

Abstract
Amyotrophic lateral sclerosis (ALS) pathology is linked to the aberrant aggregation of specific proteins, including TDP‐43, FUS, and SOD1, but it is not clear why these aggregation events cause ALS. In this issue of The EMBO Journal, Mateju et al (2017) report a direct link between misfolded proteins accumulating in stress granules and the phase transition of these stress granules from liquid to solid. This discovery provides a model connecting protein aggregation to stress granule dysfunction.
Subject Categories: Membrane & Intracellular Transport, Protein Biosynthesis & Quality Control, RNA Biology
How does stress kill neurons? This seemingly straightforward question has been at the heart of the neurodegenerative disease field for decades, and has proven surprisingly difficult to answer. The genetics and tissue pathology of aging‐associated neurodegenerative diseases such as ALS point to protein aggregation and the subsequent disruption of the protein folding environment as causative agents of cell death. On the other hand, cells are wired to withstand stress and aggregation, with only a small subset of aggregation‐prone proteins leading to a disease state. In fact, aggregation is surprisingly widespread in different cell types as a common and functional mode of protein interaction. Why then is the aggregation of certain proteins so toxic to a subset of neurons? In this issue of The EMBO Journal, Mateju et al (2017) make the breakthrough discovery that protein misfolding and aggregation directly affect stress granule properties and that this may be the initial step on the way to ALS pathology.
In recent years, the stress granule (SG) has emerged as the missing piece that makes the seemingly disparate genetic, cell biological, and pathological observations in the ALS puzzle fit together (Li et al, 2013). SG components, including TDP‐43, FUS, TAF15, EWSR1, hnRNPs, and others, are hot spots for ALS‐causing mutations and are cell biologically associated with the pathology of ALS‐FTD spectrum diseases (Ling et al, 2013; Aguzzi & Altmeyer, 2016). The SG is itself a type of aggregate, one that is formed when RNAs and RNA‐binding proteins (RBPs) that are homogenously distributed throughout the cytoplasm undergo liquid–liquid demixing (Molliex et al, 2015; Aguzzi & Altmeyer, 2016). The result is a RNA–protein (RNP) granule that is not a solid precipitate (the way aggregates are commonly thought to be) but rather has the physical properties of a viscous liquid droplet (Brangwynne et al, 2015).
Much is known about the liquid‐like properties of SGs: They have high internal mobility and readily exchange proteins with the cytoplasm; they flow and fuse much like drops of water or honey; they shear under flow stress, and they are made up mainly of RBPs (Hyman et al, 2014). Less is known about the function of SGs. Since they predominantly contain silenced mRNAs and transcription factors, they are thought to regulate protein synthesis in response to changing stress conditions and to regulate RNA degradation in concert with P‐bodies (Protter & Parker, 2016). SGs may also play a key role in mRNA targeting and delivery (such as to the synapse in neurons). Moreover, since SGs can be triggered by dozens of stress‐related signals and can sequester and release a diverse set of factors, they have also been proposed to play a much wider role as signaling hubs, simultaneously mediating splicing, translation, proliferative and apoptotic signaling, silencing, and degradation (Kedersha et al, 2013). A fascinating new hypothesis, advanced by Mateju et al (2017), is that SG function is intimately linked to their material properties. By following individual SGs in live cells, the authors demonstrate that healthy SGs can transition to an aberrant state with altered physical properties. The authors show that cells put significant effort into repairing or eliminating aberrant SGs, implying that a failure to do so may bring about the pathology observed in ALS.
Previous work has shown that SG components are found in ALS‐associated aggregates that resemble aberrant SGs. Mutant and normal variants of many SG components, including FUS and hnRPPs, have also been shown to phase separate into liquid‐like structures in vitro, and then slowly mature into solid‐like aggregates (Patel et al, 2015). The maturation process is significantly faster for ALS‐associated mutants. The key question, therefore, that Mateju et al (2017) set out to answer is whether aggregates containing SG components are aberrant SGs that have solidified due to stress and aging, or whether the defective SG components simply aggregate on their own, without a direct connection to SGs. In order to address this critical issue, the authors designed a system in which they simulate stress and aging by heat‐shocking the cells (a process that is known to generate widespread protein misfolding). They use a variety of cell biological and biophysical approaches to follow SG dynamics in live cells, with the SG markers FUS and G3BP1 expressed at physiological levels. Remarkably, the authors find that misfolded proteins “contaminate” healthy SGs during stress. What is important here is that the authors use misfolding‐prone proteins (Kaganovich et al, 2008), Ubc9ts, VHL, and SOD1A4V, which have no a priori cell biological or functional association with SGs. The SOD1A4V is a mutation known to cause ALS, but SOD1 is not generally thought to be a SG component. Ubc9ts and VHL are proteins that have been commonly used in previous studies to model misfolding and aggregation. These proteins co‐localize with some SGs during stress, providing a general model for how the folding environment impacts the dynamics of healthy and aberrant SGs.
Normal SGs (those not containing misfolded proteins like SOD1 and Ubc9ts) are transient, while SGs “contaminated” with misfolded proteins show dramatically reduced recovery. Moreover, these aberrant SGs exhibited altered physical properties. Photobleaching studies showed that the internal mobility of aberrant SGs was highly reduced; these SGs no longer fused together in the way that healthy SGs do, and they clearly differed in their morphology.
