

Abstract
Sphingolipids are membrane lipids globally required for eukaryotic life. The sphingolipid content varies among endomembranes with pre‐ and post‐Golgi compartments being poor and rich in sphingolipids, respectively. Due to this different sphingolipid content, pre‐ and post‐Golgi membranes serve different cellular functions. The basis for maintaining distinct subcellular sphingolipid levels in the presence of membrane trafficking and metabolic fluxes is only partially understood. Here, we describe a homeostatic regulatory circuit that controls sphingolipid levels at the trans‐Golgi network (TGN). Specifically, we show that sphingomyelin production at the TGN triggers a signalling pathway leading to PtdIns(4)P dephosphorylation. Since PtdIns(4)P is required for cholesterol and sphingolipid transport to the trans‐Golgi network, PtdIns(4)P consumption interrupts this transport in response to excessive sphingomyelin production. Based on this evidence, we envisage a model where this homeostatic circuit maintains a constant lipid composition in the trans‐Golgi network and post‐Golgi compartments, thus counteracting fluctuations in the sphingolipid biosynthetic flow.
Keywords: ceramide, lipid territories, lipid‐transfer protein, membrane contact sites, PtdIns(4)P
Subject Categories: Membrane & Intracellular Transport, Metabolism
Introduction
Sphingolipids (SLs) are bioactive components of eukaryotic membranes (Hannun & Obeid, 2008). Several stimuli activate SL metabolic enzymes to produce SL species that act as first or second messengers in signalling programmes involved in irreversible cell fate decisions (Hannun & Obeid, 2008) and where their derangements often result in the development of cancer (Morad & Cabot, 2013). Not surprisingly then, SL metabolism is subjected to control modules that maintain its balance and prevent unintended drifts away from the steady state (Vacaru et al, 2009; Breslow & Weissman, 2010; Siow & Wattenberg, 2012; Cantalupo et al, 2015). While these controls account for global SL homeostasis, SL biosynthesis involves the regional production and intracellular transport of different metabolites. As a consequence, SLs are unevenly distributed among cell membranes with post‐Golgi membranes being rich and pre‐Golgi membranes being poor in SLs (van Meer et al, 2008; Holthuis & Menon, 2014). Importantly, enrichment of SLs and cholesterol at post‐Golgi membranes increases membrane thickness and promotes the formation of membrane domains, both of which influence protein trafficking, and signalling and thus affect cell functions (Hannun & Obeid, 2008; Patterson et al, 2008; Lingwood & Simons, 2010; Sharpe et al, 2010; van Galen et al, 2014; Holthuis & Menon, 2014).
A hub in the intracellular SLs trafficking is the Golgi complex (De Matteis & Luini, 2008; Hannun & Obeid, 2008; Holthuis & Menon, 2014). In fact, newly synthesized ceramide (Cer) in the endoplasmic reticulum (ER) is either transported in vesicles to the cis‐Golgi pole, where it is converted to glucosylceramide (GlcCer), or delivered to the trans‐Golgi network (TGN) by the Cer‐transfer protein (CERT), for the production of sphingomyelin (SM) (Hanada et al, 2003). Similarly, cis‐Golgi produced GlcCer progresses through Golgi cisternae to be converted to ganglio‐series glyco‐SLs (GSLs) or is shunted to the TGN by the GlcCer‐transfer protein FAPP2 (D'Angelo et al, 2007, 2013) where it is used to produce globo‐series GSLs (D'Angelo et al, 2013; Fig EV1).
Figure EV1. Intracellular organization of SL metabolism.
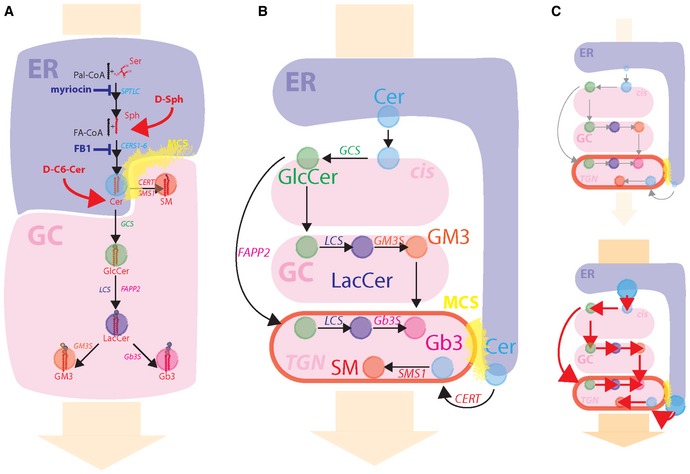
- Schematic representation of SL synthetic reactions happening at the ER (blue shaded area) and at the Golgi complex (GC; red shaded area). The metabolic positions where SL synthetic inhibitors act (blue) and where cell‐permeable SL precursors enter SL metabolism (red) are indicated. The regions highlighted in yellow indicate ER–TGN MCSs. The orange arrow in the background indicates the direction of the biosynthetic flow.
- Schematic representation of the sub‐Golgi distribution of SL metabolic enzymes and associated reactions.
- Experimental scheme for decreased (up) and increased (down) SL flow obtained by the use of cell‐permeable SL precursors or SL synthetic inhibitors.
CERT and FAPP2 are recruited to the TGN by the phosphoinositide phosphatidylinositol‐4‐phosphate (PtdIns(4)P; D'Angelo et al, 2008; Yamaji & Hanada, 2015). As a consequence, PtdIns(4)P is required for both SM and GSL syntheses and enrichment at post‐Golgi membranes (Toth et al, 2006; D'Angelo et al, 2013). PtdIns(4)P levels at the TGN depend on its production (by PtdIns‐4‐kinases) and consumption (by the ER‐localized PtdIns‐4‐phosphatase Sac1; De Matteis et al, 2013). The lipid‐transfer protein OSBP1 (Mesmin et al, 2013) operates the transport of PtdIns(4)P from the TGN to the ER for its dephosphorylation by Sac1. This trafficking/metabolic step is accomplished at specific sites of close apposition between ER and TGN defined as ER–TGN membrane contact sites (MCSs) where it is coupled to the transport of cholesterol from the ER to the TGN (Mesmin et al, 2013).
Once at the TGN, SLs and cholesterol are incorporated in membrane carriers and transported to post‐Golgi destinations, thus contributing to the establishment of the post‐Golgi membrane territory (De Matteis & Luini, 2008; Klemm et al, 2009; Deng et al, 2016). Nevertheless, carriers formation at the TGN occurs continuously so that TGN membranes are subjected to constant dissipation and rebuilding according to secretory flux volume to the TGN (De Matteis & Luini, 2008). Moreover, fluctuations in SL de novo synthesis are reported under a variety of signalling and stress conditions (Hannun & Obeid, 2008). Thus, it is not fully understood how cells keep the local TGN lipid composition (and as a consequence that of post‐Golgi membranes) controlled in spite of uncoordinated changes in membrane trafficking and SL precursor supply.
Here, we have acutely modulated the SL flow to the Golgi complex and measured the effect on TGN composition and metabolic capacity. Our results indicate that the SL flow controls the PtdIns(4)P levels at the TGN. Specifically, we describe a SL‐dependent signalling leading to PtdIns(4)P consumption and consequent release of PtdIns(4)P binding proteins from TGN membranes. Provided that PtdIns(4)P is required for SL and cholesterol transport to the TGN (Toth et al, 2006; D'Angelo et al, 2013), our findings reveal a negative feedback circuit that ensures the homeostatic control of SL levels at the TGN and at post‐Golgi membranes.
Results
Adaptive metabolic response to SL flow
To investigate how cells respond to acute changes in SL flow, HeLa cells were subjected to controlled perturbations in the SL metabolic input. Cells were first treated with fumonisin B1, an inhibitor of Cer synthesis and SL recycling (Wang et al, 1991; Ogretmen et al, 2002) (FB1; 50 μM for 24 h), or myriocin, a serine‐palmitoyl transferase inhibitor (Horvath et al, 1994; 2.5 μM for 24 h) to obliterate SL production (Fig EV1). Subsequently, cells were fed for 2 h with increasing concentrations of cell‐permeable SL precursors N‐hexanoyl‐D‐erythro‐sphingosine (C6‐D‐Cer; for FB1‐treated cells) or D‐erythro‐sphingosine (D‐Sph; for myriocin‐treated cells) and the amount of SL precursor converted either to SM or to GlcCer was quantified. As shown in Fig 1A, the fractions of SL precursors converted to SM or GlcCer were constant over a wide range (from 5 nM to 1 μM) of C6‐D‐Cer and D‐Sph concentrations. In contrast, addition of higher amounts of SL precursors resulted in a sudden decrease in SL flux directed to SM synthesis, which was counterbalanced by an increase in GlcCer synthesis and Cer accumulation (Fig 1A). When the dynamics of this phenomenon were investigated by measuring C6‐D‐Cer (10 μM) conversion to C6‐D‐SM or C6‐D‐GlcCer after short treatments (0–60 min), SM production was found to be very efficient at early time points (0–15 min) to be then reduced at later times. On the contrary, GlcCer production was constant during the time course (Appendix Fig S1).
Figure 1. Metabolic cell response to SL flow.
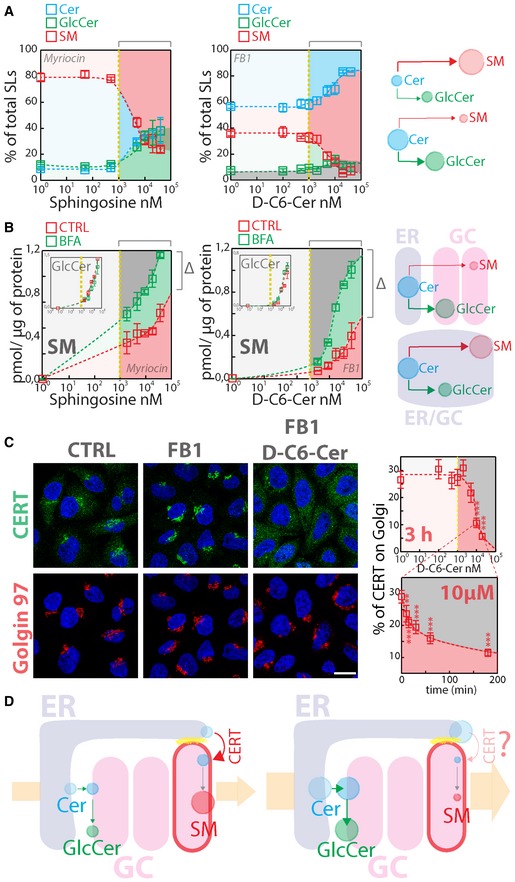
- HeLa cells treated with myriocin (2.5 μM) or FB1 (50 μM) for 24 h (left and middle panels, respectively) were fed with increasing concentrations of D‐Sph (left panel) or C6‐D‐Cer (middle panel) for 2 h. 3H‐D‐Sph and 3H‐C6‐D‐Cer (≈5 nM) were mixed as tracers with their non‐radioactive counterparts. The percentage of total radioactivity associated with Cer (cyan), GlcCer (green) and SM (red) in the different conditions was quantitated after lipid extraction and HPTLC separation. The right panel schematizes the metabolic effect of sustained SL flow.
- (Left and middle panels) Cells treated as in (A) were subjected to brefeldin A (BFA) administration (5 μg/ml) before (30‐min pre‐treatment) and during SL administration for 2 h. The amount (pmol) of SM and GlcCer (inset) produced under control (red squares) or BFA (green squares) per μg of protein lysate was calculated as detailed in Materials and Methods and indicated at increasing SL precursor concentrations. Δ indicates the difference in SM production at saturating SL precursor amounts. The right panel schematizes the effect of BFA on SM production.
- CERT localization in HeLa cells control (CTRL), treated with FB1 (50 μM, 24 h) or with 10 μM C6‐D‐Cer for 2 h after 24‐h treatment with FB1. Scale bar, 10 μm. Left panels indicate the percentage of CERT associated with the Golgi complex at increasing C6‐D‐Cer concentrations (upper graph) and at the indicated times (lower graph). **P < 0.01; ***P < 0.001; according to two‐tailed Student's t‐test.
- Schematic representation of SL flow‐induced redistribution of CERT to the cytosol and consequent inhibition of SM synthesis.
The fact that SM synthesis is promptly repressed at high SL precursor concentrations can be due to either saturation of enzymes synthesizing SM (i.e. mostly SM synthase 1 [SMS1]) (Huitema et al, 2004) or of the system that supplies Cer to SMS1 [i.e. CERT (Hanada et al, 2003)]. To discriminate between these possibilities, we performed the experiment described in Fig 1A in cells pre‐treated with brefeldin A (BFA; 5 μg/ml). BFA redistributes Golgi membranes to the ER (Lippincott‐Schwartz et al, 1989) and relocates SMS1 and GlcCer synthase (GCS) to this mixed ER–Golgi compartment (Appendix Fig S2). Under these conditions, Cer is readily available to both GCS and SMS1 with no need for either vesicular or non‐vesicular Cer transport (D'Angelo et al, 2013). As shown in Fig 1B, BFA treatment resulted in increased SM synthesis at precursor concentrations > 1 μM with no effects on GlcCer production, suggesting that the observed drop in SM synthesis (Fig 1A) is not simply due to saturation of SMS1.
We then analysed CERT localization in response to SL perturbations. As already reported by others (Kumagai et al, 2007), we found that SL depletion induced CERT recruitment to the TGN (Figs 1C and EV2, Appendix Fig S3A). Interestingly, when cells were treated with C6‐D‐Cer or D‐Sph, CERT shifted to a cytosolic distribution at concentrations inducing the drop in SM production with no measurable changes in CERT mRNA and protein levels (Figs 1C and EV2, Appendix Fig S3A). CERT redistribution was indeed sufficient to impair Cer transport to the Golgi as assessed by FL‐BODIPY‐C5‐Cer transport assay (Appendix Fig S3D) and was complete within 60 min of treatment (Figs 1C and EV2A, and Movie EV1), indicating that SL flow acutely counteracts CERT recruitment, Cer transport to the TGN, and as a consequence SM synthesis (Fig 1D). We thus wondered what mechanism could be responsible for this phenomenon.
