

Abstract
The induction of host cell autophagy by various autophagy inducers contributes to the antimicrobial host defense against Mycobacterium tuberculosis (Mtb), a major pathogenic strain that causes human tuberculosis. In this study, we present a role for the newly identified cyclic peptides ohmyungsamycins (OMS) A and B in the antimicrobial responses against Mtb infections by activating autophagy in murine bone marrow-derived macrophages (BMDMs). OMS robustly activated autophagy, which was essentially required for the colocalization of LC3 autophagosomes with bacterial phagosomes and antimicrobial responses against Mtb in BMDMs. Using a Drosophila melanogaster–Mycobacterium marinum infection model, we showed that OMS-A-induced autophagy contributed to the increased survival of infected flies and the limitation of bacterial load. We further showed that OMS triggered AMP-activated protein kinase (AMPK) activation, which was required for OMS-mediated phagosome maturation and antimicrobial responses against Mtb. Moreover, treating BMDMs with OMS led to dose-dependent inhibition of macrophage inflammatory responses, which was also dependent on AMPK activation. Collectively, these data show that OMS is a promising candidate for new anti-mycobacterial therapeutics by activating antibacterial autophagy via AMPK-dependent signaling and suppressing excessive inflammation during Mtb infections.
Introduction
Mycobacterium tuberculosis (Mtb) is an important intracellular bacterial pathogen and the causative agent of human tuberculosis, which remains a serious global burden worldwide1. Mtb is able to survive in the hostile environment of host cells by preventing phagolysosomal fusion2. Autophagy is a self-digesting process that degrades cytoplasmic aggregates and damaged organelles to maintain homeostasis during metabolic and infectious diseases3. Accumulating evidence has suggested that antibacterial autophagy (xenophagy) is a cell-autonomous host defense that leads to antimicrobial responses against Mtb infections4–8. To date, numerous agents or signaling pathways have been shown to activate antibacterial autophagy against Mtb4, 5, 9, 10.
Among the autophagy-stimulating signals, we focused on the activation of AMP-activated protein kinase (AMPK), a key energy-sensing kinase in the maintenance of metabolic homeostasis and intracellular quality control in response to various stresses11. Our recent studies revealed that AMPK activation plays an important role in the antimicrobial responses against Mtb by inducing autophagy-related genes (ATG) and enhancing phagosomal maturation12. In addition, AMPK activation is often associated with inducing anti-inflammatory responses in immune cells by directly ameliorating pro-inflammatory signaling and limiting the synthesis of certain lipid intermediates relevant to inflammation13–15.
We previously reported that ohmyungsamycins (OMS) A and B are novel cyclic peptides that were isolated from a marine bacterial strain belonging to the Streptomyces genus collected from Jeju Island in Korea16. We previously showed that OMS-A and -B are cyclic peptides that exhibit inhibitory effects against a diverse range of cancer cells and bacteria such as Bacillus subtilis, Kocuria rhizophila, and Proteus hauseri 16. In this study, we demonstrated that OMS-A and -B are robust activators of autophagy that lead to antimicrobial responses against Mtb. We used a Drosophila melanogaster (D. melanogaster)–Mycobacterium marinum infection model to show that OMS treatment elicited anti-mycobacterial effects through autophagy activation in vivo. We further investigated the mechanisms by which OMS activated antimicrobial responses and showed that AMPK-dependent signaling was involved in the OMS-mediated activation of autophagy in murine bone marrow-derived macrophages (BMDMs). In addition, OMS treatment inhibited macrophage inflammatory responses during Mtb infection by activating the AMPK pathway. Together, these data suggest that the OMS-induced activation of autophagy and suppression of excessive pathologic inflammation may contribute to the innate host defenses against mycobacterial infection.
Results
OMS-A and OMS-B stimulate the killing of mycobacteria in vitro and in vivo
OMS-A and OMS-B have inhibitory activities against Bacillus subtilis, Kocuria rhizophila, and Proteus hauseri 16. To examine the anti-mycobacterial effects of OMS-A and OMS-B, we first assessed the antibacterial properties of OMS-A and OMS-B against Mtb. The minimum inhibitory concentration (MIC) of OMS-A and OMS-B against Mtb was determined using the resazurin microtiter assay (REMA) plate method. Isoniazid and ethambutol (known antimicrobial drugs) and SQ109 (an anti-Mtb drug candidate) were selected as positive controls17. After incubating Mtb for 5 days with the compounds, a decrease in fluorescence was observed, indicating a dose-dependent killing effect. As shown in Fig. 1a, OMS-A and OMS-B were very potent, with MICs lower than those of isoniazid, ethambutol, and SQ109. The MIC50 values for OMS-A and OMS-B were 57 nM and 117 nM, respectively.
Figure 1.
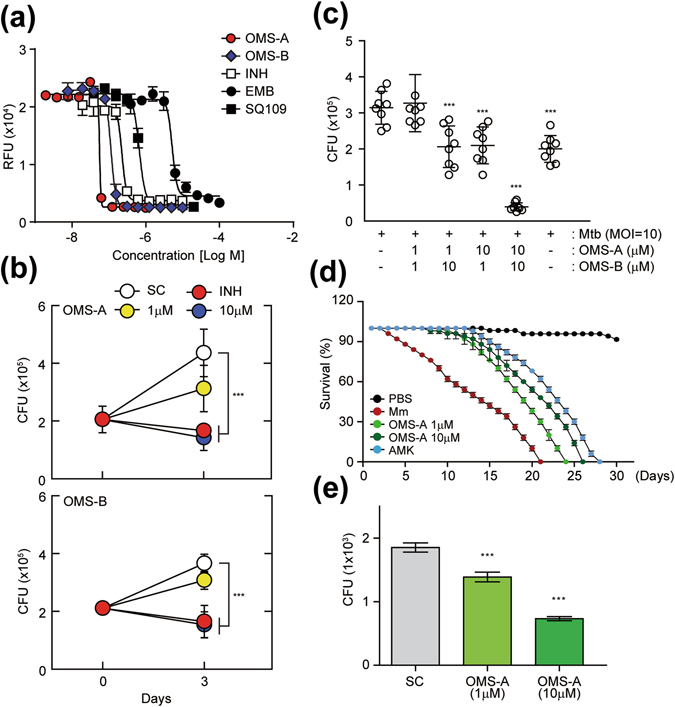
OMS shows antimycobacterial activity in vitro and in vivo. (a) Activity of OMS-A and OMS-B against Mtb replicating in culture broth medium. Mycobacterial growth was measured in relative fluorescence units (RFU). INH, EMB, and SQ-109 were used as positive controls. (b) BMDMs were infected with Mtb (moi = 10) for 4 h and then treated with OMS-A (1, 10 μM; Top), OMS-B (1, 10 μM; Bottom) or INH (0.5 μg/ml). (c) BMDMs were infected with Mtb and then co-treated with OMS-A and OMS-B or INH. (b and c) After 3 days, intracellular bacterial loads were determined by CFU assay. (d) M. marinum (CFU = 500)-infected W1118 flies were incubated with or without OMS-A (1, 10 μM) and AMK (1 μg/ml) medium. Dead flies were counted at 24 h intervals. The error bars indicate 95% confidence intervals. Log-rank analysis of the survival curves indicated that each group (n = 50) was significantly different. (e) W1118 flies were injected with M. marinum and then incubated with or without OMS-A (10 μM). After 12 days, each group (n = 20) of flies was harvested, homogenized, and quantified by CFU assay. All data represent the means ± SD of triplicates from each sample. ***p < 0.001, compared with Mtb-infected/untreated (c) and SC (b,e). INH, isoniazid; EMB, ethambutol; AMK, amikacin; SC, solvent control.
We next assessed whether OMS-A and OMS-B could elicit antimicrobial responses against Mtb in macrophages. Treating Mtb-infected BMDMs with OMS-A (1, 10 μM) and OMS-B (1, 10 μM) inhibited the survival of intracellular Mtb in a dose-dependent manner after 3 days of infection (Fig. 1b). The number of colony forming units (CFUs) was profoundly reduced in BMDMs treated with OMS-A (10 μM) or OMS-B (10 μM), similar to those treated with isoniazid (0.5 μg/ml), compared with untreated cells (Fig. 1b, Fig. S1). Additionally, OMS-A (1, 10 μM) and OMS-B (1, 10 μM) exerted synergistic effects against Mtb (Fig. 1c). OMS-A and OMS-B had nearly equal inhibitory potential against Mtb; however, OMS-A exhibited slightly higher antimicrobial effects than did OMS-B (Fig. 1b). These data suggest that OMS-A and OMS-B induce antimicrobial responses against Mtb in vitro and in macrophages.
