

Abstract
Assembly of the SNARE proteins syntaxin1, SNAP25, and synaptobrevin into a SNARE complex is essential for exocytosis in neurons. For efficient assembly, SNAREs interact with additional proteins but neither the nature of the intermediates nor the sequence of protein assembly is known. Here, we have characterized a ternary complex between syntaxin1, SNAP25, and the SM protein Munc18‐1 as a possible acceptor complex for the R‐SNARE synaptobrevin. The ternary complex binds synaptobrevin with fast kinetics, resulting in the rapid formation of a fully zippered SNARE complex to which Munc18‐1 remains tethered by the N‐terminal domain of syntaxin1. Intriguingly, only one of the synaptobrevin truncation mutants (Syb1‐65) was able to bind to the syntaxin1:SNAP25:Munc18‐1 complex, suggesting either a cooperative zippering mechanism that proceeds bidirectionally or the progressive R‐SNARE binding via an SM template. Moreover, the complex is resistant to disassembly by NSF. Based on these findings, we consider the ternary complex as a strong candidate for a physiological intermediate in SNARE assembly.
Keywords: intermediates, Munc18, NSF, SM‐protein, SNARE
Subject Categories: Membrane & Intracellular Transport, Neuroscience
Introduction
Exocytotic membrane fusion in presynaptic nerve terminals is mediated by the assembly of the SNARE proteins synaptobrevin (also referred to as VAMP‐2) residing in the synaptic vesicles, and syntaxin1 and SNAP25 residing in the presynaptic plasma membrane. Assembly is exergonic and thought to proceed from the membrane‐distal N‐terminal ends toward the C‐terminal membrane anchors, thus pulling the membranes together and overcoming the energy barrier for membrane fusion. The end result is a stable bundle of four α‐helices, in which each helix is contributed by a different SNARE motif (one each from syntaxin1 and synaptobrevin, also termed Qa‐SNARE and R‐SNARE respectively, and two from SNAP25, also termed Qb and Qc‐SNARE) (Stein et al, 2009).
Although in vitro SNARE assembly can mediate fusion without the need for cofactors, it is generally thought to be regulated by accessory proteins in intact cells (Südhof & Rizo, 2011). These include members of the conserved SM and CATCHR protein families (Munc18 and Munc13, respectively, in neurons) that function in all SNARE‐mediated fusion events, and synaptotagmin and complexin that function in the calcium‐regulated triggering of neuronal exocytosis (Jahn & Fasshauer, 2012). However, despite major efforts by many laboratories, the precise sequence and mechanism of these proteins still remain unclear. In particular, the SM protein Munc18‐1 continues to baffle scientists because of contradicting observations (Toonen & Verhage, 2007). For instance, genetic ablation of Munc18‐1 completely blocks neurotransmitter release (Verhage et al, 2000). On the other hand, Munc18‐1 binds with high affinity to syntaxin1, locking the SNARE in a cleft of Munc18‐1, in a “closed” conformation such that it is unable to enter SNARE complexes (Misura et al, 2000).To resolve this conflict, it was proposed that under certain conditions, Munc18‐1 loosens its grip on syntaxin1, allowing it to partially open as needed for SNARE assembly. For instance, Munc18‐1 appears to bind less tightly to syntaxin1 when it contains the transmembrane domain (Lewis et al, 2001; Zilly et al, 2006). Moreover, there is a spatially separate binding site between syntaxin1 and Munc18‐1 involving the N‐terminal peptide of syntaxin1, which binds to the outer surface of Munc18‐1 (Burkhardt et al, 2008), and may play a role in syntaxin1 opening. Intriguingly, disruption of N‐peptide binding does not affect synaptic transmission while it allows syntaxin1 to readily enter SNARE complexes (Burkhardt et al, 2008; Meijer et al, 2012), suggesting that “locking” of syntaxin1, at whichever point in the SNARE activation pathway, is not required for function. Recently, it has been suggested that Munc13‐1 may help in opening syntaxin1 (Ma et al, 2011; Yang et al, 2015), thus resulting in a reactive syntaxin1 intermediate, with both Munc18‐1 and Munc13‐1 being bound. Indeed, acceleration of SNARE‐mediated liposome fusion was observed in the presence of Munc18‐1 and Munc13‐1 (also requiring synaptotagmin in one case; Ma et al, 2013; Liu et al, 2016) but neither the structural intermediates nor the rate‐limiting steps are known. Finally, fatty acids like arachidonic acid may alter the interaction of Munc18‐1 with syntaxin1 to favor SNARE assembly (Rickman & Davletov, 2005), either by exposing an additional binding site or by loosening the closed syntaxin1: Munc18‐1 structure (Connell et al, 2007).
The next open question is to understand the binding sequence of the other two SNAREs (SNAP25 and synaptobrevin). Again, the suggestions are conflicting. According to the first concept, Munc18‐1 forms a metastable complex with SNAP25 and syntaxin1, thus providing a pre‐assembled acceptor site for synaptobrevin and facilitating SNARE complex formation. Intuitively, such an activated Qabc‐SNARE acceptor complex is appealing since it can be pre‐formed at the plasma membrane and does not require previous docking of the vesicle, in agreement with the observation that under certain conditions, vesicles can undergo exocytosis directly after arriving at the plasma membrane, with no measurable delay (Ninomiya et al, 1996, 1997; Takahashi et al, 1997; Liu et al, 2005). Evidence for such a ternary interaction was derived from studies using purified proteins reconstituted in liposomes or supported bilayers (Guan et al, 2008; Weninger et al, 2008) and from high‐resolution microscopy of the proteins in intact neuronal plasma membranes (Pertsinidis et al, 2013). Moreover, it has recently been shown by electron paramagnetic resonance spectroscopy that syntaxin1 can associate with SNAP25 while being bound to Munc18‐1, thereby yielding a 1:1:1 complex containing syntaxin1, SNAP25 and Munc18‐1 (Dawidowski & Cafiso, 2016). This observation, however, disagrees with an earlier study reporting that the addition of Munc18‐1 to syntaxin1:SNAP25 complexes results in complete dissociation of SNAP25 from syntaxin1, resulting in the formation of syntaxin1:Munc18‐1 complexes (Ma et al, 2013). According to the second concept, the SM protein may initiate SNARE complex assembly by simultaneously interacting with Qa‐ and R‐SNAREs, thus forming an acceptor site for the subsequent binding of Qb/c SNAREs. Support for such a scenario is derived from experiments in which N‐terminally cross‐linked SNARE complexes were pulled apart with optical tweezers (Ma et al, 2015). Moreover, crystal structures of the Munc18‐1 orthologue Vps33 have recently been published in which either the corresponding Qa‐SNARE or the R‐SNARE was bound (Baker et al, 2015), lending further support to the second model (Baker & Hughson, 2016).
The difficulties in pinpointing the intermediates of SNARE assembly are not surprising, considering the fact that the acceptor complexes need to be maintained at a high energy level in order to avoid siphoning off too much energy from the final zippering reaction. The reactive SNAREs easily assemble into stable partial complexes that may be “off‐pathway” such as the syntaxin1:SNAP25 complex (in a 2:1 stoichiometry; Fasshauer et al, 1997), which are both kinetically and thermodynamically trapped. Therefore, in this paper, we have investigated whether a ternary complex between full‐length syntaxin1 (containing its transmembrane domain), SNAP25, and Munc18‐1 can be assembled in vitro and whether synaptobrevin is capable of binding to such a complex, resulting in the formation of a fully zippered SNARE complex.
Results
A syntaxin1:SNAP25:Munc18‐1 complex allows for fast and efficient formation of SNARE complexes with synaptobrevin
In the first set of experiments, we investigated whether a ternary complex between syntaxin1, SNAP25, and Munc18‐1 (all full‐length proteins, with syntaxin1 containing its transmembrane domain) can be assembled and purified in vitro. To this end, we purified the three monomeric proteins and allowed them to assemble in vitro (see Materials and Methods). Next, we purified the complex by ion‐exchange chromatography, followed by size‐exclusion chromatography (Fig 1A). Analysis by SDS–PAGE (Fig 1B) revealed that in the first peak of Fig 1A, all the three proteins comigrate, suggesting that a ternary complex containing syntaxin1, SNAP25, and Munc18‐1 had indeed been formed.
Figure 1. Syntaxin1:SNAP25:Munc18‐1 tripartite complex sets the stage for SNARE complex assembly.
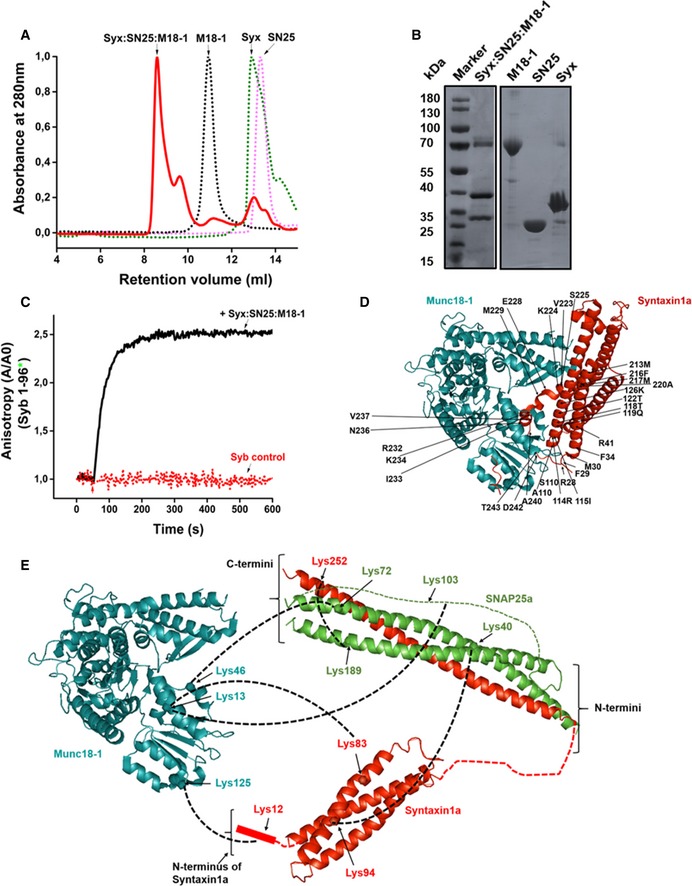
- Separation of the syntaxin1:SNAP25:Munc18‐1 complex from the monomers. Purification was performed on a Superdex 10/300 Increase column (GE Healthcare).
- SDS–PAGE analysis of the syntaxin1:SNAP25:Munc18‐1 complex and the corresponding monomers.
- Binding of fluorescently labeled synaptobrevin (1‐96) to the unlabeled syntaxin1:SNAP25:Munc18‐1 acceptor complex. Upon addition of the acceptor complex, a very fast increase in anisotropy was observed. This increase was prevented by adding excess of unlabeled synaptobrevin to the reaction mixture (1‐96; syb control).