Figure EV2. The association of GFP‐CERT with TGN membranes depends on SL flow.
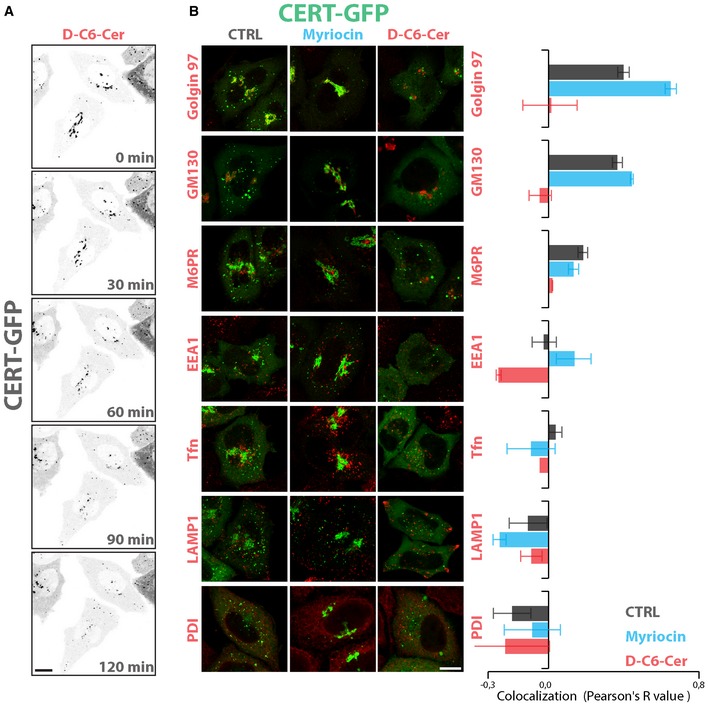
- GFP‐CERT localization in HeLa cells, pre‐treated with myriocin (2.5 μM, 24 h) and then treated with D‐C6‐Cer (10 μM) for 2 h. Images are selected frames from Movie EV1. Scale bar, 10 μm.
- GFP‐CERT co‐localization with the indicated organelle markers in HeLa cells either non‐treated (CTRL, grey), treated with myriocin (2.5 μM, 24 h) (myriocin, cyan) or pre‐treated with myriocin (2.5 μM, 24 h) and then treated with D‐C6‐Cer (10 μM) for 2 h (D‐C6‐Cer; red). Left panels: representative images. Right panel: co‐localization values calculated as Pearson's correlation coefficients (R‐values) between GFP‐CERT and the indicated markers under the different treatments. Scale bar, 10 μm. Data are means ± SEM from at least 3 independent experiments where at least 10 cells per experiment were considered.
Sustained SL flow induces the consumption of the TGN pool of PtdIns(4)P
First we looked for membrane‐associated proteins, which behaved similar to CERT in response to sustained SL flow. While most of the tested proteins remained associated with membranes after C6‐D‐Cer treatment, a specific subset of TGN‐associated proteins was displaced from membranes. This subset contained the proteins FAPP2, OSBP1, GOLPH3 and AP‐1 (γ‐adaptin) (Fig 2A and Appendix Fig S4).
Figure 2. The localization of TGN PtdIns(4)P effectors is sensitive to SL flow.
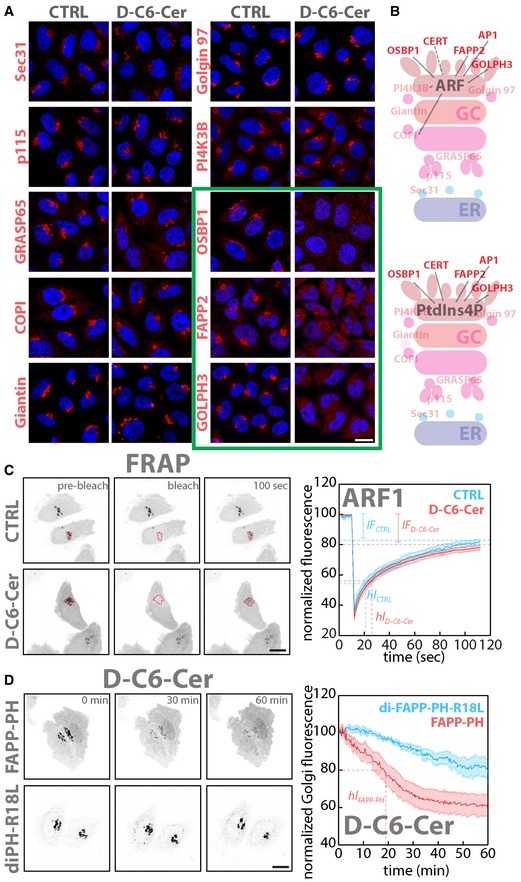
- Cells were treated with either vehicle (EtOH) or D‐C6‐Cer (10 μM) for 2 h, fixed and stained for nuclei (DAPI; blue) and with antibodies to different Golgi‐associated proteins (red).
- Schematic representation of Golgi proteins localization. Upper panel shows proteins that require ARF, and lower panel proteins that require PtdIns(4)P for their Golgi localization. In solid red are proteins sensitive to sustained SL flow.
- FRAP‐based assessment of ARF1‐GFP dynamics of association/dissociation from Golgi membranes in EtOH‐ or D‐C6‐Cer (10 μM)‐treated cells (see Materials and Methods for details; left panels). Mean normalized fluorescence intensity ± SEM over time under CTRL (cyan) (n = 10) and D‐C6‐Cer (red) (n = 10) treatment is reported (hl: half‐life; IF: immobile fraction).
- Live imaging analysis (1 h) of FAPP‐PH‐GFP (FAPP‐PH) or diFAPP‐PH‐R18L‐GFP (diPH‐R18L) during D‐C6‐Cer treatment (10 μM). Mean normalized fluorescence intensity ± SEM over time is reported for FAPP‐PH‐GFP (red) (n = 6) and diFAPP‐PH‐R18L‐GFP (cyan) (n = 5) (hl: half‐life).
Given that these proteins require the coincident binding of ARF and PtdIns(4)P for their recruitment to the TGN (Fig 2B; De Matteis et al, 2013), the effect of SL flow on ARF1 and PtdIns(4)P was investigated. As shown in Appendix Fig S5, C6‐D‐Cer treatment did not perturb ARF1 localization to the Golgi membranes. Moreover, when the dynamics of ARF1‐GFP association to the Golgi complex were examined by fluorescence recovery after photobleaching (FRAP) experiments in cells treated with C6‐D‐Cer (10 μM for 2 h; Figs 2C and EV3, and Movies EV2), no differences were observed.
We thus used the GFP‐tagged pleckstrin homology domain of FAPP2 (FAPP‐PH‐GFP) as a TGN PtdIns(4)P probe (Dowler et al, 2000; Godi et al, 2004; D'Angelo et al, 2013). As shown in Fig 2C and in Movie EV4, C6‐D‐Cer administration (10μM) induces the redistribution of FAPP‐PH from the Golgi complex to the cytosol. On the other hand, a tandem FAPP‐PH mutant (diFAPP‐PH‐R18L‐GFP), which localizes to the Golgi complex in spite of being unable to bind PtdIns(4)P (Godi et al, 2004), was much less sensitive to this treatment (Fig 2C and in Movie EV5), suggesting that SL flow to the Golgi complex controls the TGN phosphoinositide composition.
Indeed, both C6‐D‐Cer and D‐Sph treatments induced the disappearance of the PtdIns(4)P signal from the Golgi region (Figs 3A and EV3) as assessed by the use of anti‐PtdIns(4)P antibody (Hammond et al, 2009) at concentrations and times of treatment that induce CERT relocation to the cytosol (compare Figs 1C and 3B).
Figure 3. SL flow controls PtdIns(4)P levels at the Golgi.
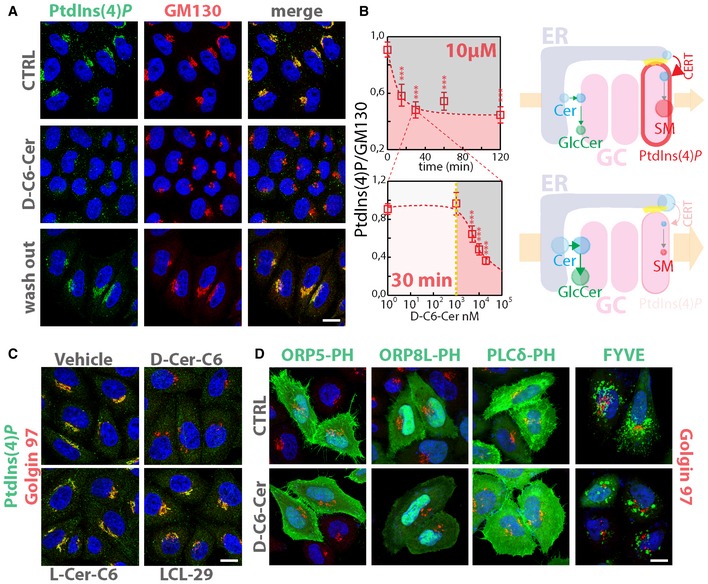
- Cells treated either with vehicle (EtOH), D‐C6‐Cer (10 μM) for 30 min or treated with D‐C6‐Cer (10 μM) for 30 min and washed out for 4 h were stained with a specific anti‐PtdIns(4)P antibody as detailed in Materials and Methods.
- HeLa cells treated with D‐C6‐Cer (10 μM) for the indicated times (upper left panel) or with increasing D‐C6‐Cer concentrations for 30 min (lower left panel) were processed and stained as in (A). Confocal images were acquired, segmented and analysed by CellProfiler software, as detailed in Materials and Methods. Average of normalized PtdIns(4)P/GM130‐integrated intensities ratio is reported for each time or concentration point. Data are means ± 5 × SEM of values obtained from at least 500 cells per experimental point. Right panels schematize the effect of SL flow on PtdIns(4)P levels, CERT recruitment to the Golgi and SM synthesis. Yellow dotted line indicates the concentration of SL precursor where effects of sustained SL flow on metabolism start to be observed. ***P < 0.001; according to two‐tailed Student's t‐test.
- HeLa cells treated either with vehicle (EtOH), D‐C6‐Cer (10 μM), L‐C6‐Cer (10 μM) or LCL‐29 (10 μM) for 30 min were fixed, permeabilized and stained as in (A).
- Cells transfected with GFP‐tagged ORP5‐PH, ORP8L‐PH, PLCδ‐PH or FYVE were treated either with vehicle (EtOH), D‐C6‐Cer (10 μM) for 2 h, fixed and stained with DAPI (blue) and an anti‐Golgin97 antibody (red).
Figure EV3. SL flow controls PtdIns(4)P levels at the Golgi.
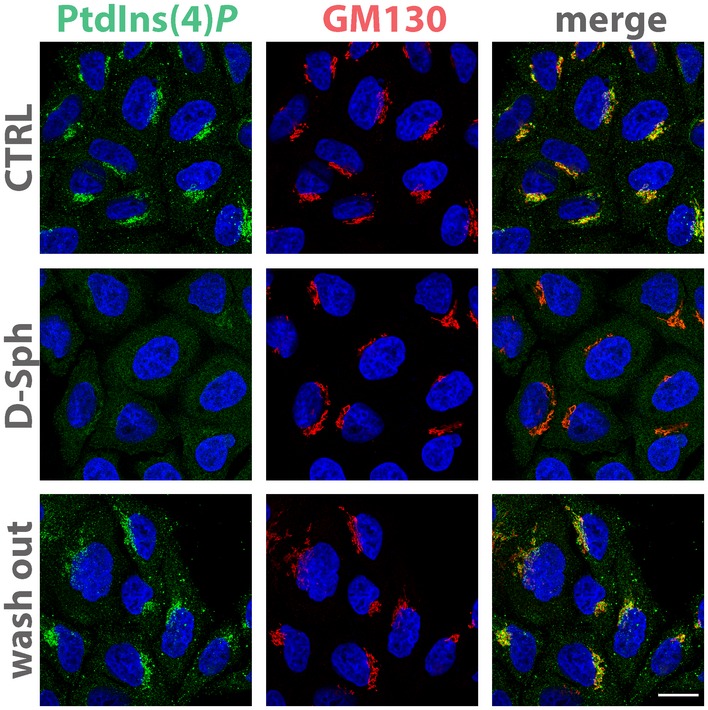
Cells treated either with vehicle (EtOH), with D‐Sph (30 μM) for 30 min or with D‐Sph (30 μM) and washed out for 4 h were fixed and permeabilized as in Fig 3A and stained with DAPI (blue), an anti‐GM130 antibody (red) and anti‐PtdIns(4)P antibody (green). Scale bar, 10 μm.
While C6‐D‐Cer‐induced PtdIns(4)P loss (Fig 3B) explains the inhibition of CERT‐dependent SM synthesis (Fig 1A), it should also hamper cholesterol transport to the TGN and globo‐series GSL production as these depend on OSBP1 and FAPP2, respectively, and to their ability to bind PtdIns(4)P at the TGN (D'Angelo et al, 2013; Mesmin et al, 2013). Accordingly, we observed a significant increase in cholesterol esters production (indicative of redirection of cholesterol flux towards lipid droplets; Ikonen, 2008) and a strong inhibition of globo‐series GSLs (Gb3) synthesis in cells treated with D‐C6‐Cer (10μM), which was counterbalanced by a significant accumulation of GlcCer (Appendix Fig S6).