It was previously reported that the M. marinum-D. melanogaster infection system is an alternative model host for evaluating Mycobacterium infections18. Therefore, we evaluated whether this infection model could be used to effectively assess the antimicrobial effects of OMS-A in vivo. As shown in Fig. 1d, flies challenged with M. marinum died within ~20 days (500 CFU/50 nL), consistent with previous reports by Kim et al.19. We then monitored the survival of flies treated with OMS-A (1, 10 μM), which exhibited a significant decrease in lethality compared with those treated with the solvent control. Antibiotic control flies, which received food containing amikacin (1 μg/ml), showed comparable survival to those treated with OMS-A after M. marinum injection (Fig. 1d). In addition, the viable bacterial counts in surviving flies infected with M. marinum were monitored in the control and OMS-treated groups. The in vivo bacterial counts were consistently higher in control flies compared with flies treated with OMS-A 12 days after infection with M. marinum (n = 20 per group; Fig. 1e). These findings suggest that OMS-A exhibits in vivo antimicrobial activities against mycobacteria.
OMS-A and OMS-B increase autophagy activation and autophagic flux in murine macrophages
We previously showed that treatment with antibiotics against Mtb infection resulted in autophagy, which is required for antimicrobial effects in the host19. Recent studies demonstrated that the important immunosuppressive agent cyclosporine A, a cyclic peptide, induces autophagic cell death in canine lens epithelial cells20. Thus, we assessed whether OMS-A or OMS-B enhances the activation of autophagy in BMDMs. Treatment with OMS-A (10 μM) or OMS-B (10 μM) caused no hazardous effects in BMDMs (data not shown). We next assessed LC3 puncta formation and lipidation, which are well-known indicators of autophagy induction21, in BMDMs treated with OMS-A (10 μM) or OMS-B (10 μM). As shown in Fig. 2a, fluorescent staining of LC3 puncta was increased in BMDMs treated with OMS-A (10 μM) or OMS-B (10 μM) for 24 h. Quantitative analysis showed that LC3 puncta formation was significantly increased by OMS-A and OMS-B treatment (Fig. 2b). It was noted that OMS-A and OMS-B had comparable effects on autophagy activation in BMDMs, similar to those observed in BMDMs treated with rapamycin (200 nM) or Torin1 (10 μM), the known autophagy activators4, 22 (Fig. S2).
Figure 2.
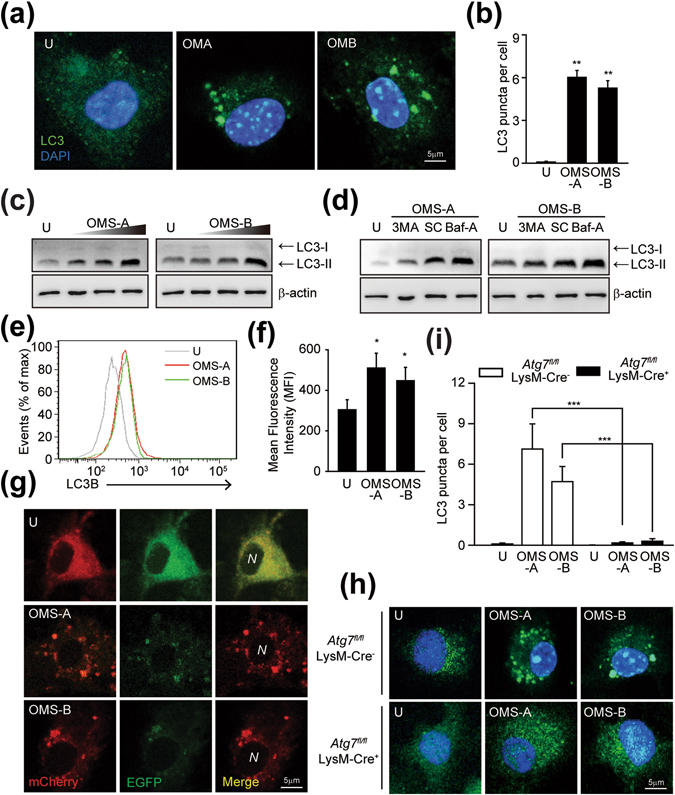
OMS induces autophagy activation in macrophages. (a and b) The Alexa 488-conjugated LC3 (green) and DAPI (blue) were detected by confocal microscopy at 24 h of OMS-A (10 μM; OMA) or OMS-B (10 μM; OMB) in BMDMs. (a) Representative of LC3 images. Scale bar, 5 μm. (b) Quantitative analysis of LC3 puncta per cell. Representative confocal microscopic images from three independent samples are shown, with each experiment including at least 100 cells scored from 6 random fields. (c) BMDMs were treated with OMS-A (1, 5, 10 μM) or OMS-B (1, 5, 10 μM) for 24 h. (d) BMDMs were pretreated with 3-MA (10 μM, for 2 h), or bafilomycin A1 (100 nM, for 1 h), and then treated with OMS-A or OMS-B for 24 h. (c and d) Cell lysates were subjected to immunoblot analysis of LC3 and Actin. The entire blots are shown in Supplementary Fig. 6. (e and f) Flow cytometric analysis of LC3B expression at 24 h of OMS-A or OMS-B treatment. (f) Quantitative analysis of LC3B expression. (g) BMDMs were transduced with retrovirus expressing a tandem-tagged mCherry-EGFP-LC3B and then treated with OMS-A or OMS-B for 24 h. Cells were tandem LC3B plasmid was detected by confocal microscopy. Scale bar, 5 μm. (h,i) Atg7fl/fl LysM-Cre− and Atg7fl/fl LysM-Cre+ BMDMs were treated with OMS-A (10 μM) and OMS-B (10 μM) for 24 h. The Alexa 488-conjugated LC3 (green) and DAPI (blue) were detected by confocal analysis. (h) Representative of LC3 images. Scale bar, 5 μm. (i) Quantitative analysis of LC3 puncta per cell. Representative confocal microscopic images from three independent samples are shown, with each experiment including at least 100 cells scored from 6 random fields. Data shown are from one representative of at least three independent experiments (means ± SD of triplicates [b,f,i] samples). *p < 0.05, **p < 0.01, ***p < 0.001, compared with untreated control or Atg7 fl/fl LysM-Cre− (b,e). U, untreated; Baf-A, bafilomycin A1; SC, solvent control; N; nucleus.
Western blotting of cell lysates showed that OMS-A (1, 5, 10 μM) or OMS-B (1, 5, 10 μM) treatment induced LC3 lipidation in a dose-dependent manner, as indicated by an increase in the autophagosomal membrane-associated LC3-II fraction (Fig. 2c; Fig. S3a,b). In addition, the OMS-A or OMS-B-induced LC3-II fractions were significantly decreased by pretreatment with 3-methyladenine (3-MA; 10 μM), an inhibitor of autophagy, and increased by pretreatment with bafilomycin A1 (100 nM), a vacuolar H+-ATPase inhibitor (Fig. 2d). We previously used flow cytometric analysis using anti-LC3B-specific antibodies to assess activation of autophagy19. In the current study, flow cytometry showed a significant increase in LC3B levels in OMS-A (10 μM) or OMS-B (10 μM)-treated BMDMs (Fig. 2e,f). We further assessed the ability of OMS-A or OMS-B to induce autophagic flux21 in macrophages using a retroviral vector containing mCherry-enhanced green fluorescent protein (EGFP)-LC3B. Cells treated with OMS-A (10 μM) or OMS-B (10 μM) exhibited an increase in red puncta, suggesting that mCherry-EGFP-LC3B was delivered to lysosomes following OMS-A or OMS-B treatment (Fig. 2g).
We then examined the effects of autophagy in OMS-induced LC3 punctate formation. To examine this, BMDMs from Atg7 fl/flLysM-Cre+ (Atg7 KO) mice and their Atg7+ littermates control (Atg7 wildtype; Atg7 WT) were treated with OMS-A (10 μM) or OMS-B (10 μM), and LC3 punctate formation was compared. As shown in Fig. 2h and i, the OMS-A or OMS-B-induced increase in LC3-positive autophagosome formation in Atg7 WT BMDMs was significantly decreased in Atg7 KO BMDMs. Together, these data suggest that OMS-A or OMS-B treatment enhances autophagy activation and autophagic flux in BMDMs.