- Crystal structure of the syntaxin1:Munc18‐1 binary complex (Burkhardt et al, 2008; PDBID: 3c98). The labeled residues indicate the regions on syntaxin1 that form contacts with the “cleft” of Munc18‐1. In this structure, syntaxin1 is present in a “closed” conformation.
- Chemical cross‐linking of the syntaxin1:SNAP25:Munc18‐1 complex showed direct cross‐links between Munc18‐1 and both SNAP25 and syntaxin1, as indicated. Munc18‐1 is shown in cyan, SNAP25 in green, and syntaxin1 in red. The structure of the syntaxin1:SNAP25 complex has been adopted from the crystal structure of the SNARE complex (Stein et al, 2009) (PDBID:3HD7), and the Habc domain has been adopted from the structure of the N‐terminal domain of syntaxin1 (Fernandez et al, 1998) (PDBID:1BR0).
Next, we tested whether synaptobrevin can bind to this ternary complex. To this end, we used a point mutant (S28C) of the cytoplasmic fragment of synaptobrevin (1‐96), labeled with the fluorescent dye Oregon Green, as reporter. The protein was added to freshly prepared ternary complex, and changes in fluorescence anisotropy were monitored as an indicator for binding (Pobbati et al, 2006). An increase in anisotropy of synaptobrevin was observed upon the addition of the syntaxin1:SNAP25:Munc18‐1 complex (Fig 1C). In contrast to this, no such increase was observed when an excess of unlabeled synaptobrevin was added to the reaction mixture. We also tried to assemble this complex using a syntaxin1 mutant lacking its transmembrane domain (syntaxin1–262; Zhang et al, 2015). Although assembly of a ternary complex was observed, fast binding of synaptobrevin was not detectable (Fig EV1A).
Figure EV1. A syntaxin1:SNAP25:Munc18‐1 complex containing the transmembrane domain of syntaxin1 serves as a labile, yet efficient acceptor for synaptobrevin binding.
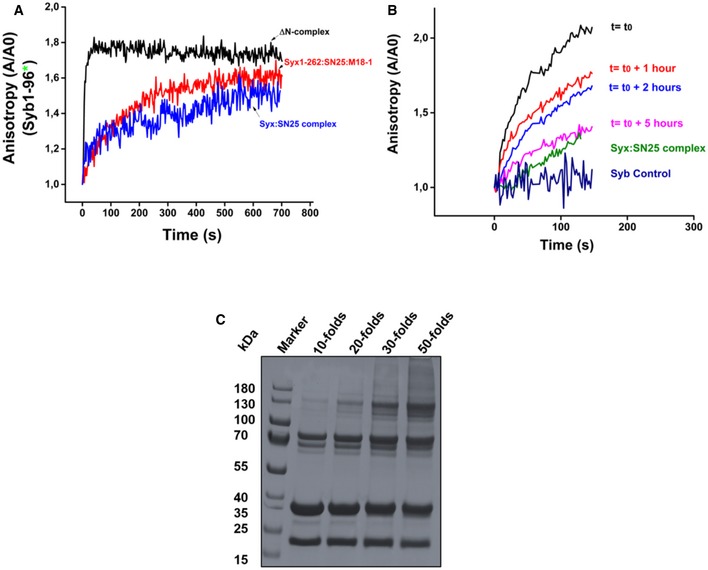
- Binding of synaptobrevin to the syntaxin1 (1‐262):SNAP25:Munc18‐1 complex (red curve) was slower as compared to the ΔN complex (black curve) and resembled binding to the binary syntaxin1a:SNAP25 (2:1) complex (blue curve). This was in contrast to the complex containing the transmembrane domain of syntaxin1. Precise time‐point measurements using the syntaxin1 (1‐262):SNAP25:Munc18‐1 complex were, however, not performed.
- Binding of fluorescently labeled synaptobrevin (1‐96) to the syntaxin1 (1‐288): SNAP25:Munc18‐1 complex was measured over increasing time intervals using fluorescence anisotropy. The complex showed a decrease in the rate of synaptobrevin binding with increasing periods of time.
- The syntaxin1 (1‐288):SNAP25:Munc18‐1 complex could be cross‐linked by the chemical cross‐linker BS3. A titration with increasing amounts of the cross‐linker showed that a 50‐fold excess of the cross‐linker was optimum for cross‐linking the syntaxin1:SNAP25:Munc18‐1 complex. The “folds” indicate the molar excess of BS3. The cross‐linked band appeared around a molecular weight of 130 kDa.
Since the ternary complex lost its activity over extended incubation times (Fig EV1B), we resorted to chemical cross‐linking of the purified complex in order to obtain insights into its architecture. To this end, we used the cross‐linker bis(sulfosuccinimidyl)suberate (BS3), a homobifunctional N‐hydroxysuccinimide ester (spacer arm length 11.4 Å) that reacts with primary amino groups in lysine side chains or at the N‐terminus. The cross‐linked fractions (see Fig EV1C) were analyzed by mass spectrometry (MS/MS; see Materials and Methods). The cross‐links obtained were compared to the previously reported crystal structure of the syntaxin1:Munc18‐1 complex (Fig 1D). In the syntaxin1:SNAP25:Munc18‐1 complex, cross‐links were obtained between all three proteins namely syntaxin1, SNAP25, and Munc18‐1 (see Table EV1). Most importantly, we observed cross‐links between lysine 46 of Munc18‐1 and lysine 72 of SNAP25a (Fig 1E). In Munc18‐1, lysine 46 is localized to the cleft that is occupied by syntaxin1 in the binary syntaxin1:Munc18‐1 complex and in SNAP25, the cross‐linked residue (lysine 72) is positioned close to the C‐terminal end of the Qb‐SNARE motif.
These observations indicate that the structure of the syntaxin1:SNAP25:Munc18‐1 complex is different from that of the binary syntaxin1:Munc18‐1 complex. Moreover, cross‐links were also found between the Habc domain of syntaxin1 (Lys 83) and residues in the cleft of Munc18‐1 (Lys 13). Interestingly, intramolecular cross‐links between the Habc domain and the H3 motif of syntaxin1 were very low (Fig EV2), suggesting that in the ternary syntaxin1:SNAP25:Munc18‐1 complex, syntaxin1 is shifted toward the “open” conformation.
Figure EV2. Intra‐cross‐links between the monomeric constituents of the syntaxin1:SNAP25:Munc18‐1 complex.
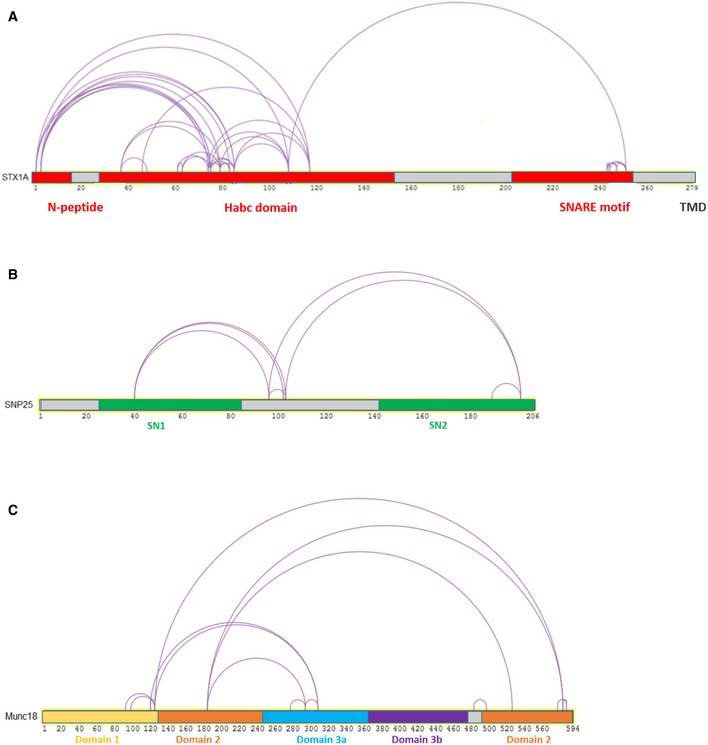
-
A‐CRepresentative intra‐cross‐links between (A) syntaxin1, (B) SNAP25, and (C) Munc18‐1, respectively, in the syntaxin1:SNAP25:Munc18‐1 complex.
Next, we compared synaptobrevin binding to the ternary complex with its binding to two previously characterized SNARE acceptor complexes: the syntaxin1:SNAP25 (2:1) complex (Fasshauer et al, 1999) and the C‐terminally stabilized ΔN complex in which a C‐terminal fragment of synaptobrevin is bound to SNAP25 and syntaxin1, leaving an N‐terminal SNARE binding site free (Pobbati et al, 2006). Synaptobrevin binding to the syntaxin1:SNAP25 (2:1) complexes is known to be slow due to the need for displacing syntaxin1 from the binding site (Margittai et al, 2001). In contrast, binding of synaptobrevin to the ΔN complex is fast due to the accessible N‐terminal binding site (Pobbati et al, 2006). We again performed fluorescence anisotropy measurements using the different acceptor complexes and monitored the binding of labeled synaptobrevin (Syb1‐96). A representation of the complexes used for these experiments has been shown in Fig 2A. As shown in Fig 2B, binding of synaptobrevin to the ternary syntaxin1:SNAP25:Munc18‐1 complex was much faster as compared to the syntaxin1:SNAP25 (2:1) complex, but occurred at a similar timescale as compared to the ΔN complex (Fig 2C). Quantifications of these measurements are shown in Fig EV3A and B. Additionally, no increase in anisotropy was observed when monomeric syntaxin1, SNAP25, or Munc18‐1 was added to fluorescently labeled synaptobrevin (Fig EV3C).
Figure 2. Synaptobrevin binds to both syntaxin1:SNAP25:Munc18‐1 complex and ΔN complex with similar kinetics, leading to SNARE complex assembly.
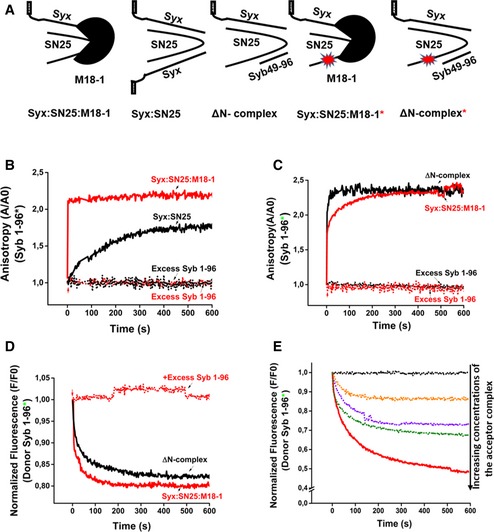
- Cartoons showing the different complexes used in the experiments. Fluorescent and non‐fluorescent versions of the syntaxin1:SNAP25:Munc18‐1 complex and the ΔN complex, as well as the non‐fluorescent version of syntaxin1:SNAP25 complex, were assembled in vitro and purified.