To exclude that the effects observed under SL challenges were caused by non‐specific cell poisoning, control experiments were performed. According to these experiments, (i) the effect of C6‐D‐Cer and D‐Sph is reversible as cells reacquire normal PtdIns(4)P staining after wash‐outs (Figs 3A and EV3). (ii) When the non‐metabolizable C6‐L‐Cer enantiomer (Duran et al, 2012) was added to the cells, no effect was observed on PtdIns(4)P staining (Fig 3C). (iii) Since Cer exerts cytostatic and pro‐apoptotic effects (Hannun & Obeid, 2008) and growth conditions influence PtdIns(4)P levels at the TGN (Blagoveshchenskaya et al, 2008; Bajaj Pahuja et al, 2015), the possibility that the observed effect is an indirect consequence of Cer on cell proliferative/apoptotic signalling was considered. Thus, cells were treated with a Cer analogue D‐erythro‐2‐N‐[6′‐(1″‐pyridinium)hexanoyl]‐sphingosine bromide (LCL29) that inhibits cell growth and potently induces apoptosis (Appendix Fig S7) without being concentrated and metabolized at the Golgi complex (Hou et al, 2011). Under these conditions, we observed no changes in PtdIns(4)P staining at the Golgi (Fig 3C), suggesting that Cer needs to be transported and metabolized at the Golgi complex to influence PtdIns(4)P levels.
Recent studies have highlighted the existence of multiple pools of PtdIns(4)P at the TGN, PM and endosomal/lysosomal compartments (Hammond et al, 2014). By the use of a PtdIns(4)P probe able to detect the different subcellular PtdIns(4)P pools (i.e. diP4M‐mCherry; Hammond et al, 2014), we observed that C6‐D‐Cer treatment affects uniquely the PtdIns(4)P pool at the Golgi (Fig EV4A and B, and Movie EV6). Moreover, the PtdIns(4)P pool at the plasma membrane is specifically recognized by the PH domains of OSBP family proteins ORP5 and ORP8L (Chung et al, 2015). As shown in Fig 3D, C6‐D‐Cer did not influence the plasma membrane association of GFP‐tagged versions of ORP5 and ORP8L PH domains. Furthermore, C6‐D‐Cer treatment did not induce appreciable changes in the membrane association of probes for other phosphoinositides (i.e. GFP‐PH‐PLCδ and GFP‐FYVE, recognizing PtdIns(4,5)P 2 and PtdIns(3)P, respectively (Lemmon et al, 1995; Burd & Emr, 1998; Stauffer et al, 1998; Varnai & Balla, 1998; Fig 3D) even after prolonged treatments (Fig EV4C). This indicates that the TGN pool of PtdIns(4)P is acutely and specifically sensitive to sustained SL flow with consequences on local SL and sterol composition.
Figure EV4. Sustained SL flow specifically controls PtdIns(4)P at the TGN .
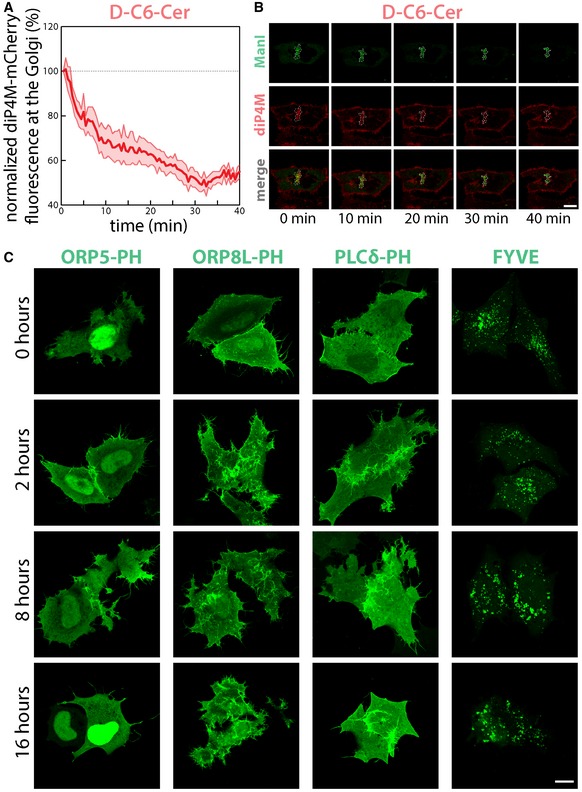
- HeLa cells transfected with plasmids encoding the PtdIns(4)P binding protein diP4M‐SidM‐mCherry (diP4M‐mCherry, red) and the Golgi enzyme Man1‐YFP (MANI, green) were treated with D‐C6‐Cer (10 μM) and imaged for the indicated time. The amount of diP4M‐mCherry associated with the Golgi region was quantified. Data are mean ± SEM.
- Selected frames from Movie EV6 are reported; dashed line indicates Golgi perimeter. Scale bar, 10 μm.
- Cells transfected with GFP‐tagged ORP5‐PH, ORP8L‐PH, PLCδ‐PH or FYVE were treated with D‐C6‐Cer (10 μM) for the indicated times, fixed and imaged. Scale bar, 10 μm.
By recruiting effector proteins with membrane remodelling and deforming properties, the PtdIns(4)P is required for the maintenance of Golgi complex structure and function (D'Angelo et al, 2012). Consequently, SL‐induced PtdIns(4)P consumption is expected to perturb Golgi morphology. Accordingly, we observe that C6‐D‐Cer treatment results in the alteration of the Golgi structure with tubulation of the cis‐side (GM130) and vesiculation of the TGN (TGN46) as assessed by immunofluorescence (Appendix Fig S8A). When the ultrastructure of the Golgi complex was analysed by electron microscopy and tomography, C6‐D‐Cer treatment was found to induce the “curling” of late cisternae and development of characteristic multivesicular structures at the trans‐Golgi pole (Appendix Fig S8B), where their formation possibly depends on inward budding events (Appendix Fig S8C,Movies EV7, EV8, EV9 and EV10). Thus, SL flow controls the morpho‐functional status of the TGN possibly by regulating the membrane association of key PtdIns(4)P effectors at the TGN.
SM synthesis induces the consumption of PtdIns(4)P at the TGN
According to the data so far reported, Cer requires to be transported to the Golgi complex to induce its effect on TGN PtdIns(4)P. At the Golgi, Cer is metabolized to GlcCer by GCS (Ichikawa et al, 1998) or to SM by SMS1 (Huitema et al, 2004). To investigate the specific role of these reactions in PtdIns(4)P consumption, GCS and SMS1 were silenced and cells were treated with D‐C6‐Cer for 2 h. As shown in Fig 4A SMS1‐KD specifically protected TGN PtdIns(4)P from C6‐D‐Cer treatment. As a consequence of this effect, SMS1‐KD prevents PtdIns(4)P binding proteins (here we show GOLPH3) from being redistributed to the cytosol after C6‐D‐Cer treatment (Appendix Fig S9A). Interestingly, untreated SMS1‐KD cells showed increased association of CERT, OSBP1 and GOLPH3‐GFP with the TGN (Appendix Fig S9B and C), possibly due to increased local PtdIns(4)P levels upon reduced basal SM synthesis. In agreement with these data, C6‐D‐Cer treatment did not induce TGN morphological alteration as assessed by immunofluorescence or electron microscopy in SMS1‐KD cells (Appendix Fig S10). Along similar lines, silencing of CERT, which mediates Cer transport to the TGN for SM synthesis (Hanada et al, 2003), but not of FAPP2, which mediates GlcCer transport to the TGN for globo‐series GSL synthesis (D'Angelo et al, 2013), also protected Golgi PtdIns(4)P from C6‐D‐Cer treatment (Appendix Fig S11).
Figure 4. Cer‐to‐SM conversion controls PtdIns(4)P levels at the TGN .
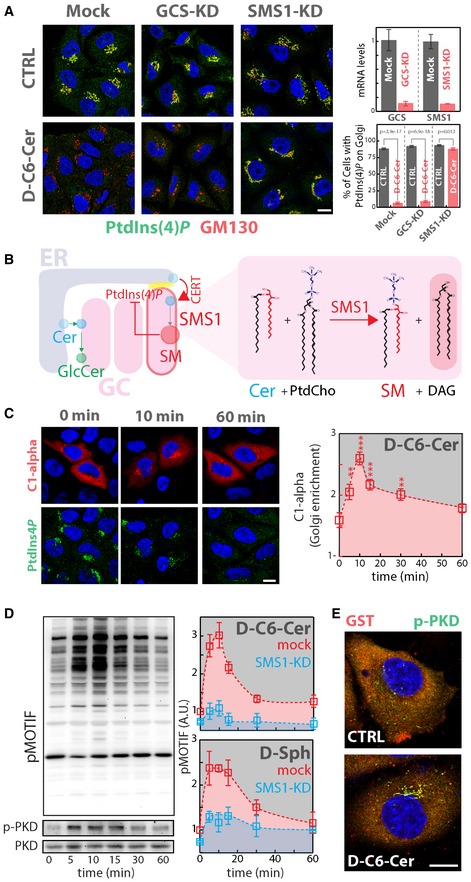
- Cells KD for GCS or SMS1 were treated with D‐C6‐Cer (10 μM) for 2 h, fixed, permeabilized and stained as in Fig 3A. KD efficiencies are indicated as reduction in mRNA levels as assessed by RT‐qPCR (upper right panel). The percentage of cells showing PtdIns(4)P staining at the Golgi in CTRL and D‐C6‐Cer‐treated cells is reported (lower right panel). Two‐tailed Student's t‐test.
- Schematic representation of the feedback loop regulating SM synthesis through PtdIns(4)P (left panel). Biochemical depiction of Cer‐to‐SM conversion operated by SMS1.
- Cells expressing the GST‐tagged DAG sensor PKD‐C1α domain (C1‐alpha) were treated for the indicated times with D‐C6‐Cer (10 μM), fixed, permeabilized and stained with DAPI (blue), anti‐GST (red) and anti‐PtdIns(4)P (green) antibodies (left panel). The Golgi‐associated C1‐alpha fraction at the different time points is reported (right panel). Yellow dotted line indicates the concentration of SL precursor where effects of sustained SL flow on metabolism start to be observed. **P < 0.01; ***P < 0.001; according to two‐tailed Student's t‐test.
- Cells treated with D‐C6‐Cer (10 μM) for the indicated times were lysed and lysates were processed for SDS–PAGE and Western blotting (as detailed in Materials and Methods). Antibodies recognizing total PKD (PKD), phospho‐Ser916‐PKD (p‐PKD) and phosphorylated PKD substrates (pMOTIF) were used to monitor PKD activation (left panels). PKD activation in both mock (red) and SMS1‐KD (cyan) cells was evaluated by quantitating phosphorylation of PKD substrates during D‐C6‐Cer (10 μM) or D‐Sph (30 μM) administration. Yellow dotted line indicates the concentration of SL precursor where effects of sustained SL flow on metabolism start to be observed.
- Cells transfected with a plasmid encoding GST‐PKD2 were treated with D‐C6‐Cer (10 μM) for 15 min, fixed, permeabilized and stained with DAPI (blue), anti‐GST (red) and anti‐p‐PKD (green).
The SMS1‐mediated conversion of Cer to SM is coupled to the production of diacylglycerol (DAG; Fig 4B; Hannun & Obeid, 2008). Thus, DAG production at the TGN was evaluated in relation to PtdIns(4)P loss in response to SL flow. HeLa cells were, thus, transfected with a vector encoding the C1α domain of PKD (PKD‐C1α), which is recruited to membranes by DAG (Baron & Malhotra, 2002). PKD‐C1α localization was then followed during C6‐D‐Cer administration along with that of PtdIns(4)P. As shown in Fig 4C, C6‐D‐Cer treatment induced a fast recruitment of PKD‐C1α to the Golgi region that peaked at 10–15 min and that preceded the disappearance of PtdIns(4)P. Similar results were obtained by using the high‐affinity DAG and PtdIns(4)P probes YFP‐DBD and diFAPP‐PH‐mCherry (Villani et al, 2008; D'Angelo et al, 2013) in live imaging experiments (Movie EV11).
Since DAG at the TGN induces PKD recruitment and activation (Baron & Malhotra, 2002), we treated cells with C6‐D‐Cer for different times and monitored PKD auto‐phosphorylation (indicative of activation) by Western blotting and immunofluorescence. As shown in Fig 4D, C6‐D‐Cer treatment induced PKD activation that peaked at 10‐ to 15‐min treatment, followed by a decrease in PKD phosphorylation at 30 and 60 min. As a consequence of this dynamics, also PKD substrates (as recognized by a specific anti‐PKD phospho‐motif, [pMOTIF]; Doppler et al, 2005) were collectively phosphorylated at early time points of C6‐D‐Cer treatment, and then dephosphorylated after 30 and 60 min of treatment (Fig 4D). The phosphorylation of PKD substrates induced by C6‐D‐Cer was largely dependent on Cer‐to‐SM conversion as SMS1‐KD inhibited their response to C6‐D‐Cer treatment. Similar results were obtained by D‐Sph, which also induced a transient and SMS1‐dependent PKD substrates phosphorylation (Fig 4D). When the subcellular location of PKD activation upon C6‐D‐Cer treatment was visualized by immunofluorescence, a strong activation signal in the perinuclear region was noticed (Fig 4E).
Given the association between PKD signalling and PtdIns(4)P consumption induced by SM production, we wondered whether PKD itself could be involved in the control of PtdIns(4)P levels at the TGN. We thus overexpressed a wt or dominant‐negative form of PKD (PKD‐KinDead, PKD‐K618N) (Liljedahl et al, 2001) in HeLa cells before C6‐D‐Cer treatment. As shown in Fig 5A, PKD‐KinDead expression protected PtdIns(4)P from C6‐D‐Cer‐induced consumption, while PKD‐wt did not. Along similar lines, the knockdown of all three PKD isoforms (Rykx et al, 2003) induced protection towards C6‐D‐Cer treatment (Fig 5B), indicating that PKD is required for the consumption of the TGN PtdIns(4)P pool under C6‐D‐Cer treatment. When the individual contribution of each isoform to this phenotype was considered, PKD2 was found to be responsible for C6‐D‐Cer‐induced PtdIns(4)P consumption (Fig 5C). Altogether, these data indicate that SM synthesis, through PKD activation, controls PtdIns(4)P levels at the TGN.