OMS-A and OMS-B enhance Mtb phagosome maturation in BMDMs
Mtb is a highly adapted pathogen that arrests the maturation of phagosomes into phagolysosomes23. Recent studies also showed that the Mtb virulent strain H37Rv significantly inhibited autophagic flux in macrophages24. The activation of antibacterial autophagy leads to mycobacterial phagosomal maturation, thus reducing the bacterial burden in macrophages25. To further evaluate OMS-A (10 μM) or OMS-B (10 μM) treatment-induced phagosome maturation, BMDMs were infected with enhanced red fluorescent protein (ERFP)-Mtb and then treated with OMS-A (10 μM) or OMS-B (10 μM). As shown in Fig. 3a–d, treating Mtb-infected BMDMs with OMS-A or OMS-B increased the co-localization of LC3, an autophagosomal marker, with Mtb phagosomes (Fig. 3a,b). Next, the co-localization of Mtb phagosomes with lysosomes was assessed in Mtb-infected BMDMs after OMS-A (10 μM) or OMS-B treatment (10 μM). Treating Mtb-infected BMDMs with OMS-A or OMS-B significantly enhanced the co-localization of Mtb phagosomes with LAMP2 (Fig. 3c,d).
Figure 3.
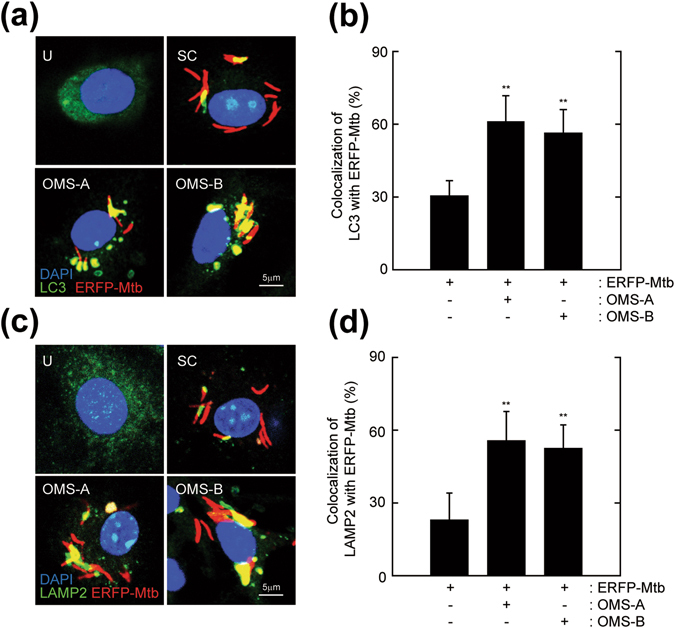
OMS-A and OMS-B activate phagosomal maturation against Mtb in macrophages. (a–d) BMDMs were infected with ERFP-Mtb (moi = 10) for 4 h and then treated with OMS-A (10 μM) or OMS-B (10 μM) for 24 h. (a) Mtb-ERFP (red), Alexa 488-conjugated-LC3 (green), and DAPI (blue) were detected by confocal analysis. Scale bar, 5 µm. (b) Quantitative analysis of Mtb-ERFP and LC3 colocalization per cell. (c) Mtb-ERFP (red), Alexa 488-conjuated LAMP2 (green), and DAPI (blue) were detected by confocal microscopy. Scale bar, 5 µm. (d) Quantitative analysis of Mtb and LAMP2 colocalization per cell. Representative confocal microscopic images from three independent samples are shown, with each experiment including at least 100 cells scored from 6 random fields (a,c). Data shown are from one representative of at least three independent experiments (means ± SD of triplicates [b, d] samples). **p < 0.01, compared with ERFP-Mtb-infected control (b,d). U, uninfected/untreated; SC, solvent control.
In addition, we infected Atg7 WT and Atg7 KO BMDMs with ERFP-Mtb in the presence or absence of OMS-A or OMS-B. Notably, the LC3 punctate formation in Mtb-infected/OMS-A/OMS-B (10 μM) treated Atg7 WT BMDMs was markedly decreased in Atg7 KO BMDMs (Fig. S4a). In addition, the co-localization of bacterial phagosomes with the autophagosomal marker LC3 was significantly decreased in Atg7 KO BMDMs compared to Atg7 WT BMDMs (Fig. S4a,b). Collectively, these data indicate that OMS-A and OMS-B induce the phagosomal maturation of Mtb through autophagy activation in BMDMs.
OMS-A and OMS-B activate the AMPK pathway, which is required for Mtb phagosomal maturation and antimicrobial responses in macrophages
Our previous studies showed that activation of AMPK contributes to autophagy activation and antimicrobial responses against Mtb12. We thus examined whether OMS-A or OMS-B activates AMPK by measuring the phosphorylation of Thr172 of the catalytic α-subunit of AMPK26. As shown in Fig. 4a,b, OMS-A and OMS-B induced the phosphorylation of AMPK at Thr172, as well as its downstream target acetyl-CoA carboxylase (ACC), in a time-dependent manner. Treating BMDMs with OMS-A (10 μM) or OMS-B (10 μM) rapidly increased AMPK phosphorylation within 0.5–1 h after stimulation. The AMPK phosphorylation levels were then slightly decreased in BMDMs after OMS-B stimulation, however, they were sustained in BMDMs in response to OMS-A and OMS-B at later time points (Fig. 4a,b).
Figure 4.

AMPK activation is required for OMS-induced phagosomal maturation and antimicrobial responses. (a and b) BMDMs were treated with OMS-A (10 μM; for a) or OMS-B (10 μM; for b) for indicated times (0–18 h). The cell lysates were subjected to immunoblot analysis of p-AMPKα, p-ACC, and Actin. The entire blots are shown in Supplementary Fig. 7. The densitometric values for p-AMPKα were normalized to Actin (bottom). (c and d) BMDMs were transduced with non-specific shRNA (shNS) or Ampk-specific shRNA (shAmpk)-expressing lentivirus for 48 h and then infected with ERFP-Mtb (moi = 10) for 4 h, followed by treatment with OMS-A or OMS-B for 24 h. (c and d) Mtb-ERFP (red), Alexa 488-conjugated-LC3 (green), and DAPI (blue) were detected by confocal analysis. Scale bar, 5 µm. (c) Representative confocal microscopic images from three independent samples are shown. (d) Quantitative analysis of cells showing the colocalization between LC3 and Mtb-ERFP. For each experiment, at least 100 cells were scored from 6 random fields. (e) After 3 days of infection, intracellular bacterial loads were determined by CFU assay. (inset) RT-PCR analysis of Ampk mRNA expression of transduction efficiency. Data shown are from one representative of at least three independent experiments (means ± SD of triplicates [a,b bottom; d] samples]). *p < 0.05, ***p < 0.001, compared with shNS. U, uninfected/untreated; SC, solvent control.
To further define the role of AMPK in enhancing phagosome maturation, BMDMs were transduced with short hairpin RNAs (shRNA) lentiviral vectors that specifically target Ampkα (shAmpk). The efficiency of Ampkα silencing was confirmed 48 h after transduction; the mRNA levels of AMPK were reduced compared with those in cells transduced with lentiviruses expressing nonspecific shRNA (shNS). To examine the effects of Ampkα silencing on the colocalization of Mtb phagosomes and LC3 autophagosomes, BMDMs were transduced with shAmpk or shNS, infected with ERFP-Mtb, and then treated with or without OMS-A (10 μM) or OMS-B (10 μM). The colocalization of LC3 autophagosomes and Mtb phagosomes was markedly increased in shNS-transduced BMDMs by treatment with OMS-A (10 μM) or OMS-B (10 μM). However, there was no significant difference between the effects of OMS-A and OMS-B on the co-localization of phagosomes and autophagosomes. In addition, silencing Ampkα dramatically attenuated OMS-induced colocalization of autophagosomes and Mtb phagosomes in BMDMs compared with those under the control conditions (Fig. 4c,d). Next, the ability of AMPK to promote the intracellular killing activities induced by OMS-A or OMS-B was examined. As shown in Fig. 4e, the inhibitory effects of OMS-A and OMS-B against Mtb in shNS-transduced macrophages were significantly counteracted by shAmpk. These findings suggest that AMPK is the major signaling pathway mediating OMS-A and OMS-B-induced Mtb phagosome maturation and antimicrobial responses in macrophages.
OMS-A and OMS-B activate autophagy to enhance antimicrobial responses against Mtb in vitro and in vivo
To further examine the effects of autophagy in the antimicrobial responses by OMS, we performed additional cfu experiments using macrophages from Atg7 KO mice. BMDMs from Atg7 WT and Atg7 KO were infected with Mtb, followed by treatment with or without OMS-A (10 μM) or OMS-B (10 μM). As shown in Fig. 5a, Atg7 WT macrophages showed significantly increased killing effects against Mtb, whereas this was significantly inhibited in Atg7 KO BMDMs following treatment with OMS-A (10 μM) or OMS-B (10 μM). It was also noted that OMS-A- or OMS-B-induced antimicrobial effects were substantially higher than those induced by another autophagy activators, such as rapamycin (100 nM) or Torin1 (10 μM) (data not shown). We next examined whether hATG5 is required for the OMS-mediated antimicrobial response against intracellular Mtb by transducing human monocyte-derived macrophages (MDMs) with shRNA against hATG5 (shATG5) and determining whether knockdown of hATG5 affected the intracellular survival of Mtb in human MDM. Silencing of ATG5 significantly counteracted the intracellular killing effects of OMS-A and OMS-B against Mtb in human MDMs (Fig. 5b; shNS vs. shATG5 under OMS-A (10 μM)- or OMS-B (10 μM)-treated conditions; P < 0.001, for both; moi = 10). These data indicate that OMS-mediated autophagy enhances the antimicrobial response in murine and human macrophages.
