

Abstract
Subarachnoid hemorrhage (SAH) can lead to delayed cerebral ischemia, which increases the rate of morbidity and mortality. The detection of microglial activation may serve as a biomarker for the identification of patients at risk of this deleterious consequence. We assessed this hypothesis in a rat model of SAH in which the exploration of neuroinflammation related to microglial activation was correlated with the degree of bleeding. We used the rat filament model and evaluated (at 48 hours postsurgery) the intensity of neuroinflammation using positron emission tomography (PET) imaging with the 18-kDa translocator protein (TSPO) tracer [18F]DPA-714, quantitative autoradiography with [3H]PK-11195, and SAH grade by postmortem brain picture. High SAH grades were strongly and positively correlated with in vivo PET imaging of TSPO in the cortex and striatum. In addition, a positive correlation was found in the cortex in TSPO, with densities determined by imaging and autoradiographic approaches. Qualitative immunofluorescence studies indicated that overexpression of TSPO was linked to astrocytic/microglial activation. In this model, PET imaging of TSPO using [18F]DPA-714 appeared to be a relevant index of the degree of bleeding, indicating that this imaging method could be used in human patients to improve the management of patients with SAH.
Keywords: animal model, neuroinflammation, PET, SAH, TSPO
Introduction
About 5% of cases with stroke involve subarachnoid hemorrhage (SAH) of aneurysmal origin. The frequency of SAH is approximately 1 per 10 000 person-years.1 Although two-thirds of patients who survive are considered to be independent, many have neurological disorders.2 Vasospasm affects approximately 70% of cases with SAH, generally 3 to 12 days after bleeding.3,4 Decreased local cerebral blood flow downstream from the site of the vasospasm was, until recently, considered to be the main cause of mortality and morbidity in the days following bleeding.5 However, angiographic findings of vasospasm do not correlate closely with SAH-associated morbidity and mortality. Consequently, more complex mechanisms are now suspected and have been grouped under the term early brain injury (EBI).6 There is increasing support for the view that these lesions are responsible for delayed cerebral ischemia (DCI) in the absence of proven angiographic vasospasm.7 Improved prevention and treatment of EBI would require a better understanding of its pathophysiological mechanisms, with none of the current hypotheses being completely satisfactory.8,9 In a rodent model of SAH, microglial activation has been observed both close to the hemorrhage and distant from subarachnoid spaces.10 Such microglial activation may serve as a biomarker for predicting the diagnosis of EBI, for the identification of patients at risk of DCI which leads to worse morbidity and mortality, and for informing patient management or evaluating the effectiveness of treatment. Indeed, the identification of an early biomarker may be useful for detecting patients with poorer outcomes who may need more aggressive treatment strategies.
The activation of microglial cells involves changes in their morphology and acquisition of new functions, including the expression and release of diverse proinflammatory molecules and the phagocytosis of dead cells.11 One of the first molecules to be overexpressed during the microglial activation process is the 18-kDa translocator protein (TSPO), formerly known as the peripheral benzodiazepine receptor.12 The TSPO is located on the outer membrane of the mitochondria and has a low level of expression in the healthy brain. Its expression increases substantially in several neurodegenerative and acute brain disorders, thereby providing a sensitive imaging biomarker for microglial activation associated with neuroinflammation.13
The most widely used positron emission tomography (PET) tracer for in vivo TSPO exploration is [11C]PK-11195, both in preclinical and in clinical studies.14 [11C]PK-11195 provided the first proof of principle that upregulation of TSPO can be detected in vivo in animal models as well as in patients with neurodegenerative diseases. However, this radiotracer exhibits limitations, such as a low ratio of specific to nonspecific binding in vivo15 and a very short half-life. Thus, a number of compounds labeled with [18F], which have the advantage of a longer half-life than [11C], are now available. Among these tracers, [18F]DPA-714 has demonstrated its utility in several animal models15,16 and in human brain disorders17 and has already been used to successfully explore the expression of TSPO associated with neuroinflammation in a rodent model of cerebral ischemia.18
Here, we report the first use of [18F]DPA-714 in a rat model of SAH. We investigated the relationships between microglial activation and the severity of SAH assessed by the degree of bleeding. Our findings suggest that neuroinflammation imaging using [18F]DPA-714 may be a useful method for identifying and better managing patients having SAH with poor outcome.