Figure 5. PKD activation is required for SL flow‐induced PtdIns(4)P consumption.
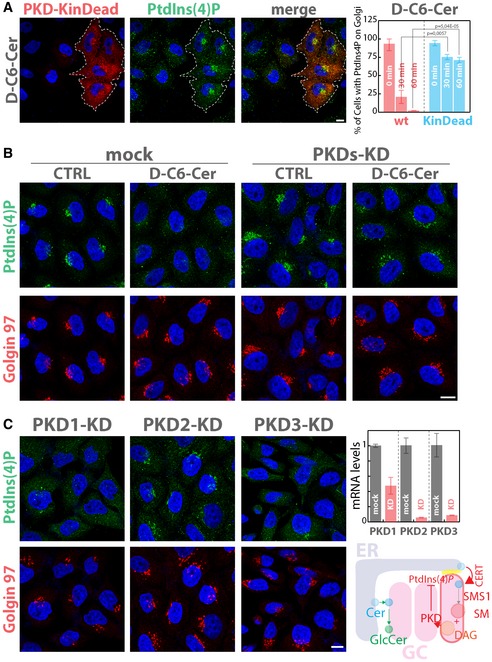
- Cells expressing GST‐tagged versions of either PKD wild type (PKD‐wt) or of a kinase‐dead mutant of PKD (PKD‐KinDead; PKD‐K618N; Liljedahl et al, 2001) were treated for 30 and 60 min with D‐C6‐Cer (10 μM). After fixation and permeabilization, cells were stained with DAPI (blue), anti‐GST (red) and anti‐PtdIns(4)P (green). Dotted line highlights transfected cells. The left panel shows a representative image of PKD‐KinDead‐transfected cells after 60 min of D‐C6‐Cer (10 μM) treatment. The percentage of PKD‐transfected (wt, red; KinDead, cyan) cells showing PtdIns(4)P staining at the Golgi at the different times of D‐C6‐Cer treatment is reported (right panel). Two‐tailed Student's t‐test.
- Cells silenced for the three PKD isoforms were treated with D‐C6‐Cer (10 μM) for 60 min, fixed, permeabilized and stained as in Fig 3A.
- Cells silenced for each of the three PKD isoforms were treated with D‐C6‐Cer (10 μM) for 60 min, fixed, permeabilized and stained as in Fig 3A. KD efficiency for each PKD isoform is reported (mRNA levels as measured by RT‐qPCR; upper right panel). Schematic representation of SL‐mediated PKD activation and PKD‐dependent PtdIns(4)P consumption at the TGN (lower right panel).
Interestingly, CERT itself is a PKD substrate and CERT phosphorylation by PKD reduces CERT affinity for PtdIns(4)P (Fugmann et al, 2007). According to this notion, SL‐induced PKD activation would counteract CERT function by reducing both the levels of PtdIns(4)P at the TGN and the ability of CERT to bind it. We evaluated the contribution of PKD phosphorylation on CERT (by the use of the CERT phospho‐depleted mutant S132A; Fugmann et al, 2007) to its localization at the Golgi under SL challenges and found it to be minor if compared with the primary effect on PtdIns(4)P levels as CERT‐S132A mutant was still sensitive to changes in the SL flow (Appendix Fig S12).
SL flow regulates phosphoinositide turnover through PI4KIIIβ and Sac1
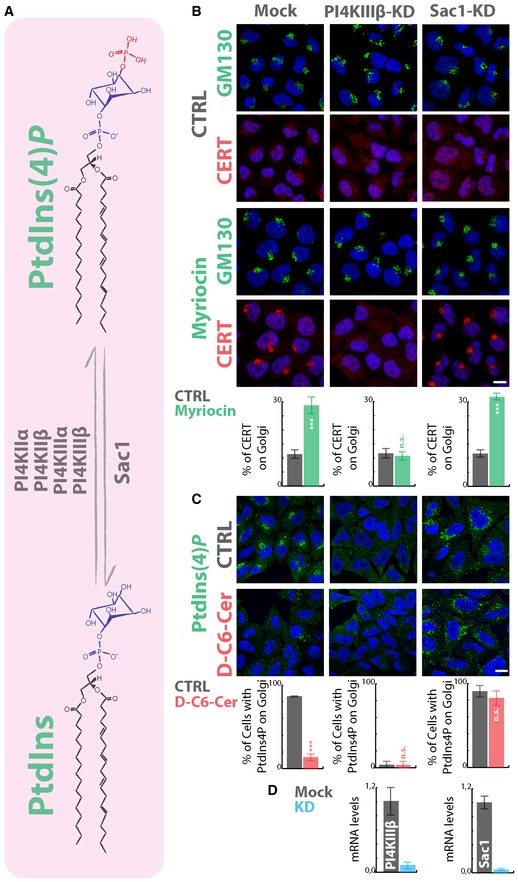
PtdIns(4)P is synthesized through the phosphorylation of PtdIns by PtdIns‐4‐kinases (PI4KIIα, PI4KIIIα, PI4KIIβ, and PI4KIIIβ), while the opposite reaction is catalysed by the PtdIns(4)P‐4‐phosphatase Sac1 (De Matteis et al, 2013) (Fig 6A). We, thus, wondered which of these enzymes is involved in the SL‐mediated modulation of PtdIns(4)P pool at the TGN. To this aim, we individually silenced each PI4K or Sac1 in HeLa cells and treated them with myriocin or C6‐D‐Cer to inhibit or stimulate the SL metabolism (Fig 6 and Appendix Fig S13). We then evaluated both myriocin‐induced CERT recruitment to the Golgi complex and C6‐D‐Cer‐triggered PtdIns(4)P consumption. As shown in Fig 6B, PI4KIIIβ‐KD specifically inhibited CERT recruitment upon myriocin treatment and PtdIns(4)P staining at the TGN in both vehicle‐ and C6‐D‐Cer‐treated cells, suggesting that PI4KIIIβ is the main PI4K involved in the production of the PtdIns(4)P pool regulated by SLs.
Figure 6. PI4KIIIβ and Sac1 control the Golgi PtdIns(4)P pool under SL flow.
-
ASchematic representation of PtdIns phosphorylation by PI4Ks and of PtdIns(4)P dephosphorylation by Sac1.
-
B, CCells silenced for the expression of PI4KIIIβ or SAC1 were treated with (B) myriocin (2.5 μM; 24 h) or with (C) D‐C6‐Cer (10 μM; 60 min). In (B), CERT recruitment to the Golgi region was evaluated by immunofluorescence and quantitated as in Fig 1C. In (C), PtdIns(4)P consumption under D‐C6‐Cer (10 μM, 1 h) was evaluated as in Fig 4A. ***P < 0.001; n.s.; non significant according to two‐tailed Student's t‐test.
-
DThe KD efficiency for PI4KIIIβ and SAC1 is assessed by RT‐qPCR (right histograms).
PI4KIIIβ can be phosphorylated by PKD on serine 294, and this phosphorylation stimulates 4‐kinase activity (Hausser et al, 2005). We thus wondered whether PI4KIIIβ is phosphorylated by PKD in response to SL flow. As shown in Appendix Fig S14A, C6‐D‐Cer treatment induced a fast and transient PI4KIIIβ phosphorylation (as recognized by the anti‐PKD pMOTIF antibody), which is completely prevented by treatment with the PKD inhibitor Gö 6976. Moreover, when the PI4KIIIβ‐S294A mutant was tested for PKD‐dependent phosphorylation following C6‐D‐Cer treatment, it was found to be non‐phosphorylated, suggesting that Ser 294 is the only phosphosite in PI4KIIIβ to be phosphorylated by PKD in response to sustained SL flow (Appendix Fig S14B). Nevertheless, while PI4KIIIβ phosphorylation by PKD on Ser 294 leads to enzyme activation and increased PtdIns(4)P production (Hausser et al, 2005; Appendix Fig S14C), the net effect of PKD activation by SLs is, in fact, a decrease in TGN PtdIns(4)P levels. We thus considered the involvement of Sac1 phosphatase.
Sac1 is a transmembrane protein cycling between the ER and the Golgi complex (Blagoveshchenskaya et al, 2008). It has been shown that signalling cues influence the distribution of Sac1 between these two compartments (Bajaj Pahuja et al, 2015) with serum starvation inducing its translocation to the Golgi complex and consequent TGN PtdIns(4)P consumption (Blagoveshchenskaya et al, 2008). We asked whether increased SL flow would mimic this situation. To test this hypothesis, cells expressing a HA‐tagged version of Sac1 were treated with C6‐D‐Cer, and changes in Sac1 subcellular distribution associated with PtdIns(4)P consumption were evaluated. As shown in Appendix Fig S15A, Sac1 overexpression sensitizes cells to C6‐D‐Cer treatment, since low C6‐D‐Cer concentrations (1 μM) caused the disappearance of PtdIns(4)P at the TGN. Nevertheless, Sac1 intracellular distribution was not influenced by the treatment with most of the protein being ER localized even after C6‐D‐Cer administration. Moreover, Sac1 overexpression was sufficient to counteract CERT recruitment to the Golgi complex induced by myriocin treatment (Appendix Fig S15B). Importantly, Sac1‐KD rendered TGN PtdIns(4)P resistant to C6‐D‐Cer treatment, indicating that C6‐D‐Cer‐induced PtdIns(4)P consumption results from its Sac1‐operated conversion to PtdIns (Fig 6C).
Thus, the metabolic couple PI4KIIIβ/Sac1 is responsible for the control of PtdIns(4)P levels at the TGN in response to SL flow and as a consequence for the maintenance of SL homeostasis. A corollary of this is that malfunction of the PI4KIIIβ/Sac1 system should translate in impaired SL metabolism. To test this, we silenced Sac1 and measured cellular SL content. As shown in Appendix Fig S14C, Sac1 silencing induced a significant increase in total Cer, GlcCer and SM levels (and of the overall SL cellular content), while sphingoid bases (i.e. Sph and sphinganine [dhSph]) were significantly decreased. This evidence is compatible with a role for Sac1 in the regulation of Cer production at the ER and confirms the importance of the PtdIns(4)P metabolic machinery in SL homeostasis by extending its involvement beyond the circuit described in this study. Nevertheless, how does PKD influence Sac1‐mediated PtdIns(4)P dephosphorylation?
SL flow induces PKD‐dependent OSBP1 phosphorylation and OSBP1‐dependent PtdIns(4)P consumption
Sac1 is not known to be a PKD substrate and our attempts to detect PKD‐dependent Sac1 phosphorylation gave negative results; thus, we considered the possibility that PKD could phosphorylate an intermediate factor that would then present PtdIns(4)P to Sac1. Recent publications have indicated that the PtdIns(4)P effector, OSBP1, interacts with Sac1 (Wakana et al, 2015); it is phosphorylated by PKD (Nhek et al, 2010) and is able to transfer PtdIns(4)P from the TGN to the ER channelling this lipid for Sac1‐dependent dephosphorylation (Mesmin et al, 2013). We, thus, decided to investigate the possible role of OSBP1 in the SL‐induced PtdIns(4)P consumption.
First, we tested whether SL flow induces OSBP1 phosphorylation by PKD. To this aim, cells expressing OSBP1‐GFP were treated with C6‐D‐Cer, and PKD phosphorylation was revealed by pMOTIF antibody after immunoprecipitation. As shown in Fig 7A, PKD‐mediated OSBP1 phosphorylation started increasing after 15 min of C6‐D‐Cer administration and remained high at 60‐min treatment while pre‐treatment of cells with the PKD inhibitor Gö 6976 completely abolished these dynamics. Noteworthy, OSBP1 is phosphorylated by PKD in response to SL flow with a delayed and persistent dynamic when compared with PI4KIIIβ (compare with Appendix Fig S14A) or with general PKD substrates (compare with Fig 4C). While the molecular reason for this difference remains unclear, the fact that OSBP1 phosphorylation is maximal at time points when PtdIns(4)P is consumed suggested that OSBP1 mediates PtdIns(4)P consumption in response to C6‐D‐Cer administration.
Figure 7. OSBP1 mediates SL flow‐induced PtdIns(4)P consumption.
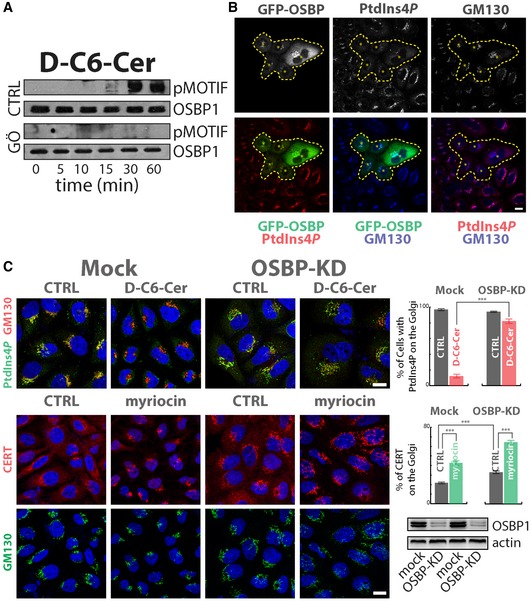
- Cells expressing OSBP1‐GFP were treated for the indicated times with D‐C6‐Cer (10 μM) and lysed, and protein lysates were subjected to immunoprecipitation by the use of anti‐GFP antibody, SDS–PAGE and Western blotting. PKD‐dependent phosphorylation of OSBP was revealed by pMOTIF antibody. The PKD inhibitor Gö 6976 (10 μM, pre‐treatment for 1 h) was used as a specificity control.