- Binding of fluorescently labeled synaptobrevin to the syntaxin1:SNAP25 complex, as monitored by fluorescence anisotropy, was much slower than to the syntaxin1:SNAP25:Munc18‐1 complex.
- Binding of synaptobrevin to the ΔN complex proceeded with a rate comparable to that of the syntaxin1:SNAP25:Munc18‐1 complex.
- Binding of synaptobrevin to the labeled syntaxin1:SNAP25:Munc18‐1 complex and the ΔN complex measured by FRET (monitored by donor quenching), resulted in kinetics similar to the anisotropy measurements.
- Dose dependence of binding of the syntaxin1:SNAP25:Munc18‐1 complex to synaptobrevin, monitored by FRET.
Figure EV3. Synaptobrevin binds efficiently to the syntaxin1:SNAP25:Munc18‐1 complex, but not to monomeric syntaxin1, SNAP25a or Munc18‐1.
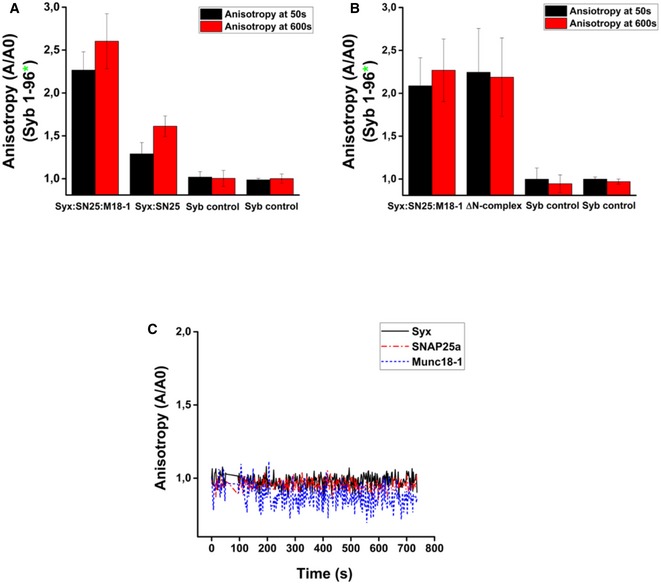
-
A, B(A) Quantification of synaptobrevin binding to the syntaxin1:SNAP25 complex and the syntaxin1:SNAP25:Munc18‐1 complex as measured by fluorescence anisotropy, and (B) quantification of synaptobrevin binding to the ΔN complex and the syntaxin1: SNAP25:Munc18‐1 complex. Error bars in both (A) and (B) indicate the range of values (n = 3).
-
CSynaptobrevin does not bind to the monomeric constituents of the syntaxin1:SNAP25:Munc18‐1 complex. No increase in anisotropy was observed upon the addition of unlabeled syntaxin1, SNAP25, or Munc18‐1 to fluorescently labeled synaptobrevin. Note that a twofold excess of the monomers with respect to synaptobrevin was used to perform this reaction.
Since fluorescence anisotropy measurements only report the local conformational flexibility of a fluorescently labeled moiety, we also measured SNARE complex formation by fluorescence resonance energy transfer (FRET). To this end, we prepared acceptor complexes with a single‐cysteine mutant of SNAP25 (C130) that was labeled with the dye Texas Red and incubated them with labeled synaptobrevin as above (Winter et al, 2009). As shown in Fig 2D, addition of synaptobrevin to both the ternary syntaxin1:SNAP25:Munc18‐1 complex and the ΔN complex resulted in a fast quenching of the donor emission, thus confirming the results obtained with fluorescence anisotropy measurements. No change in the donor fluorescence was observed when an excess of unlabeled synaptobrevin was added. Decrease in the donor emission was accompanied by a corresponding increase in the acceptor emission (Fig EV4A), confirming that donor quenching was caused due to FRET. A dose‐dependent response was observed when FRET measurements were carried over a range of concentrations of the syntaxin1:SNAP25:Munc18‐1 complex (Fig 2E).
Figure EV4. The association of synaptobrevin with the syntaxin1:SNAP25:Munc18‐1 complex does not displace Munc18‐1.
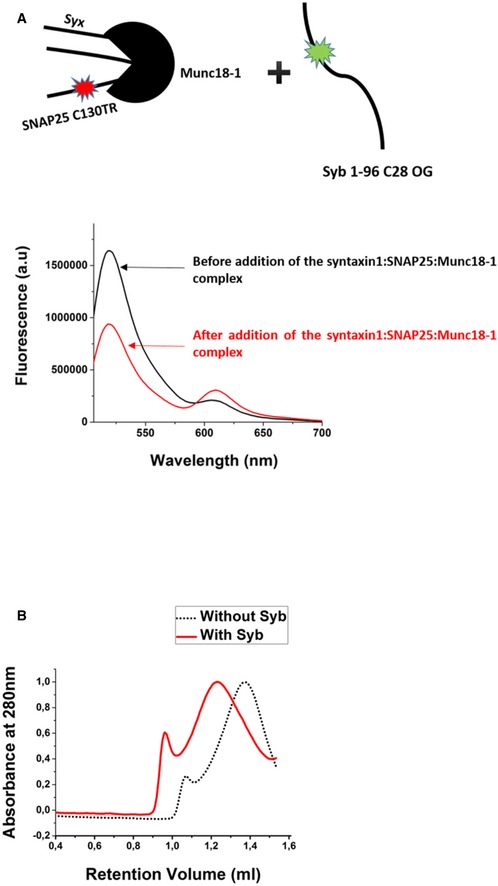
- Fluorescence spectra showing FRET upon addition of the syntaxin1:SNAP25‐TR: Munc18‐1 complex to fluorescently labeled synaptobrevin (Syb‐OG). The decrease in the donor emission was accompanied by an increase in the acceptor emission (TR indicates Texas Red, and OG indicates Oregon Green).
- The incubation of the syntaxin1:SNAP25:Munc18‐1 complex with synaptobrevin, followed by chemical cross‐linking resulted in an increase in the molecular size/hydrodynamic radius of the sample. The separation of the cross‐linked samples was performed on an analytical Superose 6 column (GE Healthcare). The cross‐linked sample after synaptobrevin incubation (red curve) showed a lower retention volume as compared to the one without synaptobrevin incubation (black curve), indicating continued association of Munc18‐1 after synaptobrevin binding.
The entire cytoplasmic domain of synaptobrevin is required for efficient binding to the syntaxin1:SNAP25:Munc18‐1 complex
It is generally believed that SNARE zippering proceeds gradually from the N‐terminal toward the C‐terminal end of the SNARE motifs, leading to a consecutive formation of the interacting side chain layers after N‐terminal nucleation of the complex. This implies that the N‐terminal end of the acceptor site is freely accessible for synaptobrevin binding, as is the case for ΔN complex. On the other hand, it is conceivable that in the ternary syntaxin1:SNAP25:Munc18‐1 complex, syntaxin1 is still partially closed and thus may render the N‐terminal end of the SNARE motifs inaccessible, at least for initial binding. To address this issue, we used single‐cysteine variants of both C‐ and N‐terminally truncated fragments of synaptobrevin (Syb1‐65 C28, Syb1‐52 C28, Syb49‐96 C79) that had been fluorescently labeled and measured their binding to acceptor complexes both by fluorescence anisotropy (using unlabeled acceptor complexes) and FRET (using labeled acceptor complexes). As shown in Fig 3A and B, progressive truncation from the C‐terminus of synaptobrevin, resulted in an impairment of binding to the syntaxin1:SNAP25:Munc18‐1 complex. A deletion of 31 residues from the C‐terminus of synaptobrevin (Syb1‐65) resulted in a significantly reduced binding to the syntaxin1:SNAP25:Munc18‐1 complex. No significant binding was detectable after truncation of the last 44 residues of synaptobrevin (Syb1‐52). Similarly, truncation of 48 residues from the N‐terminus led to a complete loss of binding. In contrast to the syntaxin1:SNAP25a:Munc18‐1 complex, binding of the synaptobrevin fragments to the ΔN complex showed a different pattern. Here, both fragments with an intact N‐terminus (Syb1‐65 and Syb1‐52) showed binding, albeit with lower efficiency as compared to the entire cytoplasmic fragment (Fig 3C and D), in agreement with previous reports (Wiedelhold & Fasshauer, 2009).
Figure 3. Full‐length synaptobrevin is required for fast binding to the syntaxin1:SNAP25:Munc18‐1 complex but not to the ΔN complex.
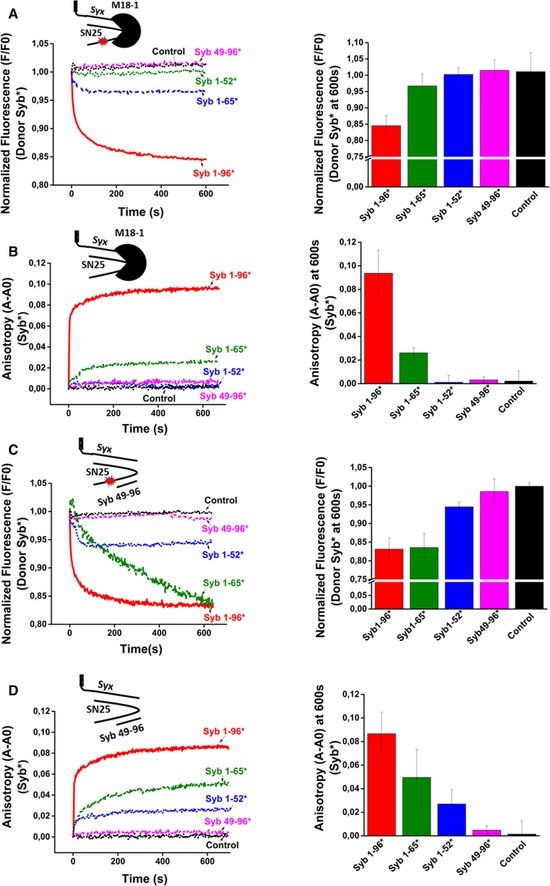
-
A, BBinding of synaptobrevin fragments to fluorescently labeled (A) or unlabeled (B) syntaxin1:SNAP25:Munc18‐1 complex.
-
C, DBinding of synaptobrevin fragments to fluorescently labeled (C) or unlabeled (D) ΔN complex.