Materials and Methods
Animals
Animals were treated in accordance with the European Community Council Directive 2010/63/EU for laboratory animal care, and the experimental protocol was validated by the Regional Ethical Committee (Authorization No 00336.01). Adult male Wistar rats weighing 300 to 350 g (Centre d’Elevage René Janvier, Le Genest-St-Isle, France) were used. Animals were housed 2 per cage, under a 12-hour light/12-hour dark cycle at 22°C with access to food and water ad libitum. Twelve rats were included in this study, 7 in the SAH group and 5 in the Sham-operated group.
Surgical Procedure
Animals were anesthetized using isoflurane (Aerrane; Baxter, France) in air mixture, at 4% to 5% in O2 for induction and then 2.5% to 3% during surgical procedure. The anesthetized rats were placed on the back, and a midline neck incision was made. The common carotid artery (CCA), the right internal artery (ICA), and the external carotid artery (ECA) were identified. Both CCA and proximal ICA were occluded with temporary aneurysm clips (right clip 10 mm, Yasargil; BBraun, Massachusetts, USA). The ECA was cut and a transparent polytetrafluoroethylene tube (SUBL-120; Braintree Scientific, Melsungen, Germany) was introduced into the end leading to the carotid bifurcation. The tube was then inserted into the ICA until a line drawn 22 mm from the end of the tube was at the ICA–pterygopalatine artery bifurcation. The tube was bound with a silk thread, and the clips were removed. For the SAH rats, a tungsten filament (diameter 0.076 mm; Scientific Instruments Services Inc) was inserted into the tube. The tube was introduced into the bifurcation of the ICA, in contact with the anterior cerebral artery (ACA). The tungsten filament was inserted until resistance was felt and then was pushed to perforate the ACA. Once the ACA was perforated, the tube was removed and the ECA–ICA bifurcation was ligated distally. The skin incision was closed with nonabsorbable monofilament nylon thread (Prolene Suture, Ethicon, USA). The rats in the Sham group were subjected to the same procedure without arterial perforation by the tungsten filament. In the case of 2 Sham rats, bleeding at the ICA–ECA bifurcation led to permanent clamping of the CCA. Postsurgery, all rats were given an injection of 0.05 mg/kg buprenorphine. The rats were monitored for 48 hours and checked for signs of suffering including prostration, ruffled fur, and weight loss.
Preparation of the Tracer
N,N-diethyl-2-(2-(4-(2-fluoroethoxy)phenyl)-5,7-dimethyl-pyrazolo[1,5-α]pyrimidin-3-yl)acetamide (DPA-714) was labeled with fluorine-18 at its 2-fluoroethyl moiety, following nucleophilic substitution of the corresponding tosylate analogue, according to slight modifications of a previously reported procedure.16 After purification, [18F]DPA-714 was eluted by injectable ethanol, and saline was added to obtain an intravenously injectable solution (10% maximum in volume for ethanol). The radiochemical yields were 50% to 65% (with decay correction). The radiochemical purity and specific activity were 98% and 80 ± 9 GBq/μmol, respectively.