Figure 5.

OMS-mediated autophagy activation is essentially required for antimicrobial responses against mycobacterial infection in vitro and in vivo. (a) Atg7 fl/fl LysM-Cre− and Atg7 fl/fl LysM-Cre+ BMDMs were infected with Mtb (moi = 10) for 4 h and then treated with OMS-A (10 μM) and OMS-B (10 μM). (b) Human MDMs were transduced with non-specific shRNA (shNS) or ATG5-specific shRNA (shATG5)-expressing lentivirus for 48 h and then infected with Mtb and then treated with OMS-A and OMS-B. (inset) RT-PCR analysis of ATG5 mRNA expression of transduction efficiency. (a and b) CFU assay. The intracellular bacterial loads were determined at 3 days after infection. (c and d) W1118 flies were infected with M. marium (CFU = 500), followed by incubation with or without OMS-A (10 μM) in the presence or absence of chloroquine (1 μM). Positive control group was treated with AMK (1 μg/ml). (e) Live flies were counted at 24 h intervals (n = 40). The error bars indicate 95% confidence intervals. Log-rank analysis of the survival curves indicated that the susceptibility to M. marinum was significantly increased in the group of OMS-A with chloroquine, when compared with that of OMS-A only (***p < 0.001). (f) Each group (n = 20) of flies was harvested at 7 days, homogenized, and quantified by CFU assay. (e and f) Atg7 control and Atg7 mutant flies were infected with M. marium (CFU = 500), followed by incubation with or without OMS-A (10 μM). (e) Flies were counted at 24 h intervals (n = 50). The error bars indicate 95% confidence intervals. Log-rank analysis of the survival curves showed that the survival rates of Atg7 control flies were significantly increased by OMS-A treatment (***p < 0.001), whereas these were not significant in Atg7 mutant flies with the same treatment. (f) Each group (n = 20) of flies was harvested at 3 days, homogenized, and quantified by CFU assay. All data represent the means ± SD of triplicates from each sample. **p < 0.01, ***p < 0.001, compared with control conditions (a,b,d,f). SC, solvent control; AMK, amikacin; CQ, chloroquine.
We further examined whether OMS-mediated antimicrobial effects were decreased in flies administered chloroquine, an inhibitor of the autophagy pathway27. We thus injected flies with M. marinum and treated them with OMS-A (10 μM) in the presence or absence of chloroquine (1 μM). We then monitored fly survival for 20 days. As shown in Fig. 5c, the flies treated with chloroquine exhibited increased lethality to M. marinum challenge compared to control flies, which were administered food containing OMS-A (10 μM) only. Consistent with these findings, viable bacterial counts in surviving flies were significantly increased by chloroquine administration relative to those in control flies treated with OMS-A only (after 7 days; n = 20 per group; Fig. 5d).
We then injected control and Atg7 mutant flies with M. marinum in the presence or absence of OMS-A (10 μM) and monitored survival for 27 days. As shown in Fig. 5e, the Atg7 mutant flies increased lethality to M. marinum challenge when compared to control flies. In addition, administration of control flies with food containing OMS-A (10 μM) led to a significant increase of survival after M. marinum injection. However, OMS-A-mediated protective effects were not significant in Atg7-mutant flies after M. marinum infection (Fig. 5e). Furthermore, viable bacterial counts of M. marinum were significantly higher in atg7 mutant flies than those in control flies infected with M. marinum (after 3 days; n = 20 per group; Fig. 5f). OMS-A treatment significantly decreased the bacterial loads in control flies, but did not decrease the viable bacteria in Atg7 mutant flies to the levels observed in control flies (Fig. 5f). These findings suggest that host autophagy activation is required for the OMS-induced antimicrobial effects against mycobacterial infection in vitro and in vivo.
OMS-A and OMS-B attenuate Mtb-induced inflammatory responses by activating AMPK in macrophages
Accumulating evidence has shown that AMPK signaling is required for anti-inflammatory responses28. However, it is unclear whether AMPK activation is essential for the inhibition of Mtb-induced inflammatory responses. We first examined cytokine production in Mtb-infected BMDMs after treatment with OMS-A (1, 5, 10 μM) or OMS-B (1, 5, 10 μM). As shown in Fig. 6a, Mtb-induced production of proinflammatory cytokines, such as tumor necrosis factor (TNF)-α, interleukin (IL)−6, IL-1β, and IL-12 p40, was dose-dependently inhibited by treatment with OMS-A (1, 5, 10 μM) or OMS-B (1, 5, 10 μM) in BMDMs. In addition, Mtb-induced nuclear factor kappa B (NF-κB) reporter gene activity was abrogated by OMS-A (1, 5, 10 μM) or OMS-B (1, 5, 10 μM) treatment in a dose-dependent manner (Fig. 6b).
Figure 6.

OMS inhibits Mtb-induced inflammatory responses through AMPK activation. (a) BMDMs were infected with Mtb (moi = 10) for 4 h and then treated with OMS-A (1, 5, 10 μM) or OMS-B (1, 5, 10 μM) for 24 h. (b) BMDMs were transduced with adenoviral NF-ĸB-luciferase reporter plasmid for 36 h, and infected with Mtb and then treated with OMS-A or OMS-B for 6 h. The cells were harvested and NF-ĸB luciferase reporter activity was determined. (c) BMDMs were transduced with non-specific shRNA (shNS) or Ampk-specific shRNA (shAmpk)-expressing lentivirus for 48 h and then infected with Mtb, followed by treatment with OMS-A or OMS-B for 24 h. (a and c) The supernatants were harvested and subjected of ELISA analysis of TNF-α, IL-6, IL-1β, and IL-12p40 production. All data represent the means ± SD of triplicates from each sample. **p < 0.01, ***p < 0.001, compared with SC (a–c). U, uninfected/untreated; SC, solvent control; ns, no significant.
Finally, we examined whether AMPK is involved in the attenuation of proinflammatory cytokine production in BMDMs. BMDMs were transduced with shAmpk or shNS, infected with Mtb, and then treated with OMS-A or OMS-B. As shown in Fig. 6c, the OMS-mediated inhibition of proinflammatory cytokine production was nearly completely counteracted by shAmpk in BMDMs. Collectively, these data suggest that OMS-induced AMPK activation is required for suppression of Mtb-induced inflammatory responses in BMDMs.
Discussion
Much of the recent focus regarding therapeutic strategies against Mtb has been on the development of host-directed therapies that enhance antimicrobial mechanisms, such as autophagy inducers, cathelicidin inducers, and anti-inflammatory agents29, 30. Novel functions and mechanisms by which various autophagy inducers enhance antibacterial autophagy and antimicrobial responses have been reported31, 32. Early studies showed that interferon (IFN)-γ5, vitamin D10, 33, 34, and both treatments synergize the elimination of Mtb via autophagy10. Various innate signals and pathogen- and damage-associated molecular patterns enhance the Mtb-induced inhibition of intracellular growth by activating autophagic machinery, interacting with innate signaling pathways, and delivering antimicrobial peptides to phagosomes10, 33, 35–37. Here, we demonstrated that the newly identified cyclic peptides OMS-A and OMS-B activate autophagy via the AMPK pathway, to promote antimicrobial effects against Mtb. In addition, OMS-induced autophagy leads to antimicrobial responses to Mtb in macrophages, as shown by intracellular survival assays.