Synaptobrevin binding to the syntaxin1:SNAP25:Munc18‐1 complex results in re‐arrangements but does not cause dissociation of Munc18‐1
Next, we investigated whether Munc18‐1 dissociates from the syntaxin1:SNAP25:Munc18‐1 complex after binding of synaptobrevin. To this end, we monitored changes in the molecular size/hydrodynamic radius of the ternary syntaxin1:SNAP25:Munc18‐1 complex upon addition of synaptobrevin using size‐exclusion chromatography. As indicated in Fig 4A, after synaptobrevin addition, the complex eluted at a slightly lower retention volume (black curve) than in the absence of synaptobrevin (red curve). This apparent size increase suggests that Munc18‐1 does not dissociate when synaptobrevin is incorporated into the syntaxin1:SNAP25:Munc18‐1 complex (see also Fig EV4B). To confirm this conclusion, we reconstituted the syntaxin1:SNAP25:Munc18‐1 complex into liposomes in the presence or absence of synaptobrevin. Then, the liposomes were separated from unbound/dissociated proteins by flotation gradient centrifugation using a Nycodenz density gradient. After centrifugation, the distribution of Munc18‐1 and synaptobrevin was monitored by immunoblotting (Fig 4B). The results showed that in both the cases, Munc18‐1 comigrates with the liposomal fraction on the top of the gradient. In contrast, when Munc18‐1 was incubated with protein‐free liposomes, it did not specifically associate with the liposomal fraction after gradient centrifugation (Fig 4B, bottom lane). These results indicate that Munc18‐1 remains bound to the SNARE complex after binding of synaptobrevin to the syntaxin1:SNAP25:Munc18‐1 complex. The orientation of the syntaxin1:SNAP25:Munc18‐1 complex on liposomes was also determined using trypsin digestion assay, which indicated reconstitution of this complex with an almost 100% “right‐side out” orientation (Fig EV5A).
Figure 4. Synaptobrevin binding does not displace Munc18‐1 from the syntaxin1:SNAP25:Munc18‐1 complex.
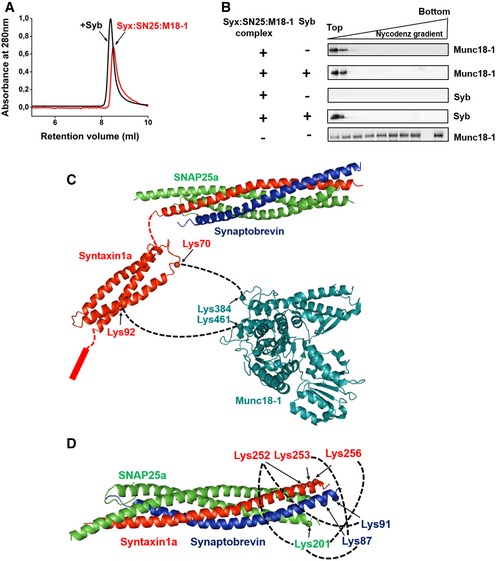
- Gel‐filtration profiles of syntaxin1:SNAP25:Munc18‐1 complex with (black curve) or without (red curve) synaptobrevin pre‐incubation. A slight shift in the elution peak toward lower retention volume was observed after incubation of the complex with synaptobrevin. Separation was performed on Superdex 10/300 Increase column (GE Healthcare).
- Syntaxin1:SNAP25:Munc18‐1 complexes were mixed with synaptobrevin and thereafter incorporated into liposomes. The liposomes were then separated from the free (or displaced) proteins using a Nycodenz density gradient; 25 μl fractions were then collected from top to bottom of the gradient and analyzed by Western blotting using α‐Munc18‐1 and α‐synaptobrevin antibodies. As a control experiment, protein‐free liposomes were treated with Munc18‐1 (bottom panel, the second last lane is blank due to the marker position) and processed as above to assess the non‐specific binding of Munc18‐1 to the liposomes.
- Cross‐links observed between syntaxin1 and Munc18‐1 after incubation of the syntaxin1:SNAP25:Munc18‐1 complex with synaptobrevin. Freshly purified complex was incubated with synaptobrevin and was subjected to chemical cross‐linking with BS3. MS/MS analysis revealed that Munc18‐1 remains associated with syntaxin1 after SNARE complex formation. Cross‐links were observed between residues lying in the Habc region of syntaxin1 (residues 70 and 92) and K384 and K461 of Munc18‐1, indicating a major conformational shift of syntaxin1 with respect to Munc18‐1 after synaptobrevin addition. No cross‐links were observable between the SNARE motifs of SNAP25 and Munc18‐1
- MS/MS analysis of the cross‐linked samples after synaptobrevin treatment also revealed cross‐links between the C‐termini of syntaxin1, SNAP25, and synaptobrevin, thereby demonstrating the formation of a fully zippered SNARE complex after synaptobrevin binding to the syntaxin1:SNAP25:Munc18‐1 complex.
Source data are available online for this figure.
Figure EV5. Assessment of orientation of the syntaxin1:SNAP25:Munc18‐1 complex on liposomes, and SNARE‐complex assembly upon synaptobrevin binding to this complex.
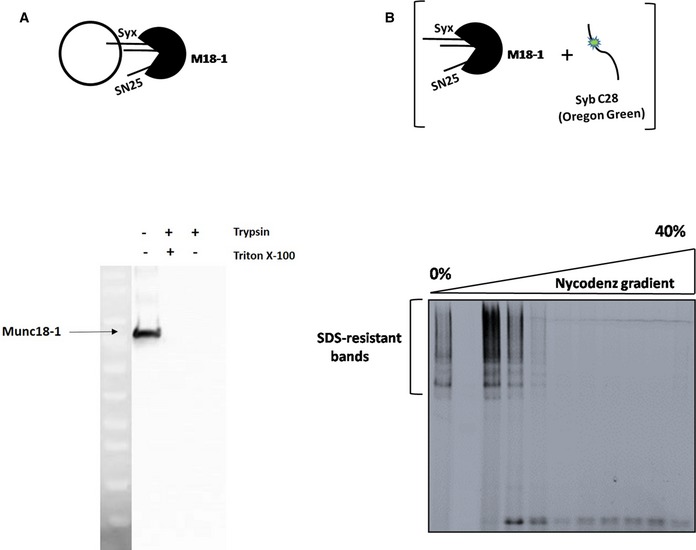
- The schematic on the top represents a liposome containing the syntaxin1:SNAP25:Munc18‐1 complex. Trypsin‐digestion assay (see Materials and Methods) followed by Western blotting against Munc18‐1 indicated that the syntaxin1:SNAP25:Munc18‐1 complex is inserted into liposomes with a nearly 100% “right‐side out” orientation.
- SDS‐resistant SNARE complexes are formed after synaptobrevin binding to the syntaxin1:SNAP25:Munc18‐1 complex. Fluorescently‐labeled synaptobrevin was added to the syntaxin1:SNAP25:Munc18‐1 complex (as depicted in the cartoon above), and the mixture was reconstituted into liposomes. SDS–PAGE analysis of the samples after co‐flotation (without prior boiling of the samples) revealed the presence of multiple high‐molecular‐weight, SDS‐resistant bands, marked by the fluorescence of synaptobrevin. The second lane from the left indicates the marker lane and hence does not show any fluorescence.
In order to obtain an understanding about the structural associations of Munc18‐1 with the SNARE complex, we cross‐linked the syntaxin1:SNAP25:Munc18‐1 complex after synaptobrevin addition, again using the chemical cross‐linker BS3 (Table EV2). Interestingly, after synaptobrevin addition, we observed cross‐links between the Habc domain of syntaxin1 and regions in domain 3b of Munc18‐1, which is positioned on the outer surface, clearly separated from the syntaxin1‐binding cleft of Munc18‐1 (Fig 4C). No cross‐links were obtained between the SNARE motifs of SNAP25 or synaptobrevin with Munc18‐1. We also observed cross‐links between the C‐terminus of synaptobrevin and the C‐termini of both syntaxin1 and SNAP25 (Fig 4D), as expected for a fully zippered SNARE complex. Apparently, binding of synaptobrevin to the syntaxin1:SNAP25:Munc18‐1 complex leads to major structural re‐arrangements of Munc18‐1 with respect to the assembled SNARE complex. The final state is represented by a completely assembled SNARE complex with Munc18‐1 being tethered to the N‐terminal region of syntaxin1, in agreement with a previous report (Burkhardt et al, 2008). Full zippering of the SNARE complex was also confirmed by the appearance of SDS‐resistant bands (Fig EV5B) that are typical hallmarks of fully assembled SNARE complexes (Hayashi et al, 1994).
The syntaxin1:SNAP25:Munc18‐1 complex but not the SNARE complex resulting from addition of synaptobrevin is resistant to disassembly by NSF‐αSNAP
In the final set of experiments, we investigated whether the ternary syntaxin1:SNAP25:Munc18‐1 complex can be disassembled by NSF‐αSNAP. It has been shown previously that NSF not only acts on fully assembled SNARE four helix bundles (Söllner et al, 1993) but also on partial complexes (syntaxin1:SNAP25; Vivona et al, 2013) and even on syntaxin1 alone. Thus, the question arises whether the association of the binary syntaxin1:SNAP25 with Munc18‐1 prevents its disassembly by NSF‐αSNAP. To this end, we used labeled variants of syntaxin1 and SNAP25 (syntaxin1 C197, labeled with Oregon Green, and SNAP25 C130, labeled with Texas Red) to prepare the ternary syntaxin1:SNAP25:Munc18‐1 complex (Fig 5A, left lane). Incubation of this complex with purified NSF and α‐SNAP, in the presence of Mg2+‐ATP, did not result in any change of the donor and acceptor fluorescence (see also Fig EV6). In contrast, some reduction in FRET was observed (here measured as an increase in donor fluorescence) when the complex was incubated in 1 M NaCl (Fig 5A). For comparison, we used the same labeled proteins to prepare a standard syntaxin1:SNAP25 (2:1) complex (see cartoon to the left of Fig 5B). When NSF‐αSNAP was added to this complex, a rapid, Mg2+‐ATP‐dependent loss of FRET was observable showing the rapid disassembly of this complex, as expected.
Figure 5. The syntaxin1:SNAP25:Munc18‐1 is resistant to disassembly by NSF‐αSNAP but becomes sensitive after binding of synaptobrevin.
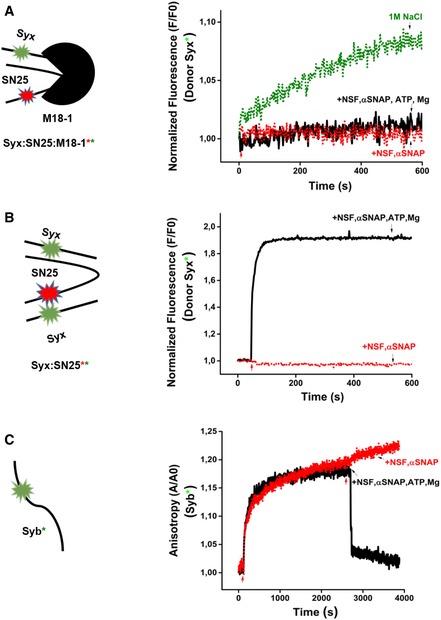
- Double‐labeled syntaxin1:SNAP25:Munc18‐1 complex containing syntaxin1 (labeled with Oregon Green) and SNAP25 (labeled with Texas Red) was purified. Incubation with NSF‐αSNAP in the presence of Mg2+‐ATP did not result in any change of donor fluorescence. Incubation with 1 M NaCl resulted in a small increase in donor fluorescence, indicating that under high‐salt conditions, some minor dissociation of the complex does occur.