Positron Emission Tomography Imaging
Positron emission tomography imaging was performed at 48 hours postsurgery. Acquisitions were performed on a microPET eXplore VISTA-CT system (GE Healthcare, France) which has an effective axial/transaxial field of view (FOV) of 4.8/6.7 cm, a spatial resolution of less than 2 mm, and a sensitivity above 2.5% in the whole FOV. Animals were anesthetized with isoflurane (Aerrane), at 4% to 5% in O2 for induction and then 1.5% to 2% during scanning. For imaging, each rat was placed on a thermoregulated bed (Minerve, France) in the prone position with a nose cone. The brain was positioned on the center of the FOV. Before PET acquisition, a 5-minute computed tomography (CT) scan was acquired for attenuation correction. A bolus injection of 37 ± 3 MBq/300 g body weight of [18F]DPA-714 in saline was administered into the tail vein. During PET acquisition, the respiratory rate and body temperature were monitored and kept as constant as possible. Each acquisition lasted 60 minutes. The PET list-mode scans were rebinned into 22 frames: Four 10-second frames followed by four 20-second frames, six 60-second frames, four 180-second frames, and four 600-second frames. Each PET scan was corrected for random, scatter, and attenuation, and the images were reconstructed using a 2D OSEM algorithm (2 iterations and 16 subsets; GE Healthcare, France) into voxels of 0.3875 × 0.3875 × 0.775 mm3. All images were analyzed using PMOD (version 3.403; PMOD Technologies, Zurich, Switzerland). The PET-corrected images were used for standard uptake value (SUV) calculations. For each PET scan, the data were summed over the first 5 minutes after radiotracer injection to create a pseudo-perfusion image. This image reflects the initial flow-dependent activity and was recorded with the CT image through a known hardware registration (PET to CT transformation). The CT scans were also recorded using a rat brain magnetic resonance imaging (MRI) template (PMOD),19 and a rat brain MRI template to CT transformation was saved. All PET images, after checking for potential head movements, were corecorded in a single interpolation to the Schiffer rat brain MRI template19 by a combination of these 2 transformations (MRI template to CT and PET to CT transformations). The inverse combined transformation was calculated. The Schiffer MRI template was processed in the PET space images using the inverse transformation applied on the original dynamic PET data and statistics for the regions of interest (ROIs) were extracted. The PET images for the ROIs of left cortex (LCO), right cortex (RCO), left striatum (LSTR), and right striatum (RSTR) were analyzed. Standard uptake value ratios in the cortex and in the striatum were calculated as SUV RCO/SUV LCO and SUV RSTR/SUV LSTR, respectively.
Grading of SAH
At the end of the imaging study, the animals were killed by decapitation under anesthesia. The brains were removed and their ventral view was photographed (Figure 1). Each picture was divided into 4 quadrants: The 2 hemispheres themselves were subdivided into 2 subparts of the branch of the middle cerebral artery and ACA division. Each picture was analyzed to grade the intensity of SAH by 2 observers blinded to the study according to the Sugawara method.20 Each quadrant was rated on a scale of 0 to 3 depending on the amount of subarachnoid blood clot in the segment as follows: grade 0 no subarachnoid blood; grade 1 minimal subarachnoid blood; grade 2 moderate blood clot with recognizable arteries; and grade 3 blood clot obliterating all arteries within the segment (Figure 1). The quadrant scores were added to obtain an SAH grade rating from 0 to 12.
Figure 1.
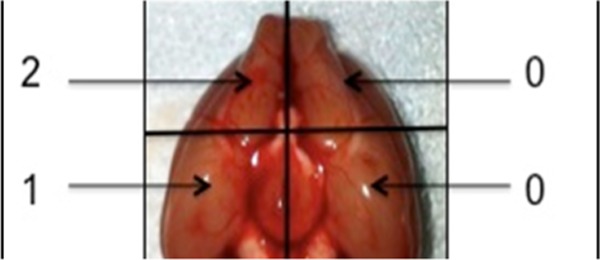
Example of a brain picture grading method. The brain was divided into 4 quadrants; quadrants were graded from 0 to 3 depending on intensity of hemorrhage. In this case, the brain was classified as grade 3 (2 + 1 + 0 + 0)
Autoradiographic Study
Immediately after being photographed, the brains were frozen and stored at −80°C. Coronal brain sections of 20 μm thickness were cut with a cryostat (CM 3050S, Leica, Germany) at −20°C, collected on gelatinized slides and stored at −80°C for at least 4 days. A total of 12 sections per brain were studied for each animal. The density of TSPO binding sites was measured by in vitro autoradiographic experiments using [3H]PK-11195 (Specific Activity 3.06 GBq/µmol; Perkin Elmer, Norwalk, Connecticut). Brain sections were allowed to equilibrate at room temperature (RT) for 3 hours and then incubated with 1 nmol/L [3H]PK-11195 in 50 mmol/L Tris-HCl buffer pH 7.4 at RT for 60 minutes. Nonspecific binding was assessed in the presence of 1 µmol/L PK-11195 (Sigma Aldrich, France). Sections were rinsed twice in ice-cold buffer (4°C) for 5 minutes, then briefly in distilled water at 4°C, and dried at RT. Dry sections were made conductive by an application of metal electric tape (3 M; Euromedex, Souffelweyersheim, Francel) on the free side and then placed in the gas chamber of the β-imager 2000 (Biospace Lab, Paris, France). Data from brain sections were collected over 90 minutes. Four anatomical ROIs, that is, the RCO, LCO, RSTR, and LSTR, were selected manually and identified in the Paxinos and Watson atlas.21 Using the β-vision software (Biospace Lab), the level of bound radioactivity was directly determined by counting the number of β-particles emitted from the delineated area. The radioligand signal in the ROIs was measured for each rat and expressed as counts per minute per square millimeter (cpm/mm2). Specific binding was determined by subtracting nonspecific binding from total binding. Ratios for both structures were calculated as RCO/LCO and RSTR/LSTR.