Since issues regarding multidrug-resistant Mtb are increasing worldwide, there is an urgent need to develop new antitubercular drugs. The current data suggest that OMS-A and OMS-B exhibit at least three attractive anti-tubercular drug activities: 1) direct antimicrobial killing activity against Mtb, 2) direct activation of autophagy, which was essential for antimicrobial effects, in host cells, and 3) inhibition of Mtb immunopathology. REMAs revealed that OMS-A and OMS-B have excellent MICs and MBCs against Mtb, comparable to those of isoniazid and ethambutol, which are the antibiotics currently used against human tuberculosis. Our previous studies revealed that OMS-A and OMS-B are cyclic peptides that harbor activity against several bacteria, as well as exhibit an inhibitory effect on cancer cell proliferation16. Recently, chemical investigations identified several cyclic peptides as promising anti-Mtb agents due to strong antitubercular activities in vitro 38. In addition, a high-throughput screening study revealed that ecumicin is an excellent drug candidate with activity against multidrug-resistant (MDR) and extensively drug-resistant (XDR) Mtb strains39. Lassomycin, a basic and ribosomally encoded cyclic peptide, also exhibits bactericidal activity against mycobacteria40. The mycobacterial ClpC1 ATPase complex was identified as a drug target for both ecumicin and lassomycin39, 40. Although we did not try to identify the targets of OMS-A and OMS-B, we speculate that the same ClpC1 ATPase complex could be a potential target. Indeed, significant effort in developing new cyclic antimicrobial peptides has been devoted to designing analogs of drugs that reduce cytotoxic side effects but promote antitubercular and antibacterial properties using conformational analyses based on structure-activity relationships41. Importantly, we did not observe any toxic effects on host macrophages and flies in vivo treated with OMS-A and OMS-B, at least at the doses and time periods evaluated. Nevertheless, further studies are needed to develop a large scale of OMS and design safe and effective chemical analogs for future clinical trials.
Both OMS-A and OMS-B activated autophagy, which is an essential part of the antimicrobial defense against Mtb. Our data demonstrated that OMS-A and OMS-B-induced autophagy is essential for phagosomal maturation, and for antimicrobial effects against Mtb infection, both in vitro and in flies. The findings of this study were in partial agreement with our previous studies in showing that isoniazid and pyrazinamide, two antibiotics used to treat tuberculosis in humans, activate the antibacterial autophagy required for successful antimicrobial activity during mycobacterial infection19. In a recent report, thiostrepton, a thiopeptide antibiotic possessing a quinaldic acid moiety, was used successfully in the treatment of M. marinum, through ER-stress-mediated autophagy42. In addition, a search for active compounds to enhance interferon-gamma responses in macrophages identified several such compounds within the flavagline (rocaglate) family, which effectively induce autophagy to combat intracellular mycobacteria and inhibit pathologic inflammation during infection43.
These OMS-A and OMS-B activities were related to their ability to activate AMPK in macrophages. AMPK is an energy sensor that regulates energy balance at a cell-autonomous level in living organisms11. Much attention has been given to promising adjunctive host-directed therapeutic candidates that affect cell signaling pathways associated mainly with immunometabolism (i.e., the core pathways intersecting immunologic and metabolic responses)44. Understanding the mechanisms that regulate AMPK and mammalian target of rapamycin signaling will likely facilitate significant advances in the development of adjunctive options for several unmet needs regarding tuberculosis, including new drugs against MDR and XDR strains, shortening the chemotherapeutic treatment duration, and targeting various clinical stages, even in combination with human immunodeficiency viral infection44, 45. Recent studies demonstrated that AMPK plays a key role in antimicrobial autophagy against mycobacteria44 and that several AMPK activators have the potential to activate autophagy and intracellular killing in response to Mtb12, 46. In addition, the widely used anti-diabetes drug metformin, which exhibits AMPK-activating activity, was identified as an excellent adjunct antituberculous therapy47. The current data demonstrate that AMPK activation by OMS-A and OMS-B could impact maturation. These studies collectively suggest that activation of AMPK activation in host cells enhances the cell-autonomous and in vivo killing effects against mycobacteria.
NF-κB activation and inflammatory responses during Mtb infections appear to be a double-edged sword, i.e., a defense reaction against microbial insults and immunopathologic responses in tuberculosis. Indeed, the design of immunomodulatory agents has been focused on host-directed therapeutics that facilitates protective immune responses while simultaneously reducing deleterious inflammatory responses45. The current data revealed that OMS-induced AMPK activation inhibited inflammatory responses in macrophages induced by Mtb infection. Activation of the AMPK signaling pathway promotes anti-inflammatory and anti-cancer effects by inducing oxidative metabolism28. AMPK activators such as metformin and salicylate suppress inflammatory responses in various cell types by modulating NF-κB or other signaling pathways28, 48, 49. In addition, autophagy controls excessive inflammatory responses and type I interferon activation, particularly during microbial infections50. Previous studies showed that autophagy modulates NF-κB signaling pathways during innate immune responses to attenuate exacerbated inflammation. It was reported that inhibition of NF-κB signaling in human macrophages increased apoptosis and autophagy, which subsequently decreased intracellular Mtb survival51. Previous studies revealed the mechanisms by which autophagy regulates NF-κB signaling. Specifically, hepatoma tumor cell-conditioned medium induced TLR2 signaling, which stimulated the degradation of NF-κBp65 by SQSTM1/p62-mediated selective autophagy52. Thus, OMS-induced anti-inflammatory responses may be the result of at least two mechanisms: AMPK-dependent- and autophagy-activating pathways.
Taken together, these results provide a novel mechanism underlying the antimicrobial effects of the novel cyclic peptides OMS-A and OMS-B, via AMPK-dependent autophagy activation, thus overcoming the inhibition of phagolysosomal fusion caused by pathogens. OMS-A and OMS-B induced a direct killing activity against Mtb and, interestingly, promoted host defenses; they did not only activate autophagy but also regulated pathologic inflammatory responses during Mtb infection. These efforts may contribute to the development of new therapeutic drugs and biologics by enhancing dual effects on both the bacteria and host immunity.
Materials and Methods
Cultivation and extraction of the actinobacterial strain SNJ042 to produce OMS-A and B
Isolation of OMS-A and OMS-B were performed as described previously16. The actinobacterial strain SNJ042 (99% identity with Streptomyces cheonanensis based on 16 S ribosomal DNA analysis) was isolated from a sand beach in Jeju Island, Republic of Korea, and was cultivated to produce OMS-A and OMS-B. It was incubated on solid YEME medium (10 g malt extract, 4 g yeast extract, 4 g glucose, and 18 g agar powder in 1 L sterilized 3.4% artificial seawater) at 25 °C to acquire spores. The spores were inoculated into 125 ml liquid A1/C medium (10 g starch, 4 g yeast extract, 2 g peptone, and 1 g CaCO3 in 1 L sterilized 2.4% artificial seawater) in a 500 ml Erlenmeyer flask. The liquid culture was incubated at 30 °C with shaking at 200 rpm. After 2 days, 10 ml of the liquid culture were transferred to 1 L liquid A1/C medium in a 2.8 L Fernbach flask and further cultivated on a rotary shaker at 30 °C and 180 rpm (108 × 1 L, total culture volume 108 L). After 7 day cultivation, 1.5 L ethyl acetate (EtOAc) were added to each 1 L bacterial culture and extracted twice using a separation funnel. The EtOAc layer was collected in an empty 2.8 L Fernbach flask, and anhydrous sodium sulfate was added to the organic extract to remove residual water. The extract in EtOAc was concentrated in vacuo to yield 12 g dry material.
Determining the MIC by REMA
The REMA was performed as described previously to determine the MICs of OMS-A or OMS-B, isoniazid (INH; I3377), ethambutol (E4630), and SQ-109 (SML1309) (all from Sigma, St. Louis, MO, USA) against H37Rv17. Briefly, a 100 µl inoculum was used to inoculate each well of the plate, and two-fold serial dilutions of each test compound were prepared in 96-well plates in triplicate. An inoculum at an optical density 600 of 0.005 was prepared by diluting mid-log cultures and then added to each well. Growth controls containing no drug and a sterile control were also prepared in each experiment. Plates were incubated at 37 °C for 5 days, and 40 µL 0.025% resazurin (Sigma, R7017) solution were then added to each well. After an overnight incubation, the fluorescence of the resazurin metabolite resorufin was determined by excitation at 560 nm and emission at 590 nm using the Synergy H1 microplate reader (Biotechnology Inc, Dallas, TX, USA). The MIC50 (the MIC required to inhibit the growth of 50% of the organism) was determined using GraphPad Prism 5.0 software.
Mycobacterial cultures
Mycobacterial cultures were performed as described previously12. Mtb H37Rv was provided by Dr. R.L. Friedmann (University of Arizona, Tucson, AZ, USA). Mycobacteria were grown in Middlebrook 7H9 medium (Difco, Detroit, MI, USA, 271310) supplemented with 10% oleic albumin dextrose catalase (OADC; BD Biosciences, Franklin Lakes, NJ, USA, 212240), 5% glycerol, and 0.05% Tween 80 (Sigma, P1754). The Mtb strains expressing ERFP were described previously53. The E. coli Mtb shuttle plasmid pMV262-RFP harboring ERFP under the control of the HSP60 promoter was used. Mtb harboring the ERFP gene was cultivated in 7H9 medium supplemented with kanamycin (Sigma, 60615). For D. melanogaster infection, the M. marinum strain Aronson (ATCC 927, fish isolate) was cultured in 7H9 medium with OADC and 0.2% Tween 80 at 30 °C for 8 weeks in the dark without agitation. Single-cell suspensions of Mtb, Mtb-ERFP, and M. marinum were aliquoted and stored at −80 °C. Mid-logarithmic-phase bacteria (OD, 0.6) were used in all assays. The CFUs were enumerated on Middlebrook 7H10 agar (Difco, 262710).