Proteins that are highly aggregation‐prone, including ALS‐associated mutants of SOD1, have previously been shown to decrease the mobility of other misfolded proteins when co‐aggregating in the same compartment (Weisberg et al, 2012). This slowdown was the result of titrating Hsp70, an essential folding and degradation chaperone, by the aggregated mutant SOD1. Here, the authors examine the relationship between the exogenous aggregating protein (SOD1A4V) and endogenous SG markers and find that SOD1A4V aggregates separately at the periphery of the SG and that it does not initially affect the mobility and properties of the RBPs inside the core of the SG. What does seem to happen over time is that the SG is depleted of its RBP content and instead accumulates more and more aggregated SOD1A4V. In addition to ectopically expressed misfolded proteins, the authors demonstrate that endogenous defective ribosomal products (DRiPs) accumulate in heat‐shock‐induced SGs as well.
The aberrant SGs were positive for chaperones such as Hsp70, Hsp27, and VCP, suggesting that these chaperones regulate the disassembly of aberrant SGs. Indeed, inhibiting Hsp70 dramatically increased the prevalence of aberrant SGs. These data indicate that the chaperone machinery is necessary not only to prevent aberrant protein misfolding and aggregation, but also for the delicate task of surveilling SGs and removing harmful aggregates from the healthy aggregate‐like SG structure (Fig 1). How chaperones distinguish potentially harmful misfolded proteins from the intrinsically disordered RBPs in the SG remains an important unanswered question. However, the authors show that aberrant SGs that cannot be “cleaned” from co‐aggregating SOD1A4V are sent for autophagic degradation.
Figure 1. Chaperone surveillance of aberrant stress granules.
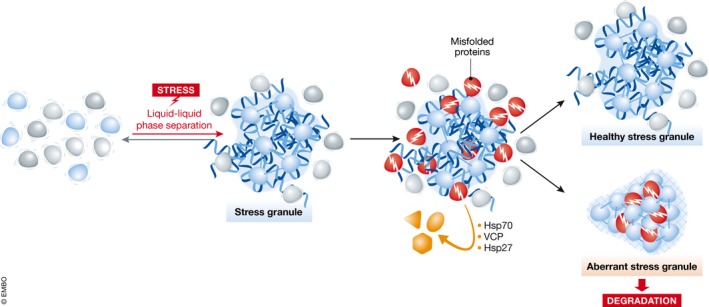
Stress causes RNAs, RBPs, and translational machinery to phase separate into liquid‐like stress granules. When misfolded proteins are abundant, they can “contaminate” the healthy stress granule and alter its physical properties, causing it to “freeze” and become more gel‐like or solid. If aberrant SGs cannot be repaired, they are targeted for degradation. The failure of this quality control process is thought to underlie onset of neurodegenerative diseases such as ALS.
The important work of Mateju et al (2017) clearly establishes the concept that healthy SGs can accumulate harmful aggregates over time, in the form of DRiPs, mutant proteins, and damaged factors that are otherwise external to SGs. What remains to be discovered in future studies is how this process impacts the functional role of SGs in the cell, and how this process brings about cell death. Additional questions will be of equal importance for linking this study to neurodegeneration in human patients. Why does the process of SG surveillance function so well for several decades, only to decline abruptly? What is the essential SG function that cells simply cannot do without? And finally, why does this pathology affect specific groups of aging neurons? The work done by Mateju et al (2017) will hopefully bring us one step closer to answering these questions.
See also: D Mateju et al (June 2017)
References
- Aguzzi A, Altmeyer M (2016) Phase separation: linking cellular compartmentalization to disease. Trends Cell Biol 26: 547–558 [DOI] [PubMed] [Google Scholar]
- Brangwynne CP, Tompa P, Pappu RV (2015) Polymer physics of intracellular phase transitions. Nat Phys 11: 899–904 [Google Scholar]
- Hyman AA, Weber CA, Jülicher F (2014) Liquid‐liquid phase separation in biology. Annu Rev Cell Dev Biol 30: 39–58 [DOI] [PubMed] [Google Scholar]
- Kaganovich D, Kopito R, Frydman J (2008) Misfolded proteins partition between two distinct quality control compartments. Nature 454: 1088–1095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kedersha N, Ivanov P, Anderson P (2013) Stress granules and cell signaling: more than just a passing phase?. Trends Biochem Sci 38: 494–506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li YR, King OD, Shorter J, Gitler AD (2013) Stress granules as crucibles of ALS pathogenesis. J Cell Biol 201: 361–372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling S‐C, Polymenidou M, Cleveland DW (2013) Converging mechanisms in ALS and FTD: disrupted RNA and protein homeostasis. Neuron 79: 416–438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mateju D, Franzmann TM, Patel A, Kopach A, Boczek EE, Maharana S, Lee HO, Carra S, Hyman AA, Alberti S (2017) An aberrant phase transition of stress granules triggered by misfolded protein and prevented by chaperone function. EMBO J 36: 1669–1687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molliex A, Temirov J, Lee J, Coughlin M, Kanagaraj AP, Kim HJ, Mittag T, Taylor JP (2015) Phase separation by low complexity domains promotes stress granule assembly and drives pathological fibrillization. Cell 163: 123–133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel A, Lee HO, Jawerth L, Maharana S, Jahnel M, Hein MY, Stoynov S, Mahamid J, Saha S, Franzmann TM, Pozniakovski A, Poser I, Maghelli N, Royer LA, Weigert M, Myers EW, Grill S, Drechsel D, Hyman AA, Alberti S (2015) A liquid‐to‐solid phase transition of the ALS protein FUS accelerated by disease mutation. Cell 162: 1066–1077 [DOI] [PubMed] [Google Scholar]
- Protter DSW, Parker R (2016) Principles and properties of stress granules. Trends Cell Biol 26: 668–679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisberg SJ, Lyakhovetsky R, Werdiger A, Gitler AD, Soen Y, Kaganovich D (2012) Compartmentalization of superoxide dismutase 1 (SOD1G93A) aggregates determines their toxicity. Proc Natl Acad Sci USA 109: 15811–15816 [DOI] [PMC free article] [PubMed] [Google Scholar]