Immunofluorescence Study
Coronal sections (20-µm thick) adjacent to those used for the autoradiographic study were used. After 15 minutes at RT, sections were thawed in a solution of 4% paraformaldehyde at RT for 30 minutes. The sections were washed 3 times in phosphate-buffered saline (PBS) 0.1 mol/L for 5 minutes at RT and were then incubated for 1 hour at RT in a buffer to enhance cell permeability and to block nonspecific sites (PBS/0.3% triton X-100/5% normal horse serum). Tissue sections were delineated using the Pencil DakoPen (Z0334; Dako, Les Ulis, France) on the glass slide before incubation overnight at 4°C with 1:500 diluted polyclonal rabbit anti-GFAP (Dako, France), 1:200 diluted polyclonal rabbit anti-Iba1 (ab108539; Abcam, France), or 1:200 diluted monoclonal mouse anti-CD11b (CBL152; Merck Millipore, France). Primary antibodies were diluted in PBS/0.3% triton X-100/1% normal horse serum. After 3 washes with PBS 0.1 mol/L at RT for 5 minutes, sections were incubated for 1 hour in a dark box at RT with secondary antibodies each at 1:500 dilution in PBS/0.3% triton X-100/1% normal horse serum: either goat anti-rabbit Dylight 549 or goat anti-mouse Dylight 488 (KPL; Eurobio, France). The sections were washed twice for 5 minutes in PBS 0.1 mol/L and twice in distilled water and incubated with DAPI 1 µg/mL (D9542; Sigma Aldrich) for 15 minutes. After 3 washes in distilled water, the sections were mounted with fluorescent-mounting medium (S3023; Dako) and kept in a dark box at 4°C until observation.
Images from immunolabeled RCO sections were captured with the ExtraNova Morphostrider software (Explora Nova; La Rochelle, France) using a fluorescence microscope with a 20× objective (Explora Nova). Multiple fluorescence images were acquired sequentially, merged as Red, Green, Blue (RGB) images; and then analyzed using the Image J software (v 1.47).
Statistical Analysis
For imaging and autoradiographic studies, results were expressed as means ± standard error of the mean. To compare the 2 groups of rats (SAH vs Sham), a Mann-Whitney U test was used. The level of significance was P < .05. Correlations between 2 quantitative parameters were estimated by Spearman tests (GraphPad Instat; GraphPad Software, San Diego, California). The level of significance was P < .05.
Results
Positron Emission Tomography Imaging
Accumulation of [18F]DPA-714 was much greater in high-grade SAH rats than in Sham rats (both in the cortex and striatum), as illustrated in Figure 2A and as shown by mean SUV time–activity curves of [18F]DPA-714 in the right brain (Figure 2B). However, the difference between the groups was not statistically significant (cortex: 0.44 ± 0.03 and 0.39 ± 0.02 in the SAH and Sham groups, respectively; striatum: 0.41 ± 0.05 and 0.31 ± 0.01 in the SAH and Sham groups, respectively).
Figure 2.

(A) Fusion sagittal positron emission tomography (PET) brain images with magnetic resonance imaging (MRI)-Template (PMOD) using [18F]DPA-714 in a Sham (left side) and high-grade subarachnoid hemorrhage (SAH) rat (right side). (B) Mean standard uptake value (SUV) time–activity curves of [18F]DPA-714 in the right cortex (RCO) and right striatum (RSTR) in Sham and SAH groups. Mean standard uptake value ratio (SUVr) of Sham and SAH groups in the cortex (C) and striatum (D). Results are expressed as mean ± standard error of the mean (SEM). (§P = .0519, Mann-Whitney U test).