Mice and Cells
C57BL/6 mice aged 6–8 weeks with a WT background were purchased from Samtako Bio Korea (Gyeonggi-do, Korea). We generated with myeloid lineage cell-specific Atg7-deficient mice (Atg7 fl/flLysM-Cre+) using the Cre/loxP recombination system, as described previously54. This study was approved by the Institutional Research and Ethics Committee at Chungnam National University School of Medicine (CNUH-01-A0008; Daejeon, Korea). All animal procedures were conducted in accordance with the guidelines of the Korean Food and Drug Administration. Mice were maintained under specific pathogen-free conditions.
BMDMs were isolated and differentiated as described previously12. BMDMs were cultured for 5 days in Dulbecco’s modified Eagle’s medium (DMEM; Lonza, 12–604 F) medium containing 25 ng/ml macrophage colony-stimulating factor (M-CSF; R&D Systems, 416-ML) and supplemented with 10% heat-inactivated fetal bovine serum (FBS; Lonza, BW14–503E) and penicillin-streptomycin-amphotericin B (Lonza, 17–745E).
Human MDMs were as described previously19. Briefly, peripheral blood mononuclear cells were isolated from healthy donors by density gradient centrifugation using Lymphoprep (Axis-Shield, Oslo, Norway). Human MDMs were cultured for 5 days in RPMI1640 (Lonza, 12–702 F) medium containing 10% pooled human serum and of 4 ng/ml human M-CSF (Sigma). Written infomed consent was obtained from all participants before enrolment in the study. The study was approved by the institutional review board of the Chungnam National University Hospital, and was done in accordance with the Declaration of Helsinki and good clinical practice guidelines.
Reagents and antibodies (Abs)
3-MA (M9281), 4,6-diamidino-2-phenylindole dihydrochloride (DAPI, D9542), LC3 (L8918), dimethyl sulfoxide (D8418), and N’-(7-chloroquinolin-4-yl)-N,N-diethylpentane-1,4-diamine (chloroquine; C6628) were purchased from Sigma. Bafilomycin A1 (196000) was purchased from Calbiochem (San Diego, CA, USA). Alexa 488-conjugated anti-rabbit IgG (A17041) was purchased from Molecular Probes (Eugene, OR, USA). Actin (sc-1616) was purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Anti-phospho-ACC (3661) and anti-phospho-AMPKα (2535) Abs were purchased from Cell Signaling Technology (Beverly, MA, USA).
Generation and transduction of small hairpin RNA (shRNA)
Lentivirus production was performed as described previously33. Briefly, the lentiviral construct vector pLKO.1 and three packaging plasmids (pRSVRev, pMD2.VSV-G, and pMDLg/pRRE) were purchased from Addgene. The pLKO.1-based target shRNA plasmids for mAmpkα (Santa Cruz Biotechnology, sc-29674-SH) or hATG5 (Santa Cruz Biotechnology, sc-41445-SH) were cotransfected into HEK293T cells using Lipofectamine 2000 (Invitrogen, 12566014, Carlsbad, CA, USA) for 72 h. Then, the lentivirus-containing supernatant was collected, filtered, and titrated. For lentivirus transduction, BMDMs or MDMs were infected with lentiviral vectors using 8 μg/mL polybrene (Sigma, 107689) for 48 h. Transduced cells were harvested, and the target gene knockdown efficiency was analyzed.
Generation of tandem LC3B retrovirus
Tandem LC3B plasmid (mCherry-EGFP-LC3B) was performed as described previously21. To produce tandem LC3 retrovirus, human Phoenix amphotropic (ATCC, CRL-3213) cells were seeded at 70–80% confluence into a 6-well plate and co-transfected with 0.75 μg of the packaging plasmid pCL-Eco (Addgene, 12371), 0.25 μg of the envelope plasmid pDM2.G (Addgene, 12259) and 1 μg pBABE-puro Tandem LC3B plasmid using Lipofectamine 2000 for 6 h. Subsequently, the media containing tandem LC3B retrovirus were replaced with fresh media and cultured for 48 h. The tandem LC3B retrovirus was then harvested and filtered through a 0.45 μm syringe filter.
CFU assay
To quantify intracellular bacteria, CFU assays were performed as described previously12. BMDMs were plated on 7H10 at a concentration of 2 × 105 cells per well and infected with Mtb for 4 h. The cells were then washed with phosphate buffered saline (PBS) to remove extracellular bacteria and treated with OMS-A, OMS-B, or INH in medium for 3 days. Thereafter, the intracellular bacteria were harvested, and the lysates were diluted two-fold in PBS. Each sample was plated on 7H10 agar plates and incubated at 37 °C in a 0.5% CO2 incubator for 3 weeks.
Western blotting and enzyme-linked immunosorbent assay (ELISA)
Western blotting and ELISA were performed as described previously55. For western blotting, cell lysates were denatured by boiling and separated on 12% acrylamide SDS-PAGE gels. Then, proteins were transferred to polyvinyl difluoride membranes (Millipore, Boston, MA, USA) and incubated with primary Abs (LC3, p-AMPKα, and p-ACC) diluted at a ratio of 1:1000. The bands were visualized using ECL (Millipore, Danvers, MA, USA) and exposure to chemiluminescence film (Fujifilm, Tokyo, Japan).
For ELISA, infected cell supernatants were used to measure mouse TNF-α (ELISA Max™ Standard Set; BioLegend, Inc., San Diego, CA, USA), IL-6 (BioLegend), IL-1β (BioLegend), and IL-12p40 (BD Pharmingen, San Diego, CA, USA) secretion according to the manufacturer’s instructions.
RNA extraction and semi-quantitative RT-PCR
RNA extraction and semi-quantitative RT-PCR were performed as described previously55. Briefly, total RNA from cells was isolated using TRIzol reagent (Thermo Fisher Scientific, 15596–026) and used for synthesis of cDNA with Superscript II reverse transcriptase (Invitrogen). PCR reactions involved 30 cycles of annealing, extension, and denaturation the following primers were used: mAmpk forward 5′-GGTGTACGGAAGGCAAAATGGC-3′, reverse 5′-CAGGATTCTTCCTTCGTACACGC-3′; mActb forward 5′-CATTGCTGACAGGATGCAGAAGG-3′, reverse 5′-TGCTGGAAGGTGGACAGTGAGG-3′, hATG5 forward 5′-GCAGATGGACAGTTGCACACAC-3′, reverse 5′-GAGGTGTTTCCAACATTGGCTCA-3′; and hACTB forward 5′-CACCATTGGCAATGAGCGGTTC-3′, 5′-AGGTCTTTGCGGATGTCCACGT-3′. Reactions were run on a Veriti 96-Well Thermal Cycler (ThermoFisher). The PCR products were analyzed by electrophoresis on 1.5% agarose gel.
Flow cytometry
Intracellular LC3B levels were analyzed as described previously12. Stimulated cells were washed with PBS, fixed with 4% paraformaldehyde for 10 min at room temperature (RT), and permeabilized using 0.25% Triton X-100 in PBS for 10 min at RT. Cells were stained with primary Abs against LC3B (2775 S; Cell Signaling, 1:100) for 1–2 h at 4 °C followed by anti-rabbit IgG secondary Abs (7074 S; Cell Signaling, 1:100) for 1 h on ice. After two washes with PBS, cells were fixed in 4% paraformaldehyde and assayed immediately. The cells were examined using a FACSCanto II flow cytometer (Becton Dickinson, San Jose, CA, USA). Data were collected from 10,000–30,000 cells and analyzed using FlowJo software (Tree Star, Ashland, OR, USA).
Confocal microscopy of autophagy analysis
LC3 punctate and LAMP2 staining and quantification were performed as described previously12. After treatment, cells on coverslips were washed twice with PBS, fixed with 4% paraformaldehyde for 20 min, permeabilized with 0.25% Triton X-100 in PBS for 10 min, and immunostained with primary Abs (anti-LC3; PM036, MBL International, 1:400) or (anti-LAMP2; sc-5571, Santa Cruz, 1:400) for 2 h. Cells were washed to remove primary Abs and incubated with the fluorescently labeled secondary Ab (Alexa 488-conjugated anti-rabbit IgG; A17041, Molecular Probes, 1:400) for 2 h. Nuclei were stained by incubation with DAPI (D9564, Sigma) for 5 min. After mounting, fluorescence images were acquired using a confocal laser-scanning microscope (LSM 710, Zeiss, Thomwood, NY, USA).