- Cells transiently expressing OSBP1‐GFP were fixed and permeabilized as in Fig 3 and stained with anti‐GM130 (blue) and anti‐PtdIns(4)P (red). Asterisks and dashed line indicate OSBP1‐GFP‐transfected cells.
- Cells KD for OSBP1 were treated with D‐C6‐Cer (10 μM) for 1 h, fixed, permeabilized and stained as in Fig 3A (upper left panels). The percentage of cells showing PtdIns(4)P at the Golgi in the different conditions tested is reported (upper right panel). Cells KD for OSBP1 were treated with myriocin (2.5 μM for 24 h), fixed, permeabilized and stained for CERT (red) and GM130 (green). CERT recruitment to the Golgi region was evaluated by immunofluorescence and quantitated as in Fig 1C (middle right panel). OSBP1 KD efficiency was evaluated by Western blotting (lower right panel). ***P < 0.001; according to two‐tailed Student's t‐test.
We thus tested whether OSBP1 has a role in PtdIns(4)P consumption induced by sustained SL flow. First we confirmed that OSBP1 overexpression induces the consumption of PtdIns(4)P at the TGN (Fig 7B) depending on its capability to contact ER membranes (by binding the ER proteins VAPs through its FFAT motif; Fig EV5A; Mesmin et al, 2013). Importantly when OSBP1‐KD cells were subjected to C6‐D‐Cer treatment, PtdIns(4)P was protected from SL flow‐induced consumption (Fig 7C). Moreover, OSBP1‐KD, similar to what observed in SMS1‐KD, induced CERT recruitment to the TGN under basal conditions (Fig 7C).
Figure EV5. OSBP1 mediates PtdIns(4)P relocation to the ER to control SL homeostasis at the TGN .
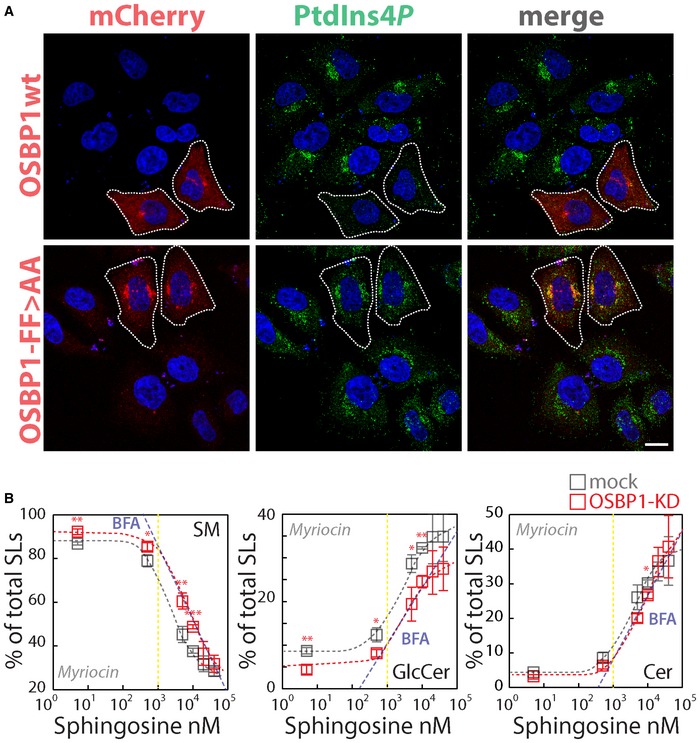
- Cells transiently expressing mCherry‐OSBP1 wt or the mCherry‐OSBP1‐FF>AA mutant were fixed and permeabilized as in Fig 3 and stained with anti‐PtdIns(4)P (green). The dashed line indicates mCherry‐OSBP1‐transfected cells. Scale bar, 10 μm.
- Cells KD for OSBP1 were treated with myriocin (2.5 μM) for 24 h and fed with increasing concentrations of D‐Sph for 2 h. 3H‐D‐Sph (≈5 nM) was mixed as a tracer with its non‐radioactive counterpart. The percentage of total radioactivity associated with SM (left panel), GlcCer (middle panel) and Cer (right panel) in mock (grey) and OSBP1‐KD cells (red) was quantitated after lipid extraction and HPTLC separation. The blue dashed line represents the values obtained under BFA treatment (from Fig 1B). Data are means ± SEM from at least 4 independent experiments. * P < 0.05; ** P < 0.01; *** P < 0.001; according to two‐tailed Student's t‐test.
Taken together, these data are compatible with a model whereby the SL‐induced PKD‐dependent phosphorylation of OSBP1 stimulates the non‐vesicular transport of PtdIns(4)P to the ER for its dephosphorylation operated by Sac1. Provided the role of PtdIns(4)P in Cer transport to the TGN and conversion to SM, a prediction of this model is that removal of OSBP1 should break this negative feedback loop. We tested this prediction and found that in OSBP1‐KD cells SM synthesis is indeed saturated at higher sphingolipid precursor levels when compared to control cells (Fig EV5B).
Along similar lines, the OSBP1/Sac1 system by consuming PtdIns(4)P (and limiting SM production) in response to PKD activation is expected to terminate PKD signalling. According to our model indeed, PKD requires PtdIns(4)P‐dependent SM and DAG production for its full activation so after PtdIns(4)P consumption PKD signalling should be terminated. To test this hypothesis, first cells were treated for 15 minutes with the DAG analogue phorbol 12,13‐dibutyrate (PdBu; 1 μM) which activates PKD (Chen et al, 2008) before or after 30 min of C6‐D‐Cer treatment. As shown in Appendix Fig S16A, PdBu induced PKD activation irrespective of C6‐D‐Cer treatment, suggesting that PKD is still responsive after C6‐D‐Cer treatment. Second, cells were treated with the PI4K inhibitor PIK93 (Toth et al, 2006) before C6‐D‐Cer administration and PKD activation was evaluated by measuring the phosphorylation of its substrates. As shown in Appendix Fig S16B, PIK93 treatment rendered cells unresponsive to C6‐D‐Cer in terms of PKD activation, supporting the hypothesis that PtdIns(4)P is required for SL‐induced PKD activation and that its consumption terminates PKD signalling.
Discussion
The post‐Golgi membranes enrichment in SLs and sterols is a shared and fundamental property of eukaryotic cells and has a key role in shaping both cell response to the environment (by influencing cell signalling) and intracellular organization (by determining intracellular protein distribution) (van Meer et al, 2008; Patterson et al, 2008; Lingwood & Simons, 2010; Holthuis & Menon, 2014). When considering the establishment of the post‐Golgi membrane territory, two traffic fluxes need to be taken into account. The first consists of material traversing the Golgi stack in a cis‐to‐trans direction, mediates the transport of proteins and of the bulk of membrane lipids and requires vesicles (Nakano & Luini, 2010). The second consists of lipids exchanged at ER–TGN MCSs (Holthuis & Menon, 2014; De Matteis & Rega, 2015) where lipid‐transfer proteins execute the non‐vesicular transport of selected lipid species (including SLs and sterols). These two fluxes are orthogonal as they are operated by distinct machineries; still they converge at the TGN and need to be coordinated in order for TGN‐derived carriers to transport the proper protein and lipid cargo to post‐Golgi destinations.
A fundamental factor regulating both non‐vesicular lipid transport and formation of TGN‐derived carriers is PtdIns(4)P (D'Angelo et al, 2008). PtdIns(4)P indeed is the recruitment factor used by the lipid‐transfer proteins CERT, FAPP2 and OSBP1 to deliver SLs and cholesterol to TGN membranes (D'Angelo et al, 2008). PtdIns(4)P also recruits to TGN cytosolic factors able to deform membranes (i.e. FAPP1, arfaptins, AP1/clathrin), thus promoting membrane budding (Wang et al, 2003; Godi et al, 2004; Lenoir et al, 2010; He et al, 2011; Cruz‐Garcia et al, 2013), and mediates the association of nascent carriers with actin cytoskeleton, which provides the tensile force required for their formation (i.e. via GOLPH3 recruitment) (Dippold et al, 2009). As a combination of these activities, PtdIns(4)P controls both the formation of TGN‐derived membrane carriers and their lipid composition.
In addition, lipid remodelling and carriers formation at the TGN are both coordinated by signalling mechanisms that converge on PKDs (Malhotra & Campelo, 2011). PKDs, indeed, are recruited to TGN by ARF1 and DAG (Malhotra & Campelo, 2011) to phosphorylate and activate PI4KIIIβ (Hausser et al, 2005) allowing its interaction with 14‐3‐3γ and CtBP1/BARS in a complex required for carriers fission (Valente et al, 2012; Pagliuso et al, 2016). At the same time, Cer transported to the TGN by CERT is converted to SM in a reaction that generates DAG. TGN‐produced DAG has, in turn, the potential to activate PKDs, thus establishing a positive feedback in SM synthesis and post‐Golgi carriers formation (Villani et al, 2008; Malhotra & Campelo, 2011; Thomaseth et al, 2013). A further PKD substrate is CERT itself, where its phosphorylation reduces its affinity for PtdIns(4)P (Fugmann et al, 2007) in a process that promotes CERT cycling and results in a further positive feedback loop between CERT transfer activity and PKD activity (Weber et al, 2015).
According to this description, the TGN is equipped with a basal lipid remodelling system accounting for the enrichment of post‐Golgi carriers (and thus post‐Golgi compartments) in SLs and cholesterol (Deng et al, 2016). But how does this system react to acute fluctuations in SL production/flow? While, indeed, both reduced and excessive SM syntheses at the TGN have been reported to impair TGN to plasma membrane trafficking (Subathra et al, 2011; Duran et al, 2012), a comprehensive picture on how SL metabolic oscillations are matched to TGN and post‐Golgi SL composition was missing. Here, we describe a molecular circuit linking SL flow to the regulation of PtdIns(4)P turnover at the TGN.
We indeed show that (i) SL flow stimulates the consumption of the TGN‐localized PtdIns(4)P; (ii) as a consequence, SL flow negatively regulates CERT, FAPP2 and OSBP1 recruitment to the Golgi complex (and thus Cer, GlcCer and cholesterol transport to the TGN); (iii) which results in SL flow repressing the production of SM and GSLs at the TGN. Specifically, we show that a sudden increase in SM synthesis at first induces the PKD‐dependent phosphorylation of PI4KIIIβ with a possible transient increase in PtdIns(4)P production. Subsequently, PKD phosphorylates OSBP1 most probably at Ser 240 (Nhek et al, 2010). OSBP1 phosphorylation by PKD is reported to inhibit its localization to the TGN possibly modulating its affinity for the ER and TGN binding factors VAPs and PtdIns(4)P, respectively. Our data suggest that OSBP1 phosphorylation fosters PtdIns(4)P relocation to the ER and Sac1‐dependent PtdIns(4)P dephosphorylation (Fig EV6A). Given the role of PtdIns(4)P in promoting SM and GSL syntheses at the TGN, sustained SM synthesis inhibits SM and GSL production at the TGN (Fig EV6B). Importantly, this circuit appears to be active also under steady‐state conditions as reductions in SL flow or SM synthesis translate in reduced basal PKD activity and recruitment of PtdIns(4)P effectors to the TGN.
Figure EV6. SL flow to the Golgi controls phosphoinositide turnover at the TGN .
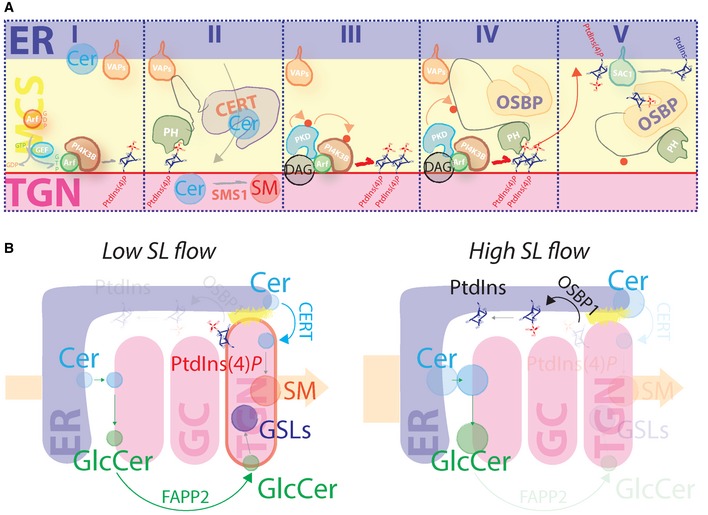
- Schematic representation of SL‐controlled phosphoinositide turnover at the ER–TGN MCSs. Phase I: ARF1 recruits PI4KIIIβ to the TGN to produce PtdIns(4)P. Phase II: CERT transfers Cer from the ER to the TGN by contacting VAPs at the ER and PtdIns(4)P at the TGN. CERT‐transferred Cer is converted to SM by SMS1. Phase III: SMS1‐produced DAG recruits (together with ARF) and locally activates PKD. PKD phosphorylates itself and PI4KIIIβ to increase PtdIns(4)P production. Phase IV: OSBP1 is recruited by PtdIns(4)P. PKD phosphorylates OSBP1. Phase V: phospho‐OSBP1 efficiently transfers PtdIns(4)P to the ER for its dephosphorylation operated by Sac1.
- Schematic representation of TGN response to sustained SL flow. At low SL flow (left scheme), Cer and GlcCer are efficiently transported to the TGN by the PtdIns(4)P binding proteins CERT and FAPP2, respectively, to yield SM and complex GSLs. At high SL flow (right scheme), PtdIns(4)P is transported to the ER by OSBP1 and dephosphorylated to PtdIns. As a consequence, CERT and FAPP2 cannot deliver further SL precursors (Cer and GlcCer) to the TGN, resulting in reduced production of SM and complex GSLs.