- Purified, double‐labeled Syx:SN25 (2:1) complex containing syntaxin1 and SNAP25 (labeled as in A) could be disassembled by NSF‐αSNAP as evident from a rapid increase in donor fluorescence (loss of FRET).
- Fluorescence anisotropy measurements showing the disassembly of the SNARE complex generated upon addition of fluorescently labeled synaptobrevin to a pre‐assembled syntaxin1:SNAP25:Munc18‐1 complex. Addition of NSF‐αSNAP led to a rapid decrease in the anisotropy as expected for SNARE disassembly. The red arrows indicate the addition of the protein(s)/reagents, whereas the black arrows indicate the reaction condition.
Figure EV6. Resistance of the syntaxin1:SNAP25:Munc18‐1 complex to disassembly by NSF‐αSNAP.
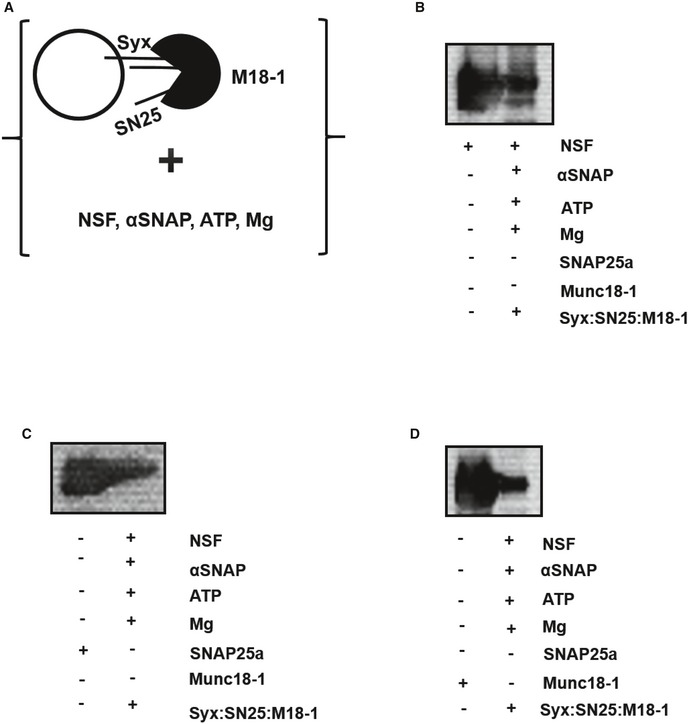
-
AFreshly prepared syntaxin1:SNAP25:Munc18‐1 complex was reconstituted into liposomes and was thereafter incubated with NSF, αSNAP, ATP, and magnesium. The mixture was then analyzed by co‐flotation analysis and subsequent immunoblotting (see Materials and Methods).
-
B–DThe top liposomal fractions were immunoblotted using (B) α‐NSF, (C) α‐SNAP25a, and (D) α‐Munc18‐1 antibodies. The presence of all these three proteins in the liposomal fractions clearly demonstrated the resistance of the syntaxin1:SNAP25:Munc18‐1 complex to disassembly by NSF‐αSNAP. The left lane in each case represents the monomeric proteins as a control, and the right lanes indicate the liposomal fractions obtained after the co‐flotation assay.
Finally, we tested whether NSF‐αSNAP disassembles the SNARE complex formed after synaptobrevin binding to the syntaxin1:SNAP25:Munc18‐1 complex. Here, we again resorted to fluorescence anisotropy using labeled synaptobrevin. As shown in Fig 5C, addition of synaptobrevin to the complex led to an increase in anisotropy. Subsequent addition of NSF‐αSNAP reverted this increase, indicative of disassembly of the SNARE complex and the subsequent release of synaptobrevin into the solution.
Discussion
In this study, we have characterized a ternary complex between the Q‐SNAREs syntaxin1 and SNAP25 with the SM protein, Munc18‐1. Our data show that this complex is protected against disassembly by NSF‐αSNAP and is highly reactive toward binding of synaptobrevin, which results in a fully zippered SNARE helical bundle, making it an attractive candidate for the still elusive SNARE acceptor complex in neuronal exocytosis.
The instability of the syntaxin1:SNAP25:Munc18‐1 complex in vitro prevented us from obtaining detailed structural information. However, several conclusions can be drawn from the biochemical characterization and from the cross‐linking experiments (see cartoon in Fig 6). First, in the ternary syntaxin1:SNAP25:Munc18‐1 complex, the N‐terminal Habc domain of syntaxin1 is associated with the syntaxin1‐binding cleft of Munc18‐1 as in the closed binary complex, but no contacts were found between the syntaxin1‐binding cleft of Munc18‐1 and the syntaxin1 SNARE motif. Second, the C‐terminal end of the Qb‐SNARE motif of SNAP25 contacts the cleft in Munc18‐1. This suggests that the structure of this complex is different from the binary syntaxin1:Munc18‐1 complex, with syntaxin1 probably being in a more open conformation. It is also different from complexes between SM proteins and fully assembled SNARE complexes, where the contact is limited to the N‐terminal peptide of the respective syntaxin (Qa‐SNARE; as is the case for Munc18‐1; Burkhardt et al, 2008) or to the surface of the four‐helical bundle (as is the case for Sec1; Togneri et al, 2006). Third, the syntaxin1:SNAP25:Munc18‐1 complex is highly reactive toward binding to synaptobrevin. Possibly, this reactivity is caused by the ability of Munc18‐1 to prevent the formation of the off‐pathway syntaxin1:SNAP25 (2:1) complex and also by induction of helicity in the syntaxin1:SNAP25:Munc18‐1 acceptor complex, similar to the role of Vc‐peptide in the ΔN complex (Ma et al, 2015; Li et al, 2016). Additionally, synaptobrevin binding to the syntaxin1:SNAP25:Munc18‐1 complex results in a fully zippered SNARE complex with Munc18‐1 remaining tethered to the N‐terminal domain of syntaxin1, with no evidence for an association with any of the SNARE motifs in the helical bundle.
Figure 6. Cartoon summarizing the different associations between Munc18‐1 and the SNAREs syntaxin1, SNAP25, and synaptobrevin that could exist at the different stages of SNARE complex assembly.
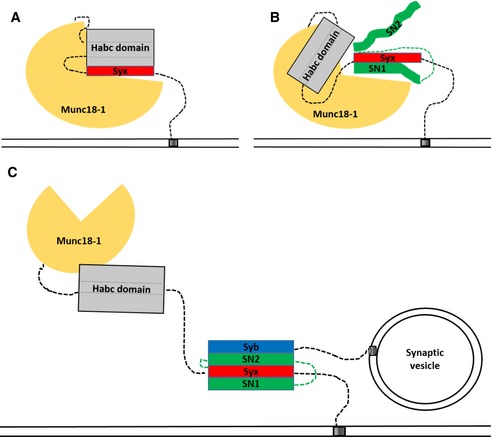
- Binary syntaxin1:Munc18‐1 complex with syntaxin in a “closed” conformation.
- Ternary complex between Munc18‐1, syntaxin1, and SNAP25. As shown by cross‐linking, the C‐terminal region of the SNAP25 Qb‐SNARE motif is associated with the cleft in Munc18‐1 together with the Habc domain. The syntaxin1:SNAP25:Munc18‐1 complex serves as an efficient acceptor complex for rapid synaptobrevin binding.
- Binding of synaptobrevin to the syntaxin1:SNAP25:Munc18‐1 complex leads to the formation of a fully zippered four‐helical SNARE complex, with syntaxin1 being in the open conformation and Munc18‐1 still tethered to the N‐terminal domain of syntaxin1.
Our results shed new light on the surprising flexibility with which SM proteins can associate with various states of SNAREs and SNARE complexes. This flexibility may explain why it has been so difficult to understand the conformational transitions and protein–protein interactions during the activation sequence of SNARE proteins. Indeed, taking the different modes of SM/SNARE interactions into account, arriving at a single, coherent mechanism for all SM proteins is challenging. However, in light of current and earlier research with Munc18‐1, we conclude that the cleft of SM proteins provides an adaptable platform for Q‐SNARE motifs to be conformationally constrained and arranged in a way that facilitates final nucleation of the SNARE complex, possibly assisted by some conformational dynamics of the Habc domain. These intermediates are probably transient and stabilized by weak interactions so that their re‐arrangement during SNARE assembly does not siphon off too much energy from the zippering reaction. Such transient states may explain why pre‐incubation of Q‐SNARE‐containing liposomes with Munc18‐1 for several hours accelerated fusion in a previous study (Shen et al, 2007).
It remains to be established whether the R‐SNARE binds to the SM protein before assembling with its SNARE partners. Support for such a scenario is provided by the presence of a conserved R‐SNARE binding site on the surface of the SM proteins (Baker et al, 2015) which also appears to be functionally involved in exocytosis of synaptic vesicles (Xu et al, 2010; Parisotto et al, 2014). Indeed, based on crystal structures of the SM protein Vps33 with either the corresponding R‐SNARE or the Qa‐SNARE, it has recently been proposed that these two SNAREs bind first and then recruit the Qb and Qc‐SNAREs to the complex. A similar mechanism for SNARE assembly has recently been also suggested for Munc18‐1 based on single‐molecule force experiments (Ma et al, 2015). Additionally, the helix 12 of Munc18‐1 has been proposed to be involved in synaptobrevin binding, thereby helping in synaptic vesicle priming (Munch et al, 2016). However, according to our data, the Q‐SNAREs need to pre‐assemble on the SM template before synaptobrevin can bind. Also, it should be noted that simultaneous binding of the Qa‐ and R‐SNAREs to Vps33 was not detectable, making the model somewhat tenuous. Intriguingly, however, structural modeling shows that the R‐SNARE binding site in Munc18‐1 only becomes accessible if the protein is in the open conformation. Thus, it is possible that R‐SNARE binding to the SM protein is required for assembly (as suggested by Baker et al, 2015) even though the sequence of SNARE assembly on the SM template is different than that proposed for Vps33 (Baker et al, 2015).