LC3 punctate dot fluorescence intensity was measured using ImageJ analysis software. For colocalization analysis, the co-distribution of LC3 autophagosomes or LAMP2 phagolysosomes with bacterial phagosomes were quantified using the ImageJ analysis software. Each condition was assayed in triplicate, and at least 100 cells per well were counted.
M. marinum infection and intracellular bacterial growth in D. melanogaster
The culture, M. marinum infection, and intracellular bacterial growth of W1118, CG5335 d30 /Atg7 d14 (Atg7 control), and Atg7 d77 /Atg7 d14 (Atg7 mutant) files were performed as described previously12, 56. For M. marinum infections, 3–5 days old male W1118, Atg7 control, and Atg7 mutant flies were anesthetized with CO2 and injected with 500 CFU M. marinum in 50 nl (total volume) PBS using an individually calibrated pulled-glass needle attached to a microinjector (IM-5B; Narishige, Japan). Then, M. marinum-infected flies were incubated with standard cornmeal medium, OMS-A (1, 10 μM), amikacin (1 μg/ml) or co-treated with chloroquine (1 μM) and OMS-A (10 μM) containing medium and maintained at 25 °C and 60% humidity. All flies were transferred to fresh vials at least twice per week.
To determine bacterial growth, M. marinum-infected live W1118, Atg7 control, and Atg7 mutant flies were incubated with various containing medium (OMS-A or co-treated with chloroquine and OMS-A) for indicated days (n = 20/group, three independent experiments). The homogenized M. marinum-infected flies were serially diluted and plated onto 7H10 agar. After 14 days, the number of colonies on each plate was counted.
NF-κB luciferase reporter assay
NF-κB luciferase reporter assays were performed as described previously55. Briefly, BMDMs were transduced with adenoviruses harboring a NF-κB-luciferase reporter plasmid (Genetransfer Vector Core, Iowa, IA, USA) for 36 h. The cells were then infected with Mtb for 4 h and stimulated with OMS-A or OMS-B for 6 h. The cells were washed three times in PBS and lysed in luciferase lysis buffer (E1531, Promega, Madison, WI, USA). A luciferase assay system (E1501, Promega) was used according to the manufacturer’s instructions.
Statistical analyses
All data obtained from independent experiments are presented as means ± SD. Group-wise post-hoc comparisons were performed using the Student’s t-test in GraphPad Prism 5.0. Differences were considered statistically significant if P < 0.05.
Electronic supplementary material
Acknowledgements
This research was supported by a grant of the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare, Republic of Korea (HI15C0395) and research fund of Chungnam National University. This work was also supported by a National Research Foundation of Korea (NRF) grant funded by the Korean government (Ministry of ICT and Future Planning; 2009–0083533) and a grant of the Marine Biotechnology Program (Genome Analysis of Marine Organisms and Development of Functional Applications, PJT200620) at Seoul National University.
Author Contributions
T.S.K., Y.H.S., and H.M.L. contributed equally to this work. T.S.K., Y.H.S., H.M.L., J.K.K., J.H.C., J.C.J., S. U., H.S.J., and M.K. performed the experiments in this work. Y.H.S., S.U., M.K., G.H.C., H.J.C., D.C.O., and E.K.J. contributed the reagents. T.S.K., H.M.L., J.C.J., Y.H.S., S.U., J.H.C., G.H.C., H.J.C., D.C.O., and E.K.J. analysed the data. T.S.K., H.M.L., G.H.C., D.C.O., and E.K.J. wrote the paper. E.K.J. and D.C.O. supervised the experimental design and corresponding authors. All authors reviewed the manuscript.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Tae Sung Kim, Yern-Hyerk Shin and Hye-Mi Lee contributed equally to this work.
Electronic supplementary material
Supplementary information accompanies this paper at doi:10.1038/s41598-017-03477-3
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Dong-Chan Oh, Email: dongchanoh@snu.ac.kr.
Eun-Kyeong Jo, Email: hayoungj@cnu.ac.kr.
References
- 1.Committee, W. G. A. b. t. G. R. Guidance for National Tuberculosis Programmes on the Management of Tuberculosis in Children. 2nd edition. Geneva: World Health Organization; 2014 (2014). [PubMed]
- 2.Nguyen L, Pieters J. The Trojan horse: survival tactics of pathogenic mycobacteria in macrophages. Trends Cell Biol. 2005;15:269–276. doi: 10.1016/j.tcb.2005.03.009. [DOI] [PubMed] [Google Scholar]
- 3.Levine B, Mizushima N, Virgin HW. Autophagy in immunity and inflammation. Nature. 2011;469:323–335. doi: 10.1038/nature09782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bradfute SB, et al. Autophagy as an immune effector against tuberculosis. Curr Opin Microbiol. 2013;16:355–365. doi: 10.1016/j.mib.2013.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gutierrez MG, et al. Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell. 2004;119:753–766. doi: 10.1016/j.cell.2004.11.038. [DOI] [PubMed] [Google Scholar]
- 6.Deretic V, Saitoh T, Akira S. Autophagy in infection, inflammation and immunity. Nat Rev Immunol. 2013;13:722–737. doi: 10.1038/nri3532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Münz C. Regulation of innate immunity by the molecular machinery of macroautophagy. Cell Microbiol. 2014;16:1627–1636. doi: 10.1111/cmi.12358. [DOI] [PubMed] [Google Scholar]
- 8.Watson RO, Manzanillo PS, Cox JS. Extracellular M. tuberculosis DNA Targets Bacteria for Autophagy by Activating the Host DNA-Sensing Pathway. Cell. 2012;150:803–815. doi: 10.1016/j.cell.2012.06.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Singh SB, Davis AS, Taylor GA, Deretic V. Human IRGM induces autophagy to eliminate intracellular mycobacteria. Science. 2006;313:1438–1441. doi: 10.1126/science.1129577. [DOI] [PubMed] [Google Scholar]
- 10.Fabri M, et al. Vitamin D is required for IFN-gamma-mediated antimicrobial activity of human macrophages. Sci Transl Med. 2011;3:104ra102. doi: 10.1126/scitranslmed.3003045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hardie DG, Ross FA, Hawley SA. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol. 2012;13:251–262. doi: 10.1038/nrm3311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yang CS, et al. The AMPK-PPARGC1A pathway is required for antimicrobial host defense through activation of autophagy. Autophagy. 2014;10:785–802. doi: 10.4161/auto.28072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mounier R, et al. AMPKα1 regulates macrophage skewing at the time of resolution of inflammation during skeletal muscle regeneration. Cell Metab. 2013;18:251–264. doi: 10.1016/j.cmet.2013.06.017. [DOI] [PubMed] [Google Scholar]
- 14.O’Neill LA, Hardie DG. Metabolism of inflammation limited by AMPK and pseudo-starvation. Nature. 2013;493:346–355. doi: 10.1038/nature11862. [DOI] [PubMed] [Google Scholar]
- 15.Fullerton MD, Steinberg G, Schertzer JD. Immunometabolism of AMPK in insulin resistance and atherosclerosis. Mol Cell Endocrinol. 2013;366:224–234. doi: 10.1016/j.mce.2012.02.004. [DOI] [PubMed] [Google Scholar]
- 16.Um S, et al. Ohmyungsamycins A and B: cytotoxic and antimicrobial cyclic peptides produced by Streptomyces sp. from a volcanic island. J Org Chem. 2013;78:12321–12329. doi: 10.1021/jo401974g. [DOI] [PubMed] [Google Scholar]
- 17.Stehr M, Elamin AA, Singh M. Filling the pipeline - new drugs for an old disease. Curr Top Med Chem. 2014;14:110–129. doi: 10.2174/1568026613666131113152908. [DOI] [PubMed] [Google Scholar]
- 18.Dionne MS, Pham LN, Shirasu-Hiza M, Schneider DS. Akt and FOXO dysregulation contribute to infection-induced wasting in Drosophila. Curr Biol. 2006;16:1977–1985. doi: 10.1016/j.cub.2006.08.052. [DOI] [PubMed] [Google Scholar]
- 19.Kim JJ, et al. Host cell autophagy activated by antibiotics is required for their effective antimycobacterial drug action. Cell Host Microbe. 2012;11:457–468. doi: 10.1016/j.chom.2012.03.008. [DOI] [PubMed] [Google Scholar]
- 20.Chandler HL, et al. Cyclosporine A prevents ex vivo PCO formation through induction of autophagy-mediated cell death. Exp Eye Res. 2015;134:63–72. doi: 10.1016/j.exer.2015.03.020. [DOI] [PubMed] [Google Scholar]