Based on these data, we envisage a model where acute fluctuations in SL provision are buffered by a circuit that acts as a homeostatic device to keep SM and GSL synthetic rates constant irrespective of instant changes in the SL flow. In other words, the circuit described here would work in a way similar to a voltage stabilizer in a power supply, which generates a fixed output voltage of a pre‐set magnitude regardless of changes to its input. The intuitive outcome of the operation of this circuit would be that of providing robustness and stability to the TGN lipid biosynthetic system and, as a consequence, that of maintaining constant the SM, GSL and cholesterol composition of TGN and post‐Golgi compartments. Interestingly, when considered in the general frame of cellular lipid homeostatic systems, the circuit described here presents two unique features: (i) it relies on the local sensing of a transient event (i.e. SM synthetic rate at the TGN) and not on the detection of global mass levels of a given metabolite (Holthuis & Menon, 2014); (ii) the architecture of this circuit wires together with the local metabolism of lipid mediators with roles in metabolic and trafficking TGN functions (namely Cer, SM, DAG, PtdIns(4)P, GSLs and cholesterol; De Matteis & Luini, 2008; Holthuis & Menon, 2014). It is thus tempting to speculate that by these properties this circuit preforms a harmonizing function on TGN lipid remodelling during the dynamic event of post‐Golgi carriers formation.
In conclusion, decades of research in membrane biology have shown how membrane composition impacts on membrane properties (van Meer et al, 2008; Lingwood & Simons, 2010). Specifically, SLs and cholesterol at the TGN and post‐Golgi membranes exert an organizing function on other lipids and membrane proteins influencing their lateral distribution, their trafficking and ultimately their specific functions (Holthuis & Menon, 2014). Previous studies have described homeostatic circuits devoted to maintain the global SL content in eukaryotic cells by regulating the reactions initiating SL production in the ER (Vacaru et al, 2009; Breslow et al, 2010; Cantalupo et al, 2015). In this study, we provide evidence for the existence (and dissect the architecture) of a homeostatic circuit accounting for the localized and acute control of SL metabolism at the TGN and thus of post‐Golgi membrane composition.
Materials and Methods
Reagents
N‐C6:0‐D‐erythro‐sphingosine (D‐C6‐ceramide), N‐hexanoyl‐L‐erythro‐sphingosine (L‐C6‐ceramide) and D‐erythro‐2‐N‐[6′‐(1″‐pyridinium)hexanoyl]‐sphingosine bromide (LCL29) were from Matreya, USA. D‐erythro‐Sphingosine (D‐Sph) was from Avanti Polar, USA. Phorbol 12,13‐dibutyrate (PdBU) and GÖ 6976 were from Sigma‐Aldrich. PIK93 inhibitor was from Echelon Bioscience, USA. Fumonisin B1 (FB1) and myriocin were from Enzo Life Sciences, USA. Digitonin was from Calbiochem, USA. FL‐BODIPY‐C5‐Cer complexed to BSA, and human transferrin conjugated to Fluor 568 were from Thermo Fisher Scientific, USA. All other unmentioned reagents were from Sigma‐Aldrich, USA. Antibodies used in this study are listed in Appendix Table S1.
Cell lines and culture conditions
HeLa cells were obtained from the American Tissue Type Collection (ATTC, USA). HeLa cells were grown in RPMI‐1640 medium (Gibco, USA) supplemented with 10% (v/v) foetal calf serum (FCS) (Gibco), containing 4.5 g/l glucose, 2 mM l‐glutamine, 100 U/ml penicillin and streptomycin. Cells were grown under controlled atmosphere (5% CO2 and 95% air) at 37°C.
Construct and plasmids
To generate the human Sac1 (NM_014016)‐, SMS1 (NM_147156)‐ and GCS (NM_003358)‐expressing plasmids, total mRNA was purified, from HeLa cells, and retrotranscribed and cDNAs were amplified by PCR using the following primers:
- Sac1
- Forward: 5′‐ATAGGATTCATTGAGAGAGAAGGAAGGAGGTG‐3′
- Reverse: 5′‐ATAGATATCATTAAAAGTATGCCTGCTAATAGTG‐3′
- SMS1
- Forward: 5′‐GCGCGCGAATTCATGAAGGAAGTGGTTTATTGGTC‐3′
- Reverse: 5′‐TGTGTCATTCACCAGCCGGCT‐3′
- GCS
- Forward: 5′‐GCGCGCGAATTCATGGCGCTGCTGGACCTGG‐3′
- Reverse: 5′‐TACATCTAGGATTTCCTCTGC‐3′
PCRs were performed as follows: 1 min at 98°C, followed by 30 cycles at 98°C for 20 s, 60°C for 30 s, 72°C for 2 min and a final extension at 72°C for 7 min. The PCR‐generated fragment for Sac1 was digested with BamHI and EcoRV restriction enzymes (New England Biolabs, USA) to be cloned into pcDNA4b‐3xHA‐expression vector (Clontech, USA). The PCR‐generated fragment for SMS1 and GCS was digested with EcoRI and XhoI (New England Biolabs, USA) and cloned into pcDNA4b‐3xHA‐expression vector (Clontech, USA). All the obtained plasmids were sequenced before use.
PKD wild type, PKD‐C1α and PKD‐KinDead were provided by Vivek Malhotra (Centre Genomic Regulation‐CRG, Barcelona); GOLPH3‐GFP was provided by Alberto Luini (Institute of Protein Biochemistry, National Research Council, Naples, Italy).
FAPP2‐PH‐GFP and diFAPP‐PH‐R18L‐GFP were from Antonella De Matteis laboratory (Telethon Institute for Genetics and Medicine); ORP5‐PH‐GFP and ORP8L‐PH‐GFP were from Pietro De Camilli laboratory (Yale Medical School); PLCδ‐PH‐GFP was from Roman Polishchuk laboratory (Telethon Institute for Genetics and Medicine).
PI4KIIIβ‐GFP‐, FYVE‐GFP‐, CERT‐GFP‐ and OSBP‐GFP‐expressing plasmids were obtained from Daniela Corda laboratory (Institute of Protein Biochemistry, National Research Council, Naples, Italy); PI4KIIIβ‐S294A‐GFP, Flag‐CERT‐wt and Flag‐CERT‐S132A were obtained from Angelika Hausser laboratory (University of Stuttgart, Institute of Cell Biology and Immunology, Germany). The diP4M‐SidM‐mCherry construct was provided by Tamas Balla (Section on Molecular Signal Transduction within the Program for Developmental Neuroscience, Bethesda). mCherry‐C1‐OSBP‐FF>AA mutant was obtained from Bruno Mesmin and Bruno Antonny laboratory (Institut de Pharmacologie Moléculaire et Cellulaire, Université Nice Sophia Antipolis and CNRS, Valbonne, France). YFP‐DAG binding domain construct was from Chiara Luberto laboratory (Department of Physiology and Biophysics, Stony Brook University, Stony Brook, NY).
siRNA treatments and transfections
The siRNAs for human SMS1 (NM_147156.3), CERT (NM_001130105.1), GCS (NM_003358), PKD1 (NM_002742.2), PKD2 (NM_001079880.1), PKD3 (NM_005813.4), OSBP1 (NM_002556.2), PI4KIIα (NM_018425), PI4KIIIα (NM_058004), PI4KIIβ (NM_018323), PI4KIIIβ (NM_001198773) and Sac1 (NM_014016) were obtained from Sigma‐Aldrich (Italy; Appendix Table S2). HeLa cells were plated at 20% confluence in 24‐well plates on 24‐mm coverslips, or in 12‐well plates, and transfected with 120 pmol of siRNAs with oligofectamine (Invitrogen, USA), according to the manufacturer's protocol. 72 h after the initial treatment with the siRNAs, the cells were processed for the different experiments. The silencing efficiencies were evaluated either by Western blotting or by qPCR using specific primers (listed below).
All the constructs mentioned in this study were transfected into HeLa cells using TransIT‐LT1 Transfection Reagent (Mirus Bio LLC, USA), according to the manufacturer's instructions.
RNA extraction and real‐time PCR
Total RNA was isolated from HeLa cells using RNeasy Mini kits (Qiagen, Germany) according to the manufacturer's instructions. The yield and the integrity of the RNA were determined using a spectrophotometer (NanoDrop 2000c; Thermo Scientific, USA), by TAE agarose gel electrophoresis and with an Agilent 2100 Bioanalyser (Agilent Technologies, USA). RNA (1 μg) was reverse‐transcribed using QuantiTect Reverse Transcription kits (Qiagen, Germany) according to the manufacturer's instructions and subjected to real‐time qPCR with gene‐specific primers in the presence of LightCycler® 480 SYBR Green I Master Mix (Roche, Switzerland) on a LightCycler® 480 II detection system (Roche, Switzerland). Relative abundance of mRNA was calculated by normalization to hypoxanthine phosphoribosyltransferase‐1 (HPRT1). Real‐time PCR primers used in this study are listed in Appendix Table S3.
Immunoblotting
Proteins were detected using the mentioned primary antibodies. The signals were visualized using a goat anti‐rabbit or goat anti‐mouse IgG‐HRP secondary antibody (Santa Cruz, USA) at a 1:10,000 dilution.
Immunoprecipitation assays
HeLa cells transfected with the indicated GFP‐tagged plasmids were lysed in a buffer consisting of 20 mM MOPS pH 7.0, 2 mM EGTA, 5 mM EDTA, 60 mM β‐glycerophosphate, 30 mM NaF, 1 mM Na3VO4, 1 mM DTT, 1% (v/v) Triton X‐100 and protease inhibitor. Total lysates (1 mg) were mixed with 1 μg of anti‐GFP IgGs overnight and incubated for 1 h with protein A–Sepharose (Sigma). After centrifugation and washing steps, proteins associated with the precipitated fraction were eluted by adding sample buffer (0.125 M Trizma base, 4% SDS, 20% glycerol, 10% β‐mercaptoethanol, pH 6.8), and the eluted fraction was analysed by SDS–PAGE and Western blotting.
Sterol measurements
HPTLC
HeLa cells pre‐treated overnight (18 h) with vehicle only or myriocin (2.5 μM) were pulse‐labelled in serum‐free RPMI‐1640 with 20 μCi/ml 3H‐acetate for 2 h in the presence of vehicle only, myriocin or myriocin + 10 μM D‐C6‐Cer. After labelling, the cells were further incubated in RPMI‐1640 + 10% FCS in the presence or absence of myriocin and D‐C6‐Cer for 4 h. Cells were then harvested and processed for lipid extractions. Lipids were spotted on silica gel high‐performance–TLC (HPTLC) plates (Merck, Germany) and resolved with a mixture of petroleum ether, diethyl ether and acetic acid (60:40:1 v/v/v), and unlabelled and radiolabeled lipids were analysed as described before (Holtta‐Vuori et al, 2012).
Gas chromatography–mass spectrometry
Lipid extracts were analysed by GC‐MS as described before (Loizides‐Mangold et al, 2012). Briefly, samples were injected into a VARIAN CP‐3800 gas chromatograph equipped with a FactorFour Capillary Column VF‐5ms 15 m × 0.32 mm i.d. DF = 0.10 and analysed by a Varian 320 MS triple quadrupole with electron energy set to – 70 eV at 250°C. Samples were applied to the column oven at 45°C, held for 4 min, then raised to 195°C (20°C/min). Sterols were eluted with a linear gradient from 195 to 230°C (4°C/min), followed by rising to 320°C (10°C/min). Finally, the column temperature was raised to 350°C (6°C/min) to elute sterol esters. Cholesterol and cholesterol esters were identified by their retention times (compared with standards) and fragmentation patterns, which were compared with the NIST library.
Sphingolipid measurements
HPTLC
HeLa cells pre‐treated with myriocin (2.5 μM) or FB1 (50 μM) were pulse‐labelled in RPMI‐1640 10% FCS, with 0.1 μCi/ml (≈5 nM) 3H‐D‐erythro‐sphingosine or with [3H]‐D‐erythro‐C6‐ceramide mixed with increasing amounts of non‐radioactive D‐Sph and C6‐D‐Cer for 2 h, respectively. After 2 h, cells were harvested and processed for lipid extractions. Lipids were spotted on silica gel high‐performance–TLC (HPTLC) plates (Merck, Germany) and resolved with a mixture of chloroform, methanol and water (65:25:4 v/v/v). To visualize the unlabelled standards (i.e. Cer, GlcCer and SM), the TLC plates were placed in a sealed tank saturated with iodine vapours, while the radiolabelled lipids were analysed using a RITA® TLC Analyser (Raytest, Germany) and quantified using GINA® (Raytest, Germany) software analysis. The percentage of total C.P.M. associated with Cer, GlcCer and SM peaks for each of the three lipids is reported. For activity measurements, cells labelled as described above were harvested; 1/5th of total cell fraction was used for protein content determination, while the rest was used for lipid extraction and analysis. For each analysed sample, pmol of SM produced per μg of protein was calculated according to the following formula:
where C.P.M.SM represents the counts per minute associated with the SM peak; C.P.M.0.1μCi SL represents the counts per minute associated with 0.1 μCi of 3H‐labelled SL precursor; SL [pmol] represents the amount of SL precursors fed to cells (pmoles); and protein lysate [μg] represents the amount of proteins obtained by lysing the sample (μg).
HPLC–mass spectrometry
Sphingolipids were analysed by liquid chromatography–tandem mass spectrometry (LC–MS/MS) as described earlier (Bielawski et al, 2006). Briefly, extracts were analysed with a Quantum Ultra triple quadrupole mass spectrometer connected to an Accela HPLC and Accela autosampler using a solvent gradient. Ceramides identity was achieved through MRM analysis with soft fragmentation. Quantitative analysis is based on calibration curves generated for each lipid. Levels of SLs were normalized to inorganic phosphate (Pi) released from total phospholipids.