Intriguingly, a truncation of 44 residues from the C‐terminus of the cytoplasmic fragment of synaptobrevin (Syb1‐52) resulted in a complete loss of binding to the syntaxin1:SNAP25:Munc18‐1 complex, in stark contrast to the artificial ΔN complex containing a free N‐terminal nucleation site. Additionally, a truncation of 48 residues from the N‐terminus (Syb49‐96) also resulted in a complete loss of binding to the syntaxin1:SNAP25:Munc18‐1 complex. It is conceivable that the N‐terminal ends of the Q‐SNARE motifs are not accessible in the syntaxin1:SNAP25:Munc18‐1 complex. Such an occlusion may also explain why SM proteins shield SNAREs from NSF‐driven disassembly (see also Ma et al, 2013; Baker et al, 2015). Accordingly, this behavior can be explained by an alternative zippering mechanism where nucleation of SNARE assembly commences somewhat downstream of the N‐terminal end of the SNARE motifs, with zippering then progressing in a cooperative fashion both toward the N‐ and C‐terminal end of the SNARE bundle. On the other hand, the truncated versions may have reduced affinities for the SM protein (as shown for Nyv1 binding to Vps33), thereby preventing efficient binding to the syntaxin1:SNAP25:Munc18‐1 complex. Presently, it cannot be decided which of the two scenarios is correct. Moreover, it needs to be determined whether the assembly pathways of SNAREs on different SM proteins are always the same or whether the versatile SM templates permit different binding sequences for different sets of SNAREs.
Our data lend strong support for Munc18‐1 being the central catalyst for SNARE nucleation. The rapid and coordinated binding of synaptobrevin suggests that the ternary syntaxin1:SNAP25:Munc18‐1 complex is the direct precursor of the fully zippered SNARE complex and thus precedes fusion. Our data agree with single‐molecule experiments suggesting that Munc18‐1 stabilizes the acceptor complexes for SNARE zippering (Weninger et al, 2008) and with the presence of assemblies of syntaxin1:SNAP25:Munc18‐1 on the neuronal plasma membrane (Pertsinidis et al, 2013). They also agree with a recent work showing that the association of syntaxin1:Munc18‐1 complexes with SNAP25 shifts the equilibrium toward the “open”‐state of syntaxin1 (Dawidowski & Cafiso, 2016), and with the observation that Munc18b, a relative of Munc18‐1, forms ternary complexes with syntaxin3 and SNAP25 for regulated epithelial secretion (Liu et al, 2007).
It remains to be established how the ternary syntaxin1: SNAP25:Munc18‐1 complex fits into the overall pathway of SNARE activation and zippering. In particular, it needs to be clarified by which mechanisms this energized complex is assembled and stabilized. A possible starting point could be the binary syntaxin1:Munc18‐1 complex which is energetically trapped, since syntaxin1 mutants assuming a more open conformation promote both exocytosis and SNARE complex assembly (Rizo & Xu, 2015). Thus, a first step would involve activation of this complex, with a strong candidate for this role being Munc13‐1, a protein essential for regulated exocytosis. Munc13‐1 is known to interact both with the binary syntaxin1:Munc18‐1 complex (Ma et al, 2011) and with free syntaxin (Betz et al, 1997). A similar activation can also be brought about by arachidonic acid (Connell et al, 2007). Finally, it needs to be clarified at which point the SNARE assembly is arrested before exocytosis gets triggered by calcium, and how the arrest is relieved by the combined action of synaptotagmin and complexin.
Materials and Methods
Expression constructs
Wild‐type pET28a expression constructs containing sequences encoding full‐length syntaxin (1‐288), a cysteine‐free variant of full‐length SNAP25a (1‐206), full‐length synaptobrevin (1‐116), the cytoplasmic fragment of synaptobrevin (1‐96), and full‐length Munc18‐1 (1‐584) were used for the study [as described earlier (Fasshauer et al, 1997, 1999; Margittai et al, 1999; Burkhardt et al, 2008)]. For anisotropy and FRET experiments, we used the single‐cysteine mutants of syntaxin (C197), SNAP25a (C130), synaptobrevin 1‐96 (C28), synaptobrevin 1‐65 (C28), synaptobrevin 1‐52 (C28), and synaptobrevin 49‐96 (C79; Margittai et al, 2001; Pobbati et al, 2006; Burkhardt et al, 2008; Wiedelhold & Fasshauer, 2009; Walter et al, 2010). All the proteins used were from Rattus norvegicus.
Protein purification
Monomeric syntaxin1, SNAP25, and synaptobrevin were purified by Ni2+‐NTA affinity chromatography, followed by ion‐exchange chromatography (as described in Fasshauer et al, 1999; Fasshauer & Margittai, 2004). Munc18‐1 was purified by affinity chromatography followed by gel‐filtration chromatography on Superdex 200 16/60 (GE Healthcare).For complex formation, the purified monomers were mixed and incubated overnight at 4°C. Unless indicated otherwise, all proteins were full‐length, with syntaxin1 containing its transmembrane domain. The assembled complexes were further purified either by ion‐exchange chromatography or by size‐exclusion chromatography. All complexes (syntaxin1:SNAP25, syntaxin1:SNAP25:Munc18‐1, or ΔN complex) were purified in one of the following detergents: 1% (w/v) CHAPS, 1% (w/v) octyl β‐D‐glucopyranoside, or 0.03% (w/v) n‐dodecyl β‐D‐maltoside (DDM).
Assembly of syntaxin1:SNAP25a:Munc18‐1 complex
For complex assembly, purified monomeric syntaxin (1‐288) and SNAP25a (1‐206) were mixed overnight at 4°C, followed by the addition of a twofold molar excess of full‐length Munc18‐1, and a further incubation of 4 h at room temperature. The assembled complex was further purified by ion‐exchange chromatography or (and) size‐exclusion chromatography. The buffer composition used for purification of the complex by size‐exclusion chromatography included 20 mM HEPES, 200 mM NaCl, 0.5 mM TCEP, 10% glycerol (with the corresponding percentage of detergent) at a pH of 7.4. The complex could be purified in either 0.03% (w/v) n‐dodecyl β‐D‐maltoside (DDM) or 1% (w/v) octyl β‐D‐glucopyranoside. The freshly purified complex was immediately used for all the experiments.
Proteoliposome preparation
1,2‐dioleoyl‐sn‐glycero‐3‐phosphocholine (DOPC), 1,2‐dioleoyl‐sn‐glycero‐3‐phosphoethanolamine (DOPE), 1,2‐dioleoyl‐sn‐glycero‐3‐phospho‐L‐serine (DOPS), cholesterol, 1‐oleoyl‐2‐{12‐[(7‐nitro‐2‐1,3‐benzoxadiazol‐4‐yl)amino]dodecanoyl}‐sn‐glycero‐3‐phosphoserine (NBD‐PS), and 1,2‐dioleoyl‐sn‐glycero‐3‐phosphoethanolamine‐N‐(lissamine rhodamine B sulfonyl) (Rhodamine‐PE) were ordered from Avanti Polar lipids for proteoliposome preparation (Fasshauer et al, 1998, 1999). The molar (%) ratio between the different lipids was 50% (DOPC):20% (DOPE):20% (DOPS):10% (cholesterol). For the preparation of labeled liposomes, the ratio of the lipids mixed was 50% (DOPC): 18.5% (DOPE): 1.5% (Rhodamine‐PE): 18.5% (DOPS): 1.5% (NBD‐PS): 10% (cholesterol). After drying the lipids, the lipid film was resuspended in 20 mM HEPES, 150 mM KCl, and 5% (w/v) sodium cholate solution and vortexed thoroughly. The lipid mix was then mixed with the respective protein(s) in 20 mM HEPES, 150 mM KCl, 3% (w/v) sodium cholate (Pobbati et al, 2006). Proteoliposomes were prepared by detergent removal using gel‐filtration chromatography on a column packed with Sephadex G‐50. The buffer composition for proteoliposome preparation was 20 mM HEPES, 150 mM KCl, 1 mM DTT at a pH of 7.4. The protein: lipid (n/n) ratio was 1:1,000.
Anisotropy measurements
All anisotropy measurements were carried out in a Fluorolog 3 spectrometer containing a magnetic stirrer and an in‐built T‐configuration equipped for polarization (Model FL322, Jobin Yvon; described in Pobbati et al (2006)). For monitoring, the binding of synaptobrevin to the different acceptor complexes, 200 nM of synaptobrevin labeled with Oregon Green at position C28, was used. The concentration of the acceptor complexes used was 400 nM. Anisotropy (r) was calculated using the formula r = (IVV − G × IVH)/(IVV + 2 ×G × IVH), where “I” denotes the fluorescence intensity, and the first and second subscript letters indicate the polarization of the exciting light and the emitting light, respectively. For synaptobrevin C28‐Oregon Green, the excitation wavelength was 490 nm and the emission wavelength was set to 520 nm. All experiments were performed in a reaction volume of 600 μl and at a temperature of 37°C.
Fluorescence Resonance Energy Transfer (FRET) experiments
All FRET measurements were performed in a FluoroMax 3 (Horiba Jovin Yvon) spectrometer equipped with a magnetic stirrer. In order to study SNARE zippering between synaptobrevin and the different acceptor complexes (syntaxin1:SNAP25:Munc18‐1 complex or ΔN complex), we monitored FRET between synaptobrevin labeled with Oregon Green iodoacetamide at position C28 and SNAP25a labeled with the fluorescent dye Texas Red maleimide at position C130 (Margittai et al, 2001). SNARE zippering was measured as a real‐time decrease in donor emission upon the addition of the acceptor complexes. Spectral scans were also performed to monitor the simultaneous increase in the acceptor emission with a decrease in donor emission. The spectral scans were performed over a wavelength range of 500–700 nm, with an excitation wavelength of 490 nm. The excitation maximum for Oregon Green is 490 nm, and the emission maximums for Oregon Green and Texas Red are 520 and 615 nm, respectively. All experiments were performed with freshly purified complexes in a reaction volume of 600 μl at 37°C. Fluorescence intensity is represented as F/F 0, where F 0 is the initial fluorescence.
Co‐flotation assay
Proteins bound to liposomes can be separated from the unbound fraction by using a Nycodenz density gradient (Park et al, 2014). For the assessment of synaptobrevin binding to the syntaxin1:SNAP25:Munc18‐1 complex, freshly purified complex was mixed with synaptobrevin and reconstituted into liposomes. The liposomes were then separated by ultracentrifugation on a Nycodenz density gradient. Proteoliposomes (50 μl) were mixed with 80% Nycodenz (50 μl). Another layer of 30% Nycodenz (50 μl) was layered on top of this, followed by a very gentle addition of 50 μl reconstitution buffer (20 mM HEPES, 150 mM KCl, 1 mM DTT, pH 7.4; Park et al, 2014). The density gradient was subjected to ultracentrifugation in a Beckman TL‐100 ultracentrifuge (TLS55 rotor) at 100,000 g for 1.5 h at 4°C; 25 μl fractions were carefully taken from top to bottom of the gradient and analyzed by Western blotting using α‐synaptobrevin and α‐Munc18‐1 antibodies.
Western blotting
Protein samples were separated by SDS–PAGE and were then transferred onto nitrocellulose membranes (Towbin et al, 1979). α‐Syntaxin (78.2, Synaptic Systems), α‐SNAP25a (71.1, Synaptic Systems), α‐synaptobrevin (69.1, Synaptic Systems), and α‐Munc18‐1 (polyclonal) antibodies were used for immunodetection. The primary antibodies were used at a dilution of 1:1,000. Goat α‐mouse (for syntaxin1, SNAP25, and synaptobrevin) or goat α‐rabbit (for Munc18‐1) secondary antibodies coupled to horseradish peroxidase (HRP) were used at a dilution of 1:2,000 for subsequent detection by enhanced chemiluminescence (ECL).