- 21.Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell. 2010;140:313–326. doi: 10.1016/j.cell.2010.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Andersson, A. M. et al. Autophagy induction targeting mTORC1 enhances Mycobacterium tuberculosis replication in HIV co-infected human macrophages. Sci Rep15, 6:28171 (2016). [DOI] [PMC free article] [PubMed]
- 23.Vergne I, et al. Mycobacterium tuberculosis phagosome maturation arrest: mycobacterial phosphatidylinositol analog phosphatidylinositol mannoside stimulates early endosomal fusion. Mol Biol Cell. 2004;15:751–760. doi: 10.1091/mbc.E03-05-0307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chandra P, et al. Mycobacterium tuberculosis Inhibits RAB7 Recruitment to Selectively Modulate Autophagy Flux in Macrophages. Sci Rep. 2015;5:16320. doi: 10.1038/srep16320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Deretic V. Autophagy, an immunologic magic bullet: Mycobacterium tuberculosis phagosome maturation block and how to bypass it. Future Microbiol. 2008;3:517–524. doi: 10.2217/17460913.3.5.517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stein SC, et al. The regulation of AMP-activated protein kinase by phosphorylation. Biochem J. 2000;345(Pt 3):437–443. doi: 10.1042/bj3450437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zirin J, Nieuwenhuis J, Perrimon N. Role of autophagy in glycogen breakdown and its relevance to chloroquine myopathy. PLoS Biol. 2013;11:e1001708. doi: 10.1371/journal.pbio.1001708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Grahame Hardie D. AMP-activated protein kinase: a key regulator of energy balance with many roles in human disease. J Intern Med. 2014;276:543–559. doi: 10.1111/joim.12268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wallis RS, Hafner R. Advancing host-directed therapy for tuberculosis. Nat Rev Immunol. 2015;15:255–263. doi: 10.1038/nri3813. [DOI] [PubMed] [Google Scholar]
- 30.Rayasam GV, Balganesh TS. Exploring the potential of adjunct therapy in tuberculosis. Trends Pharmacol Sci. 2015;36:506–513. doi: 10.1016/j.tips.2015.05.005. [DOI] [PubMed] [Google Scholar]
- 31.Sachan M. Opportunities and Challenges for Host-Directed Therapies in Tuberculosis. Curr Pharm Des. 2016;22:2599–2604. doi: 10.2174/1381612822666160128150636. [DOI] [PubMed] [Google Scholar]
- 32.Jo EK. Autophagy as an innate defense against mycobacteria. Pathog Dis. 2013;67:108–118. doi: 10.1111/2049-632X.12023. [DOI] [PubMed] [Google Scholar]
- 33.Yuk JM, et al. Vitamin D3 induces autophagy in human monocytes/macrophages via cathelicidin. Cell Host Microbe. 2009;6:231–243. doi: 10.1016/j.chom.2009.08.004. [DOI] [PubMed] [Google Scholar]
- 34.Campbell GR, Spector SA. Vitamin D Inhibits Human Immunodeficiency Virus Type 1 and Mycobacterium tuberculosis Infection in Macrophages through the Induction of Autophagy. PLoS Pathog. 2012;8:e1002689. doi: 10.1371/journal.ppat.1002689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Delgado MA, et al. Toll-like receptors control autophagy. EMBO J. 2008;27:1110–1121. doi: 10.1038/emboj.2008.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xu Y, et al. Toll-like receptor 4 is a sensor for autophagy associated with innate immunity. Immunity. 2007;27:135–144. doi: 10.1016/j.immuni.2007.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ponpuak M, et al. Delivery of cytosolic components by autophagic adaptor protein p62 endows autophagosomes with unique antimicrobial properties. Immunity. 2010;32:329–341. doi: 10.1016/j.immuni.2010.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Daletos G, et al. Callyaerins from the Marine Sponge Callyspongia aerizusa: Cyclic Peptides with Antitubercular Activity. J Nat Prod. 2015;78:1910–1925. doi: 10.1021/acs.jnatprod.5b00266. [DOI] [PubMed] [Google Scholar]
- 39.Gao W, et al. The cyclic peptide ecumicin targeting ClpC1 is active against Mycobacterium tuberculosis in vivo. Antimicrob Agents Chemother. 2015;59:880–889. doi: 10.1128/AAC.04054-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gavrish E, et al. Lassomycin, a ribosomally synthesized cyclic peptide, kills mycobacterium tuberculosis by targeting the ATP-dependent protease ClpC1P1P2. Chem Biol. 2014;21:509–518. doi: 10.1016/j.chembiol.2014.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pal S, et al. Tetrahydrofuran amino acid-containing gramicidin S analogues with improved biological profiles. Org Biomol Chem. 2015;13:6789–6802. doi: 10.1039/C5OB00622H. [DOI] [PubMed] [Google Scholar]
- 42.Zheng Q, et al. Thiopeptide Antibiotics Exhibit a Dual Mode of Action against Intracellular Pathogens by Affecting Both Host and Microbe. Chem Biol. 2015;22:1002–1007. doi: 10.1016/j.chembiol.2015.06.019. [DOI] [PubMed] [Google Scholar]
- 43.Bhattacharya B, et al. Fine-tuning of macrophage activation using synthetic rocaglate derivatives. Sci Rep. 2016;6:24409. doi: 10.1038/srep24409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mahon RN, Hafner R. Immune Cell Regulatory Pathways Unexplored as Host-Directed Therapeutic Targets for Mycobacterium tuberculosis: An Opportunity to Apply Precision Medicine Innovations to Infectious Diseases. Clin Infect Dis. 2015;61(Suppl 3):S200–216. doi: 10.1093/cid/civ621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hawn TR, Shah JA, Kalman D. New tricks for old dogs: countering antibiotic resistance in tuberculosis with host-directed therapeutics. Immunol Rev. 2015;264:344–362. doi: 10.1111/imr.12255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rekha RS, et al. Phenylbutyrate induces LL-37-dependent autophagy and intracellular killing of Mycobacterium tuberculosis in human macrophages. Autophagy. 2015;11:1688–1699. doi: 10.1080/15548627.2015.1075110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Singhal A, et al. Metformin as adjunct antituberculosis therapy. Sci Transl Med. 2014;6:263ra159. doi: 10.1126/scitranslmed.3009885. [DOI] [PubMed] [Google Scholar]
- 48.Yue W, Yang CS, DiPaola RS, Tan XL. Repurposing of metformin and aspirin by targeting AMPK-mTOR and inflammation for pancreatic cancer prevention and treatment. Cancer Prev Res (Phila) 2014;7:388–397. doi: 10.1158/1940-6207.CAPR-13-0337. [DOI] [PubMed] [Google Scholar]
- 49.Moiseeva O, et al. Metformin inhibits the senescence-associated secretory phenotype by interfering with IKK/NF-kappaB activation. Aging Cell. 2013;12:489–498. doi: 10.1111/acel.12075. [DOI] [PubMed] [Google Scholar]
- 50.Deretic V, et al. Immunologic manifestations of autophagy. J Clin Invest. 2015;125:75–84. doi: 10.1172/JCI73945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bai X, et al. Inhibition of nuclear factor-kappa B activation decreases survival of Mycobacterium tuberculosis in human macrophages. PLoS One. 2013;8:e61925. doi: 10.1371/journal.pone.0061925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chang CP, Su YC, Hu CW, Lei HY. TLR2-dependent selective autophagy regulates NF-κB lysosomal degradation in hepatoma-derived M2 macrophage differentiation. Cell Death Differ. 2013;20:515–523. doi: 10.1038/cdd.2012.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Abadie V, et al. Neutrophils rapidly migrate via lymphatics after Mycobacterium bovis BCG intradermal vaccination and shuttle live bacilli to the draining lymph nodes. Blood. 2005;106:1843–1850. doi: 10.1182/blood-2005-03-1281. [DOI] [PubMed] [Google Scholar]
- 54.Jung HS, et al. Loss of autophagy diminishes pancreatic beta cell mass and function with resultant hyperglycemia. Cell Metab. 2008;8:318–324. doi: 10.1016/j.cmet.2008.08.013. [DOI] [PubMed] [Google Scholar]
- 55.Kim TS, et al. Mycobacterium massiliense induces inflammatory responses in macrophages through Toll-like receptor 2 and c-Jun N-terminal kinase. J Clin Immunol. 2014;34:212–223. doi: 10.1007/s10875-013-9978-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Juhász G, Erdi B, Sass M, Neufeld TP. Atg7-dependent autophagy promotes neuronal health, stress tolerance, and longevity but is dispensable for metamorphosis in Drosophila. Genes Dev. 2007;21:3061–3066. doi: 10.1101/gad.1600707. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.