Immunofluorescence, staining and image analysis
HeLa cells grown on 24‐mm coverslips and treated according to the experimental procedure were fixed with 4% paraformaldehyde for 15 min and washed once in PBS. Blocking buffer (1× PBS, 0.5% BSA, 0.05% saponin, 50 mM NH4Cl, 0.02% NaN3) was added to the cells for 20 min, followed by 1‐h incubation with the primary antibody. Subsequently, coverslips were washed with PBS and incubated with secondary antibodies (1:400) diluted in blocking solution for 45 min, washed again and mounted on glass microscopic slides (Carlo Erba, Italy) with Mowiol.
The PtdIns(4)P staining was performed as described in Hammond et al (2009). HeLa cells were grown on 24‐mm coverslips. At 80% confluence, the cells were quenched with PBS containing 50 mM NH4Cl and fixed in 2% paraformaldehyde for 15 min. The cells were permeabilized for 5 min with 20 μM digitonin in buffer A containing 20 mM PIPES pH 6.68, 135 mM NaCl and 27 mM KCl. HeLa cells were blocked in buffer A supplemented with 5% (v/v) FBS for 45 min. Anti‐PtdIns(4)P monoclonal mouse antibody dissolved in buffer A with 5% FBS was used for incubation (1:200) for 1 h. After washing with buffer A, anti‐mouse IgM secondary antibodies dissolved in buffer A with 5% FBS (1:400) were used for incubation for 45 min. Cells were, then, post‐fixed for 5 min in 2% paraformaldehyde, washed three times with PBS containing 50 mM NH4Cl, washed once with water and mounted.
Immunofluorescence samples were examined under confocal laser microscope (Zeiss LSM 700 confocal microscope systems; Carl Zeiss, Gottingen, Germany). Optical confocal sections were taken at 1 Airy unit. Images were analysed using either ImageJ (Schneider et al, 2012) or CellProfiler (Carpenter et al, 2006) software.
Live cell imaging and FRAP assays
HeLa cells plated on 35‐mm glass bottom microwell dishes (MatTech, USA) were transfected with the indicated plasmids (1 μg) using TransIT‐LT1 Transfection Reagent (Mirus Bio LLC, USA) according to the manufacturer's instruction and analysed 24 h after transfection. For temperature control during live observation, the microscope was equipped with a temperature‐controlled chamber, and the cells were imaged on a 37°C stage in DMEM‐HEPES‐buffered pH 7.4 media using a Zeiss LSM 700 confocal microscope. Image series were exported as single lsm files and processed with ImageJ for analysis, single frame extraction and conversion into QuickTime movies.
Fluorescence recovery after photobleaching (FRAP) experiments were performed by bleaching the GFP‐ARF1‐associated fluorescence in the Golgi area (50 bleaching iterations) and then monitoring the fluorescence intensity recovery in the bleached area. For both live imaging and FRAP experiments, normalized Golgi‐associated fluorescence was calculated according to the formula:
where nGolgif(t) represents the normalized Golgi‐associated fluorescence; Golgif(t) and Golgif(0) represent the Golgi‐associated fluorescence at time (t) and 0, respectively; and Cellf(t) and Cellf(0) represent the whole cell‐associated fluorescence at time (t) and 0, respectively.
Transmission electron microscopy
For conventional electron microscopy, cells grown in 35‐mm plastic dishes were fixed with 1% glutaraldehyde (8% aqueous solution – Electron Microscopy Sciences) in HEPES 0.2 M pH 7.3 at 4°C for overnight. The fixative was replaced with 1% BSA in PBS and cells were carefully detached using a plastic cell scraper, collected into Eppendorf tubes and centrifuged to obtain the pellet. All samples were then washed three times in HEPES 0.2 M pH 7.3 and post‐fixed for 30 min in 1% OsO4 at 4°C in the same buffer. They were then washed three times in distilled water and post‐fixed for 25 min in 1% OsO4 and 1.5% potassium ferrocyanide at room temperature in HEPES 0.2 M pH 7.3. They were then washed three times in distilled water and stained with 0.5% uranyl acetate overnight at 4°C. The pellets were dehydrated in graded steps of ethanol (50, 70, 90, 100%), two times with 100% of acetone and embedded into Epon. Sections (60‐nm thick) were cut on a Leica UC7 ultramicrotome and examined with a Fei Tecnai 12 BioTwin Spirit transmission electron microscope.
EM tomography
The sample preparation and tomographic reconstruction were done essentially as described previously (Trucco et al, 2004). In brief, HeLa cells were fixed with 2% glutaraldehyde for 2 h at room temperature, and the samples were then processed, stained, dehydrated and Epon‐embedded. The samples were sectioned (200‐nm sections), and gold particles were placed on the surface of the plastic sections and examined by an electron microscope (Tecnai‐12; FEI). The Golgi profiles with well‐preserved structures were identified, and the images were acquired at tilt angles of +65° to −65° at 1° intervals, with a magnification of 26,500 using a Veletta CCD digital camera. The tomographic reconstruction was done using the Inspect 3D (FEI).
Statistics
Error bars correspond to either standard deviation (SD) or standard error of the mean (SEM) according to the different experiments and as indicated in the figure legends. Statistical evaluations were made by Student's t‐test ∗ P < 0.05, ∗∗ P < 0.01, and ∗∗∗ P < 0.001 (ns, not significant).
Author contributions
GD'A supervised the entire project, with advice from AL and SP; GD'A and SC wrote the manuscript with comments from all co‐authors; SC, with the help of LS, RR, MP and DR, designed and conducted all the experiments described. NAD designed the strategy and produced plasmid vectors for SMS1 and GCS. SC designed the strategy and produced plasmid vectors for Sac1. FC and JvG performed experiments described in Appendix Figs S4, S5 and S16A under the supervision of VM. AH provided the PI4KIIIβ‐S294A and CERT‐S132A plasmids used in Appendix Figs S12 and S14B. Electron microscopy and electron tomography experiments were performed by RR, MP and GT. EI, MH‐V, IR and HR performed cholesterol analysis; CL performed HPLC/MS sphingolipid analyses.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Movie EV1
Movie EV2
Movie EV3
Movie EV4
Movie EV5
Movie EV6
Movie EV7
Movie EV8
Movie EV9
Movie EV10
Movie EV11
Review Process File
Acknowledgements
We thank MA. De Matteis for initial discussions; D. Corda and A. Colanzi for critical reading of the manuscript; T. Balla for the diP4M‐mCherry probe; P. De Camilli for the ORP5‐PH‐ and ORP8L‐PH‐GFP constructs; R. Polishchuk for the PH‐PLCδ GFP construct; and B. Antonny and B. Mesmin for the OSBP1‐FF>AA construct. We thank the IBP‐bioimaging facility and the Stony Brook University Lipidomics Core Facility for technical support. GD'A acknowledges the financial support of AIRC (MFAG 10585), of the Italian Ministry of Health (GR‐2011‐02352256) and of MIUR (PON_00862). RR acknowledges the financial support of FIRC (Fellowship No. 15111). AL acknowledges FFC [Italian Cystic Fibrosis Research Foundation FFC #2 2014], MIUR [COSM Progetto Interomics], AIRC [Italian Association for Cancer Research, IG 15767], the MIUR Project “FaReBio di Qualità”, the PON Projects No. 01/00117 and 01‐00862, PNR‐CNR Aging Program 2012–2014, and Progetto Bandiera “Epigen”. CL acknowledges the support of NIH Grant # 5P01CA097132‐12‐NCI. EI acknowledges the support of the Academy of Finland (Grants 282192, 284667, 307415). HR acknowledges financial support from Swiss National Science Foundation and NCCR Chemical Biology. VM acknowledges support of the Ministerio de Economía y Competitividad, “Centro de Excelencia Severo Ochoa 2013‐2017”, SEV‐2012‐0208. VM is an Institució Catalana de Recerca i Estudis Avançats professor at the Center for Genomic Regulation, and the work in his laboratory is funded by grants from Ministerio de Economía y Competitividad's Plan Nacional (ref. BFU2013‐44188‐P).
The EMBO Journal (2017) 36: 1736–1754
References
- Bajaj Pahuja K, Wang J, Blagoveshchenskaya A, Lim L, Madhusudhan MS, Mayinger P, Schekman R (2015) Phosphoregulatory protein 14‐3‐3 facilitates SAC1 transport from the endoplasmic reticulum. Proc Natl Acad Sci USA 112: E3199–E3206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baron CL, Malhotra V (2002) Role of diacylglycerol in PKD recruitment to the TGN and protein transport to the plasma membrane. Science 295: 325–328 [DOI] [PubMed] [Google Scholar]
- Bielawski J, Szulc ZM, Hannun YA, Bielawska A (2006) Simultaneous quantitative analysis of bioactive sphingolipids by high‐performance liquid chromatography‐tandem mass spectrometry. Methods 39: 82–91 [DOI] [PubMed] [Google Scholar]
- Blagoveshchenskaya A, Cheong FY, Rohde HM, Glover G, Knodler A, Nicolson T, Boehmelt G, Mayinger P (2008) Integration of Golgi trafficking and growth factor signaling by the lipid phosphatase SAC1. J Cell Biol 180: 803–812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breslow DK, Collins SR, Bodenmiller B, Aebersold R, Simons K, Shevchenko A, Ejsing CS, Weissman JS (2010) Orm family proteins mediate sphingolipid homeostasis. Nature 463: 1048–1053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breslow DK, Weissman JS (2010) Membranes in balance: mechanisms of sphingolipid homeostasis. Mol Cell 40: 267–279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burd CG, Emr SD (1998) Phosphatidylinositol(3)‐phosphate signaling mediated by specific binding to RING FYVE domains. Mol Cell 2: 157–162 [DOI] [PubMed] [Google Scholar]
- Cantalupo A, Zhang Y, Kothiya M, Galvani S, Obinata H, Bucci M, Giordano FJ, Jiang XC, Hla T, Di Lorenzo A (2015) Nogo‐B regulates endothelial sphingolipid homeostasis to control vascular function and blood pressure. Nat Med 21: 1028–1037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpenter AE, Jones TR, Lamprecht MR, Clarke C, Kang IH, Friman O, Guertin DA, Chang JH, Lindquist RA, Moffat J, Golland P, Sabatini DM (2006) Cell Profiler: image analysis software for identifying and quantifying cell phenotypes. Genome Biol 7: R100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Deng F, Li J, Wang QJ (2008) Selective binding of phorbol esters and diacylglycerol by individual C1 domains of the PKD family. Biochem J 411: 333–342 [DOI] [PubMed] [Google Scholar]
- Chung J, Torta F, Masai K, Lucast L, Czapla H, Tanner LB, Narayanaswamy P, Wenk MR, Nakatsu F, De Camilli P (2015) INTRACELLULAR TRANSPORT. PI4P/phosphatidylserine countertransport at ORP5‐ and ORP8‐mediated ER‐plasma membrane contacts. Science 349: 428–432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruz‐Garcia D, Ortega‐Bellido M, Scarpa M, Villeneuve J, Jovic M, Porzner M, Balla T, Seufferlein T, Malhotra V (2013) Recruitment of arfaptins to the trans‐Golgi network by PI(4)P and their involvement in cargo export. EMBO J 32: 1717–1729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Angelo G, Polishchuk E, Di Tullio G, Santoro M, Di Campli A, Godi A, West G, Bielawski J, Chuang CC, van der Spoel AC, Platt FM, Hannun YA, Polishchuk R, Mattjus P, De Matteis MA (2007) Glycosphingolipid synthesis requires FAPP2 transfer of glucosylceramide. Nature 449: 62–67 [DOI] [PubMed] [Google Scholar]
- D'Angelo G, Vicinanza M, De Matteis MA (2008) Lipid‐transfer proteins in biosynthetic pathways. Curr Opin Cell Biol 20: 360–370 [DOI] [PubMed] [Google Scholar]
- D'Angelo G, Vicinanza M, Wilson C, De Matteis MA (2012) Phosphoinositides in Golgi complex function. Subcell Biochem 59: 255–270 [DOI] [PubMed] [Google Scholar]
- D'Angelo G, Uemura T, Chuang CC, Polishchuk E, Santoro M, Ohvo‐Rekila H, Sato T, Di Tullio G, Varriale A, D'Auria S, Daniele T, Capuani F, Johannes L, Mattjus P, Monti M, Pucci P, Williams RL, Burke JE, Platt FM, Harada A et al (2013) Vesicular and non‐vesicular transport feed distinct glycosylation pathways in the Golgi. Nature 501: 116–120 [DOI] [PubMed] [Google Scholar]
- De Matteis MA, Luini A (2008) Exiting the Golgi complex. Nat Rev Mol Cell Biol 9: 273–284 [DOI] [PubMed] [Google Scholar]
- De Matteis MA, Rega LR (2015) Endoplasmic reticulum‐Golgi complex membrane contact sites. Curr Opin Cell Biol 35: 43–50 [DOI] [PubMed] [Google Scholar]
- De Matteis MA, Wilson C, D'Angelo G (2013) Phosphatidylinositol‐4‐phosphate: the Golgi and beyond. BioEssays 35: 612–622 [DOI] [PubMed] [Google Scholar]
- Deng Y, Rivera‐Molina FE, Toomre DK, Burd CG (2016) Sphingomyelin is sorted at the trans Golgi network into a distinct class of secretory vesicle. Proc Natl Acad Sci USA 113: 6677–6682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dippold HC, Ng MM, Farber‐Katz SE, Lee SK, Kerr ML, Peterman MC, Sim R, Wiharto PA, Galbraith KA, Madhavarapu S, Fuchs GJ, Meerloo T, Farquhar MG, Zhou H, Field SJ (2009) GOLPH3 bridges phosphatidylinositol‐4‐ phosphate and actomyosin to stretch and shape the Golgi to promote budding. Cell 139: 337–351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doppler H, Storz P, Li J, Comb MJ, Toker A (2005) A phosphorylation state‐specific antibody recognizes Hsp27, a novel substrate of protein kinase D. J Biol Chem 280: 15013–15019 [DOI] [PubMed] [Google Scholar]