Electrophoretic procedures
The property of SDS resistance of SNARE complex was used as a measure to monitor SNARE complex formation upon the addition of synaptobrevin to the syntaxin1:SNAP25:Munc18‐1 complex. Fluorescently labeled synaptobrevin was added to the freshly prepared syntaxin1:SNAP25:Munc18‐1 complex, and the resulting mixture was used for proteoliposome preparation. Thereafter, the bound proteins were separated from the unbound fraction by co‐flotation assay (as described above). The samples from the co‐flotation assay were mixed with SDS sample buffer and were separated by electrophoresis, without prior boiling of the samples. The binding of synaptobrevin to the syntaxin1:SNAP25:Munc18‐1 complex and the formation of SDS‐resistant SNARE complexes was monitored by observing the fluorescence of synaptobrevin (Fig EV5B; Burkhardt et al, 2008). The scans of the fluorescent gels were made in TECAN fluorescence scanner (FLA‐7000).
Chemical cross‐linking
Freshly purified syntaxin1:SNAP25:Munc18‐1 complex was used for chemical cross‐linking with bis(sulfosuccinimidyl)suberate (BS3; Pierce, Thermo Fisher Scientific), an amine‐reactive, water‐soluble, homobifunctional protein cross‐linker (Xu et al, 2010). The syntaxin1:SNAP25:Munc18‐1 complex was titrated with increasing amounts of the cross‐linker in order to determine the optimum amount of the cross‐linker required for efficient cross‐linking (see Fig EV1C). A fifty times molar excess of the cross‐linker over the syntaxin1:SNAP25:Munc18‐1 complex seemed to be optimal for the cross‐linking reaction; 80 pmol of the syntaxin1:SNAP25:Munc18‐1 complex was incubated with 4 nmol of BS3 and incubated at room temperature for 1 h. After the incubation period, the reaction was quenched by addition of 1 μl of 1 M Tris–HCl (at a pH of 8.0). The cross‐linked samples were thereafter subjected to SDS–PAGE on a 4–12% Bis–Tris gel (Invitrogen) with MES as running buffer. The gel was stained using Coomassie blue. The band corresponding to the cross‐linked sample was excised and subjected to in‐gel trypsin digestion (Shevchenko et al, 2007). The proteolytic peptides were reconstituted in a solvent system containing 5% acetonitrile (ACN) and 0.1% formic acid (FA) in a final volume of 20 μl and subjected to liquid chromatography–tandem mass spectrometry (LC‐MS/MS) analysis.
Mass spectrometry (MS/MS)
The reconstituted proteolytic peptides were subjected to further separation using a nano‐liquid chromatography system (UltiMate™ 3000 RSLCnano system) containing a C18‐trapping column of 3 cm × 150 μm inner diameter, in line with a 30 cm × 75 μm inner diameter C18 analytical column (both packed in‐house with 1.9‐μm C18 material, Dr. Maisch GmbH). Peptides were loaded on the trapping column and desalted for 3 min at a flow rate of 10 μl/min in 95% of mobile phase A (0.1% FA in H2O, v/v) and 5% of mobile phase B (80% ACN and 0.05% FA in H2O, v/v). Thereafter, the peptides were eluted and separated on the analytical column using a 43‐min linear gradient of 15–46% mobile phase B, at a flow rate of 300 nl/min (Absmeier et al, 2015).
Orbitrap Fusion mass spectrometer (Thermo Scientific) was used for the online analysis of the separated peptides. We employed a method called “top‐20”, where the 20 most intense precursor ions with charge states 3–8 in the survey scan (380–1,580 m/z scan range) were isolated in the quadrupole mass filter (isolation window 1.6 m/z) and fragmented in the higher energy collisional dissociation (HCD) cell with normalized energy. A dynamic exclusion of 20 s was used. Both the survey scan (MS1) and the product ion scan (MS2) were performed in the Orbitrap at 120,000 and 30,000 resolution, respectively. Spray voltage was set at 2.3 kV, and 60% of S‐lens RF level was used. Automatic gain control (AGC) targets were set at 5 × 105 (MS1) and 5 × 104 (MS2).
The raw data were converted to mgf files by Proteome Discoverer 2.0.0.802 software (Thermo Scientific). The mgf files were searched against a FASTA database containing the sequences of syntaxin1, SNAP25a, synaptobrevin 2, and Munc18‐1 from Rattus norvegicus by pLink 1.23 software using a target‐decoy strategy (Elias & Gygi, 2007). Database search parameters included mass accuracies of MS1 < 10 ppm and MS2 < 20 ppm, carbamidomethylation on cysteine as a fixed modification, oxidation on methionine as variable modification. Number of residues of each peptide on a cross‐link pair was set between 4 and 40. A maximum of two trypsin missed‐cleavage site was allowed. The results were obtained with 1% false discovery rate.
Trypsin digestion assay
In order to determine the orientation of the syntaxin1:SNAP25:Munc18‐1 complex after its incorporation into liposomes, we carried out limited proteolysis. The liposomes containing syntaxin1:SNAP25:Munc18‐1 complex were incubated with 0.1 mg of trypsin (Sigma Aldrich) in the presence or absence of 0.3% (w/v) Triton X‐100 at 37°C, for 2 h. As control, the liposomes were incubated only with the reconstitution buffer (without trypsin or Triton). The reconstitution buffer consisted of 20 mM HEPES, 150 mM KCl, 1 mM DTT, at a pH of 7.4. After completion of the incubation period, the samples were analyzed by Western blotting using a polyclonal antibody against Munc18‐1.
Protein labeling
The single‐cysteine mutants (C28) of synaptobrevin 1‐96, synaptobrevin 1‐65, and synaptobrevin 1‐52 and the cysteine mutant of syntaxin (C197) were labeled with Oregon Green iodoacetamide (Thermo Fisher Scientific) by incubating the proteins with around six times molar excess of the dye, overnight at 4°C (Fasshauer & Margittai, 2004). Excess unreacted dye was removed by passing the labeled proteins through commercially available PD‐10 columns (GE Healthcare). The cysteine mutant of SNAP25a (C130) was labeled with Texas Red maleimide (Thermo Fisher Scientific) using the same procedure.
Disassembly by NSF‐αSNAP
Syntaxin1‐288 (C197) labeled with Oregon Green and SNAP25a (C130) labeled with Texas Red were used to assemble syntaxin1:SNAP25 complex and syntaxin1:SNAP25:Munc18‐1 complex. The assembled complexes were further purified by ion‐exchange chromatography, using a MonoQ column (GE Healthcare). The disassembly reaction was performed by monitoring the change in FRET between syntaxin1 and SNAP25 upon the addition of NSF and αSNAP in the presence of ATP and magnesium. The reaction was performed in FluoroMax 3 (Horiba Jovin Yvon) spectrometer equipped with a magnetic stirrer. The fluorescence signal was monitored at an excitation of 490 nm and an emission of 520 nm, which corresponds to the excitation and emission maximum, respectively, of Oregon Green (the donor fluorophore). The fluorescence signals are represented as F/F 0, where F 0 is the initial fluorescence. As a negative control, the disassembly reaction was performed in the absence of magnesium and ATP. As a positive control, 1 M NaCl solution was used to disassemble the syntaxin1:SNAP25:Munc18‐1 complex. The concentrations of the different components used were as follows: NSF (200 nM), αSNAP (1.5 μM), ATP (100 nM), and magnesium (200 nM). All the reactions were performed at 37°C, in a reaction volume of 600 μl.
Data availability
Referenced data: (Fernandez et al, 1998; Burkhardt et al, 2008; Stein et al, 2009).
Author contributions
SJ performed all the biophysical and biochemical experiments. C‐TL and HU provided the mass spectrometry facility for cross‐linking, and C‐TL performed the experiments with mass spectrometry. RJ and SJ wrote the paper.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Table EV1
Table EV2
Review Process File
Source Data for Figure 4B
Acknowledgements
The authors are indebted to Dr. Angel Perez‐Lara for critical reading of the manuscript and helpful discussions. We would also like to thank Halenur Yavuz for kindly providing the truncated synaptobrevin fragments. This work was supported by Grant P01 GM72694 from the National Institutes of Health to R.J.
