias NJ, Okuno Y, Sakakibara Y, Shindo K, Sandmann G, Bode HB. ChemBioChem. 2016;17:247. doi: 10.1002/cbic.201500474. [DOI] [PubMed] [Google Scholar]
- 23.Whidbey C, Harrell MI, Burnside K, Ngo L, Becraft AK, Iyer LM, Aravind L, Hitti J, Adams Waldorf KM, Rajagopal L. J Exp Med. 2013;210:1265. doi: 10.1084/jem.20122753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jin M, Fischbach MA, Clardy J. J Am Chem Soc. 2006;128:10660. doi: 10.1021/ja063194c. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.