- Dowler S, Currie RA, Campbell DG, Deak M, Kular G, Downes CP, Alessi DR (2000) Identification of pleckstrin‐homology‐domain‐containing proteins with novel phosphoinositide‐binding specificities. Biochem J 351: 19–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duran JM, Campelo F, van Galen J, Sachsenheimer T, Sot J, Egorov MV, Rentero C, Enrich C, Polishchuk RS, Goni FM, Brugger B, Wieland F, Malhotra V (2012) Sphingomyelin organization is required for vesicle biogenesis at the Golgi complex. EMBO J 31: 4535–4546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fugmann T, Hausser A, Schoffler P, Schmid S, Pfizenmaier K, Olayioye MA (2007) Regulation of secretory transport by protein kinase D‐mediated phosphorylation of the ceramide transfer protein. J Cell Biol 178: 15–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Galen J, Campelo F, Martinez‐Alonso E, Scarpa M, Martinez‐Menarguez JA, Malhotra V (2014) Sphingomyelin homeostasis is required to form functional enzymatic domains at the trans‐Golgi network. J Cell Biol 206: 609–618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godi A, Di Campli A, Konstantakopoulos A, Di Tullio G, Alessi DR, Kular GS, Daniele T, Marra P, Lucocq JM, De Matteis MA (2004) FAPPs control Golgi‐to‐cell‐surface membrane traffic by binding to ARF and PtdIns(4)P . Nat Cell Biol 6: 393–404 [DOI] [PubMed] [Google Scholar]
- Hammond GR, Schiavo G, Irvine RF (2009) Immunocytochemical techniques reveal multiple, distinct cellular pools of PtdIns4P and PtdIns(4,5)P(2). Biochem J 422: 23–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond GR, Machner MP, Balla T (2014) A novel probe for phosphatidylinositol 4‐phosphate reveals multiple pools beyond the Golgi. J Cell Biol 205: 113–126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanada K, Kumagai K, Yasuda S, Miura Y, Kawano M, Fukasawa M, Nishijima M (2003) Molecular machinery for non‐vesicular trafficking of ceramide. Nature 426: 803–809 [DOI] [PubMed] [Google Scholar]
- Hannun YA, Obeid LM (2008) Principles of bioactive lipid signalling: lessons from sphingolipids. Nat Rev Mol Cell Biol 9: 139–150 [DOI] [PubMed] [Google Scholar]
- Hausser A, Storz P, Martens S, Link G, Toker A, Pfizenmaier K (2005) Protein kinase D regulates vesicular transport by phosphorylating and activating phosphatidylinositol‐4 kinase IIIbeta at the Golgi complex. Nat Cell Biol 7: 880–886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He J, Scott JL, Heroux A, Roy S, Lenoir M, Overduin M, Stahelin RV, Kutateladze TG (2011) Molecular basis of phosphatidylinositol 4‐phosphate and ARF1 GTPase recognition by the FAPP1 pleckstrin homology (PH) domain. J Biol Chem 286: 18650–18657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holthuis JC, Menon AK (2014) Lipid landscapes and pipelines in membrane homeostasis. Nature 510: 48–57 [DOI] [PubMed] [Google Scholar]
- Holtta‐Vuori M, Vainio S, Kauppi M, Van Eck M, Jokitalo E, Ikonen E (2012) Endosomal actin remodeling by coronin‐1A controls lipoprotein uptake and degradation in macrophages. Circ Res 110: 450–455 [DOI] [PubMed] [Google Scholar]
- Horvath A, Sutterlin C, Manning‐Krieg U, Movva NR, Riezman H (1994) Ceramide synthesis enhances transport of GPI‐anchored proteins to the Golgi apparatus in yeast. EMBO J 13: 3687–3695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou Q, Jin J, Zhou H, Novgorodov SA, Bielawska A, Szulc ZM, Hannun YA, Obeid LM, Hsu YT (2011) Mitochondrially targeted ceramides preferentially promote autophagy, retard cell growth, and induce apoptosis. J Lipid Res 52: 278–288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huitema K, van den Dikkenberg J, Brouwers JF, Holthuis JC (2004) Identification of a family of animal sphingomyelin synthases. EMBO J 23: 33–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichikawa S, Ozawa K, Hirabayashi Y (1998) Molecular cloning and characterization of the mouse ceramide glucosyltransferase gene. Biochem Biophys Res Commun 253: 707–711 [DOI] [PubMed] [Google Scholar]
- Ikonen E (2008) Cellular cholesterol trafficking and compartmentalization. Nat Rev Mol Cell Biol 9: 125–138 [DOI] [PubMed] [Google Scholar]
- Klemm RW, Ejsing CS, Surma MA, Kaiser HJ, Gerl MJ, Sampaio JL, de Robillard Q, Ferguson C, Proszynski TJ, Shevchenko A, Simons K (2009) Segregation of sphingolipids and sterols during formation of secretory vesicles at the trans‐Golgi network. J Cell Biol 185: 601–612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumagai K, Kawano M, Shinkai‐Ouchi F, Nishijima M, Hanada K (2007) Interorganelle trafficking of ceramide is regulated by phosphorylation‐dependent cooperativity between the PH and START domains of CERT. J Biol Chem 282: 17758–17766 [DOI] [PubMed] [Google Scholar]
- Lemmon MA, Ferguson KM, O'Brien R, Sigler PB, Schlessinger J (1995) Specific and high‐affinity binding of inositol phosphates to an isolated pleckstrin homology domain. Proc Natl Acad Sci USA 92: 10472–10476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenoir M, Coskun U, Grzybek M, Cao X, Buschhorn SB, James J, Simons K, Overduin M (2010) Structural basis of wedging the Golgi membrane by FAPP pleckstrin homology domains. EMBO Rep 11: 279–284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liljedahl M, Maeda Y, Colanzi A, Ayala I, Van Lint J, Malhotra V (2001) Protein kinase D regulates the fission of cell surface destined transport carriers from the trans‐Golgi network. Cell 104: 409–420 [DOI] [PubMed] [Google Scholar]
- Lingwood D, Simons K (2010) Lipid rafts as a membrane‐organizing principle. Science 327: 46–50 [DOI] [PubMed] [Google Scholar]
- Lippincott‐Schwartz J, Yuan LC, Bonifacino JS, Klausner RD (1989) Rapid redistribution of Golgi proteins into the ER in cells treated with brefeldin A: evidence for membrane cycling from Golgi to ER. Cell 56: 801–813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loizides‐Mangold U, David FP, Nesatyy VJ, Kinoshita T, Riezman H (2012) Glycosylphosphatidylinositol anchors regulate glycosphingolipid levels. J Lipid Res 53: 1522–1534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malhotra V, Campelo F (2011) PKD regulates membrane fission to generate TGN to cell surface transport carriers. Cold Spring Harb Perspectives Biol 3: a005280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Meer G, Voelker DR, Feigenson GW (2008) Membrane lipids: where they are and how they behave. Nat Rev Mol Cell Biol 9: 112–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesmin B, Bigay J, Moser von Filseck J, Lacas‐Gervais S, Drin G, Antonny B (2013) A four‐step cycle driven by PI(4)P hydrolysis directs sterol/PI(4)P exchange by the ER‐Golgi tether OSBP. Cell 155: 830–843 [DOI] [PubMed] [Google Scholar]
- Morad SA, Cabot MC (2013) Ceramide‐orchestrated signalling in cancer cells. Nat Rev Cancer 13: 51–65 [DOI] [PubMed] [Google Scholar]
- Nakano A, Luini A (2010) Passage through the Golgi. Curr Opin Cell Biol 22: 471–478 [DOI] [PubMed] [Google Scholar]
- Nhek S, Ngo M, Yang X, Ng MM, Field SJ, Asara JM, Ridgway ND, Toker A (2010) Regulation of oxysterol‐binding protein Golgi localization through protein kinase D‐mediated phosphorylation. Mol Biol Cell 21: 2327–2337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogretmen B, Pettus BJ, Rossi MJ, Wood R, Usta J, Szulc Z, Bielawska A, Obeid LM, Hannun YA (2002) Biochemical mechanisms of the generation of endogenous long chain ceramide in response to exogenous short chain ceramide in the A549 human lung adenocarcinoma cell line. Role for endogenous ceramide in mediating the action of exogenous ceramide. J Biol Chem 277: 12960–12969 [DOI] [PubMed] [Google Scholar]
- Pagliuso A, Valente C, Giordano LL, Filograna A, Li G, Circolo D, Turacchio G, Marzullo VM, Mandrich L, Zhukovsky MA, Formiggini F, Polishchuk RS, Corda D, Luini A (2016) Golgi membrane fission requires the CtBP1‐S/BARS‐induced activation of lysophosphatidic acid acyltransferase delta. Nat Commun 7: 12148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson GH, Hirschberg K, Polishchuk RS, Gerlich D, Phair RD, Lippincott‐Schwartz J (2008) Transport through the Golgi apparatus by rapid partitioning within a two‐phase membrane system. Cell 133: 1055–1067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rykx A, De Kimpe L, Mikhalap S, Vantus T, Seufferlein T, Vandenheede JR, Van Lint J (2003) Protein kinase D: a family affair. FEBS Lett 546: 81–86 [DOI] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, Eliceiri KW (2012) NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9: 671–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharpe HJ, Stevens TJ, Munro S (2010) A comprehensive comparison of transmembrane domains reveals organelle‐specific properties. Cell 142: 158–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siow DL, Wattenberg BW (2012) Mammalian ORMDL proteins mediate the feedback response in ceramide biosynthesis. J Biol Chem 287: 40198–40204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stauffer TP, Ahn S, Meyer T (1998) Receptor‐induced transient reduction in plasma membrane PtdIns(4,5)P2 concentration monitored in living cells. Curr Biol 8: 343–346 [DOI] [PubMed] [Google Scholar]
- Subathra M, Qureshi A, Luberto C (2011) Sphingomyelin synthases regulate protein trafficking and secretion. PLoS One 6: e23644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomaseth C, Weber P, Hamm T, Kashima K, Radde N (2013) Modeling sphingomyelin synthase 1 driven reaction at the Golgi apparatus can explain data by inclusion of a positive feedback mechanism. J Theor Biol 337: 174–180 [DOI] [PubMed] [Google Scholar]
- Toth B, Balla A, Ma H, Knight ZA, Shokat KM, Balla T (2006) Phosphatidylinositol 4‐kinase IIIbeta regulates the transport of ceramide between the endoplasmic reticulum and Golgi. J Biol Chem 281: 36369–36377 [DOI] [PubMed] [Google Scholar]
- Trucco A, Polishchuk RS, Martella O, Di Pentima A, Fusella A, Di Giandomenico D, San Pietro E, Beznoussenko GV, Polishchuk EV, Baldassarre M, Buccione R, Geerts WJ, Koster AJ, Burger KN, Mironov AA, Luini A (2004) Secretory traffic triggers the formation of tubular continuities across Golgi sub‐compartments. Nat Cell Biol 6: 1071–1081 [DOI] [PubMed] [Google Scholar]
- Vacaru AM, Tafesse FG, Ternes P, Kondylis V, Hermansson M, Brouwers JF, Somerharju P, Rabouille C, Holthuis JC (2009) Sphingomyelin synthase‐related protein SMSr controls ceramide homeostasis in the ER. J Cell Biol 185: 1013–1027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valente C, Turacchio G, Mariggio S, Pagliuso A, Gaibisso R, Di Tullio G, Santoro M, Formiggini F, Spano S, Piccini D, Polishchuk RS, Colanzi A, Luini A, Corda D (2012) A 14‐3‐3gamma dimer‐based scaffold bridges CtBP1‐S/BARS to PI(4)KIIIbeta to regulate post‐Golgi carrier formation. Nat Cell Biol 14: 343–354 [DOI] [PubMed] [Google Scholar]
- Varnai P, Balla T (1998) Visualization of phosphoinositides that bind pleckstrin homology domains: calcium‐ and agonist‐induced dynamic changes and relationship to myo‐[3H]inositol‐labeled phosphoinositide pools. J Cell Biol 143: 501–510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villani M, Subathra M, Im YB, Choi Y, Signorelli P, Del Poeta M, Luberto C (2008) Sphingomyelin synthases regulate production of diacylglycerol at the Golgi. Biochem J 414: 31–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakana Y, Kotake R, Oyama N, Murate M, Kobayashi T, Arasaki K, Inoue H, Tagaya M (2015) CARTS biogenesis requires VAP‐lipid transfer protein complexes functioning at the endoplasmic reticulum‐Golgi interface. Mol Biol Cell 26: 4686–4699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang E, Norred WP, Bacon CW, Riley RT, Merrill AH Jr (1991) Inhibition of sphingolipid biosynthesis by fumonisins. Implications for diseases associated with Fusarium moniliforme. J Biol Chem 266: 14486–14490 [PubMed] [Google Scholar]
- Wang YJ, Wang J, Sun HQ, Martinez M, Sun YX, Macia E, Kirchhausen T, Albanesi JP, Roth MG, Yin HL (2003) Phosphatidylinositol 4 phosphate regulates targeting of clathrin adaptor AP‐1 complexes to the Golgi. Cell 114: 299–310 [DOI] [PubMed] [Google Scholar]
- Weber P, Hornjik M, Olayioye MA, Hausser A, Radde NE (2015) A computational model of PKD and CERT interactions at the trans‐Golgi network of mammalian cells. BMC Syst Biol 9: 9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaji T, Hanada K (2015) Sphingolipid metabolism and interorganellar transport: localization of sphingolipid enzymes and lipid transfer proteins. Traffic 16: 101–122 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Movie EV1
Movie EV2
Movie EV3
Movie EV4
Movie EV5
Movie EV6
Movie EV7
Movie EV8
Movie EV9
Movie EV10
Movie EV11
Review Process File