The EMBO Journal (2017) 36: 1788–1802
References
- Absmeier E, Wollenhaupt J, Mozaffari‐jovin S, Becke C, Lee C, Preussner M, Heyd F, Urlaub H, Lührmann R, Santos KF, Wahl MC (2015) The large N‐terminal region of the Brr2 RNA helicase guides productive spliceosome activation. Genes Dev 29: 2576–2587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker RW, Jeffrey PD, Zick M, Phillips BP, Wickner WT, Hughson FM (2015) A direct role for the Sec1/Munc18‐family protein Vps33 as a template for SNARE assembly. Science 349: 1111–1114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker RW, Hughson FM (2016) Chaperoning SNARE assembly and disassembly. Nat Rev Mol Cell Biol 17: 465–479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betz A, Okamoto M, Benseler F, Brose N (1997) Cell biology and metabolism : direct interaction of the rat unc‐13 homologue Munc13‐1 with the N terminus of syntaxin. J Biol Chem 272: 2520–2526 [DOI] [PubMed] [Google Scholar]
- Burkhardt P, Hattendorf DA, Weis WI, Fasshauer D (2008) Munc18a controls SNARE assembly through its interaction with the syntaxin N‐peptide. EMBO J 27: 923–933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connell E, Darios F, Broersen K, Gatsby N, Peak‐Chew S‐Y, Rickman C, Davletov B (2007) Mechanism of arachidonic acid action on syntaxin‐Munc18. EMBO Rep 8: 414–419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawidowski D, Cafiso DS (2016) Munc18‐1 and the syntaxin‐1N terminus regulate open‐closed states in a t‐SNARE complex. Structure 24: 392–400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elias JE, Gygi SP (2007) Target‐decoy search strategy for increased confidence in large‐scale protein identifications by mass spectrometry. Nat Methods 4: 207–214 [DOI] [PubMed] [Google Scholar]
- Fasshauer D, Otto H, Eliason WK, Jahn R, Brünger AT (1997) Structural changes are associated with soluble N‐ethylmaleimide‐sensitive fusion protein attachment protein receptor complex formation. J Biol Chem 272: 28036–28041 [DOI] [PubMed] [Google Scholar]
- Fasshauer D, Eliason WK, Brünger AT, Jahn R (1998) Identification of a minimal core of the synaptic SNARE complex sufficient for reversible assembly and disassembly. Biochemistry 37: 10354–10362 [DOI] [PubMed] [Google Scholar]
- Fasshauer D, Antonin W, Margittai M, Pabst S, Jahn R (1999) Mixed and non‐cognate SNARE complexes. Biochemistry 274: 15440–15446 [DOI] [PubMed] [Google Scholar]
- Fasshauer D, Margittai M (2004) A transient N‐terminal interaction of SNAP‐25 and syntaxin nucleates SNARE assembly. J Biol Chem 279: 7613–7621 [DOI] [PubMed] [Google Scholar]
- Fernandez I, Ubach J, Dulubova I, Zhang X, Südhof TC, Rizo J (1998) Three‐dimensional structure of an evolutionarily conserved N‐terminal domain of syntaxin 1A. Cell 94: 841–849 [DOI] [PubMed] [Google Scholar]
- Guan R, Dai H, Rizo J (2008) Binding of the Munc13‐1 MUN domain to membrane‐anchored SNARE complexes. Biochemistry 47: 1474–1481 [DOI] [PubMed] [Google Scholar]
- Hayashi T, McMahon H, Yamasaki S, Binz T, Hata Y, Südhof TC, Niemann H (1994) Synaptic vesicle membrane fusion complex: action of clostridial neurotoxins on assembly. EMBO J 13: 5051–5061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahn R, Fasshauer D (2012) Molecular machines governing exocytosis of synaptic vesicles. Nature 490: 201–207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis JL, Dong M, Earles CA, Chapman ER (2001) The transmembrane domain of syntaxin 1A is critical for cytoplasmic domain protein‐protein interactions. J Biol Chem 276: 15458–15465 [DOI] [PubMed] [Google Scholar]
- Li F, Tiwari N, Rothman JE, Pincet F (2016) Kinetic barriers to SNAREpin assembly in the regulation of membrane docking/priming and fusion. Proc Natl Acad Sci USA 113: 10536–10541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu TT, Kishimoto T, Hatakeyama H, Nemoto T, Takahashi N, Kasai H (2005) Exocytosis and endocytosis of small vesicles in PC12 cells studied with TEPIQ (two‐photon extracellular polar‐tracer imaging‐based quantification) analysis. J Physiol 568: 917–929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Seven AB, Camacho M, Esser V, Xu J, Trimbuch T, Quade B, Su L, Ma C, Rosenmund C, Rizo J (2016) Functional synergy between the Munc13 C‐terminal C1 and C2 domains. Elife 5: e13696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Wang D, Ding X, Deng H, Feng M, Yu X, Jiang K, Ward T, Guo Z, And JF, Yao X (2007) A mechanism of Munc18b‐Syntaxin 3‐SNAP25 complex assembly in regulated epithelial secretion. FEBS Lett 581: 4318–4324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma C, Li W, Xu Y, Rizo J (2011) Munc13 mediates the transition from the closed syntaxin‐Munc18 complex to the SNARE complex. Nat Struct Mol Biol 18: 542–549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma C, Su L, Seven AAB, Xu Y, Rizo J (2013) Reconstitution of the vital functions of Munc18 and Munc13 in neurotransmitter release. Science 339: 421–425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma L, Rebane AA, Yang G, Xi Z, Kang Y, Gao Y, Zhang Y (2015) Munc18‐1‐regulated stage‐wise SNARE assembly underlying synaptic exocytosis. Elife 4: e09580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margittai M, Otto H, Jahn R (1999) A stable interaction between syntaxin 1a and synaptobrevin 2 mediated by their transmembrane domains. FEBS Lett 446: 40–44 [DOI] [PubMed] [Google Scholar]
- Margittai M, Fasshauer D, Pabst S, Jahn R, Langen R (2001) Homo‐ and heterooligomeric SNARE complexes studied by site‐directed spin labeling. J Biol Chem 276: 13169–13177 [DOI] [PubMed] [Google Scholar]
- Meijer M, Burkhardt P, de Wit H, Toonen RF, Fasshauer D, Verhage M (2012) Munc18‐1 mutations that strongly impair SNARE‐complex binding support normal synaptic transmission. EMBO J 31: 2156–2168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misura KM, Scheller RH, Weis WI (2000) Three‐dimensional structure of the neuronal‐Sec1‐syntaxin 1a complex. Nature 404: 355–362 [DOI] [PubMed] [Google Scholar]
- Munch AS, Kedar GH, van Weering JRT, Vazquez‐Sanchez S, He E, André T, Braun T, Söllner TH, Verhage M, Sørensen JB (2016) Extension of helix 12 in Munc18‐1 induces vesicle priming. J Neurosci 36: 6881–6891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ninomiya Y, Kishimoto T, Miyashita Y, Kasai H (1996) Ca2+‐dependent exocytotic pathways in Chinese hamster ovary fibroblasts revealed by a caged‐Ca2+ compound*. J Biol Chem 271: 17751–17754 [DOI] [PubMed] [Google Scholar]
- Ninomiya Y, Kishimoto T, Yamazawa T, Ikeda H, Miyashita Y, Kasai H (1997) Kinetic diversity in the fusion of exocytotic vesicles. EMBO J 16: 929–934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parisotto D, Pfau M, Scheutzow A, Wild K, Mayer MP, Malsam J, Sinning I, Söllner TH (2014) An extended helical conformation in domain 3a of Munc18‐1 provides a template for SNARE (soluble N‐ethylmaleimide‐sensitive factor attachment protein receptor) complex assembly. J Biol Chem 289: 9639–9650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park Y, Vennekate W, Yavuz H, Preobraschenski J, Hernandez JM, Riedel D, Walla PJ, Jahn R (2014) α‐SNAP interferes with the zippering of the SNARE protein membrane fusion machinery. J Biol Chem 289: 16326–16335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertsinidis A, Mukherjee K, Sharma M, Pang ZP, Park SR, Zhang Y, Brunger AT, Südhof TC, Chu S (2013) Ultrahigh‐resolution imaging reveals formation of neuronal SNARE/Munc18 complexes in situ . Proc Natl Acad Sci USA 110: E2812–E2820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pobbati AV, Stein A, Fasshauer D (2006) N‐ to C‐terminal SNARE complex assembly promotes rapid membrane fusion. Science 313: 673–676 [DOI] [PubMed] [Google Scholar]
- Rickman C, Davletov B (2005) Arachidonic acid allows SNARE complex formation in the presence of Munc18. Chem Biol 12: 545–553 [DOI] [PubMed] [Google Scholar]
- Rizo J, Xu J (2015) The synaptic vesicle release machinery. Annu Rev Biophys 44: 339–367 [DOI] [PubMed] [Google Scholar]
- Shen J, Tareste DC, Paumet F, Rothman JE, Melia TJ (2007) Selective activation of cognate SNAREpins by Sec1/Munc18 proteins. Cell 128: 183–195 [DOI] [PubMed] [Google Scholar]
- Shevchenko A, Tomas H, Havlis J, Olsen JV, Mann M (2007) In‐gel digestion for mass spectrometric characterization of proteins and proteomes. Nat Protoc 1: 2856–2860 [DOI] [PubMed] [Google Scholar]
- Söllner T, Bennett MK, Whiteheart SW, Scheller RH, Rothman JE (1993) A protein assembly‐disassembly pathway in vitro that may correspond to sequential steps of synaptic vesicle docking, activation, and fusion. Cell 75: 409–418 [DOI] [PubMed] [Google Scholar]
- Stein A, Weber G, Wahl MC, Jahn R (2009) Helical extension of the neuronal SNARE complex into the membrane. Nature 460: 525–528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Südhof TC, Rizo J (2011) Synaptic vesicle exocytosis. Cold Spring Harb Perspect Biol 3: a005637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi N, Kadowaki T, Yazaki Y, Miyashita Y, Kasai H (1997) Multiple exocytotic pathways in pancreatic β cells. J Cell Biol 138: 55–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Togneri J, Cheng Y‐S, Munson M, Hughson FM, Carr CM (2006) Specific SNARE complex binding mode of the Sec1/Munc‐18 protein, Sec1p. Proc Natl Acad Sci USA 103: 17730–17735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toonen RFG, Verhage M (2007) Munc18‐1 in secretion: lonely Munc joins SNARE team and takes control. Trends Neurosci 30: 564–572 [DOI] [PubMed] [Google Scholar]
- Towbin H, Staehelin T, Gordon J (1979) Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc Natl Acad Sci USA 76: 4350–4354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhage M, Maia AS, Plomp JJ, Brussaard AB, Heeroma JH, Vermeer H, Toonen RF, Hammer RE, van den Berg TK, Missler M, Geuze HJ, Sudhof TC (2000) Synaptic assembly of the brain in the absence of neurotransmitter secretion. Science 287: 864–869 [DOI] [PubMed] [Google Scholar]
- Vivona S, Cipriano DJ, O'Leary S, Li YH, Fenn TD, Brunger AT (2013) Disassembly of all snare complexes by N‐ethylmaleimide‐sensitive factor (NSF) is initiated by a conserved 1:1 interaction between α‐soluble NSF attachment protein (SNAP) and SNARE complex. J Biol Chem 288: 24984–24991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter AM, Wiederhold K, Bruns D, Fasshauer D, Sorensen JB (2010) Synaptobrevin N‐terminally bound to syntaxin‐SNAP‐25 defines the primed vesicle state in regulated exocytosis. J Cell Biol 188: 401–413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weninger K, Bowen ME, Choi UB, Chu S, Brunger AT (2008) Accessory proteins stabilize the acceptor complex for synaptobrevin, the 1:1 syntaxin/SNAP‐25 complex. Structure 16: 308–320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiedelhold K, Fasshauer D (2009) Is assembly of the SNARE complex enough to fuel membrane fusion? J Biol Chem 284: 13143–13152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter U, Chen X, Fasshauer D (2009) A conserved membrane attachment site in α‐SNAP facilitates N‐ethylmaleimide‐sensitive factor (NSF)‐driven SNARE complex disassembly. J Biol Chem 284: 31817–31826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Su L, Rizo J (2010) Binding of Munc18‐1 to synaptobrevin and to the SNARE four‐helix bundle. Biochemistry 49: 1568–1576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Wang S, Sheng Y, Zhang M, Zou W, Wu L, Kang L, Rizo J, Zhang R, Xu T, Ma C (2015) Syntaxin opening by the MUN domain underlies the function of Munc13 in synaptic‐vesicle priming. Nat Struct Mol Biol 22: 1–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Diao J, Colbert KN, Lai Y, Pfuetzner RA, Padolina MS, Vivona S, Ressl S, Cipriano DJ, Choi UB, Shah N, Weis WI, Brunger AT (2015) Munc18a does not alter fusion rates mediated by neuronal SNAREs, synaptotagmin, and complexin. J Biol Chem 290: 10518–10534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zilly FE, Sørensen JB, Jahn R, Lang T (2006) Munc18‐bound syntaxin readily forms SNARE complexes with synaptobrevin in native plasma membranes. PLoS Biol 4: 1789–1797 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Table EV1
Table EV2
Review Process File
Source Data for Figure 4B
Data Availability Statement
Referenced data: (Fernandez et al, 1998; Burkhardt et al, 2008; Stein et al, 2009).