The mean SUVr was 22% higher in the cortex of the SAH group than in the Sham group (1.06 ± 0.06 vs. 0.87 ± 0.04; Figure 2C) and 26% higher in the striatum (1.17 ± 0.11 vs 0.93 ± 0.03; Figure 2D). However, these increases were at the limit of the statistical significance (P = .0519; Mann-Whitney U test), probably due to the small number of animals in each group.
Grading and Mortality of SAH
No significant difference in body weight was observed between the 2 groups before and after surgery. After PET imaging, brains were removed and graded for SAH (Figure 3A). The SAH grading by the 2 observers was highly correlated (Figure 3B; r = .9305, P < .0001, Spearman test). The SAH scores attributed ranged from grade 1 to 9 for the SAH group and from 0 to 2 for the Sham group. Among SAH rats, 2 had a high grade, 2 had a medium grade, and 2 had a low grade. One rat in the SAH group died after the surgical procedure. Autopsy of this rat revealed an SAH grade of 11.
Figure 3.

(A) Representative pictures of a Sham rat (a), a low-grade subarachnoid hemorrhage (SAH) rat (b) and a high-grade SAH rat showing an interhemispheric clot (c). (B) Correlation of the SAH grading between the 2 observers (ρ = .93, P < .0001).
Autoradiographic Study
The TSPO density was measured by [3H]PK-11195 binding in the 4 ROIs, that is, the LCO, RCO, LSTR, and RSTR. As illustrated in Figure 4A, TSPO binding was higher in a high-grade SAH rat than in a Sham rat. The mean TSPO density evaluated by [3H]PK-11195 binding showed no statistically significant difference between the SAH and the Sham groups, although higher values were observed in the right side of the SAH group compared to Sham group, both in the cortex (3.21 ± 0.59 vs 2.24 ± 0.38 cpm/mm2) and in the striatum (3.46 ± 1.19 vs 1.81 ± 0.40 cpm/mm2; Table 1). Moreover, the ratio of [3H]PK-11195 binding between the right and the left sides was clearly but nonsignificantly higher in the SAH group compared to the Sham group, both in the cortex (1.95 ± 0.48 vs 1.00 ± 0.11; Figure 4B) and in the striatum (2.40 ± 0.89 vs 1.03 ± 0.06; Figure 4C).
Figure 4.

(A) Representative autoradiographic images with [3H]PK-11195 obtained on 20-µm-thick coronal brain sections: Total binding in Sham (a) and subarachnoid hemorrhage (SAH; c) rats; nonspecific binding in the same Sham (b) and SAH (d) rats. Ratios of [3H]PK-11195 binding values in the Sham and SAH groups in the cortex (B) and striatum (C).
Table 1.
Autoradiographic Study with [3H]PK-11195 on Coronal Brain Sections.
| Cortex | Striatum | |||
|---|---|---|---|---|
| Left | Right | Left | Right | |
| Sham | 2.34 ± 0.40 | 2.24 ± 0.38 | 1.79 ± 0.44 | 1.81 ± 0.40 |
| SAH | 1.81 ± 0.20 | 3.21 ± 0.59 | 1.57 ± 0.15 | 3.46 ± 1.19 |
Abbreviations: SAH, subarachnoid hemorrhage; SEM, standard error of the mean.
The specific binding of the tracer is expressed as mean cpm/mm2 ± SEM in the cortex and the striatum on the left and right sides, in Sham (n = 5) and SAH (n = 6) rats.
Immunofluorescence Study
We performed a qualitative analysis of immunostaining with GFAP, Iba-1, and CD11b in the cortex of an SAH rat and a Sham rat (Figure 5A). Higher signals were observed for each marker in SAH rats compared to Sham rats (Figure 5B-D).
Figure 5.

Immunoreactivity of GFAP, Iba1, and CD11b in a Sham rat and in a high-grade subarachnoid hemorrhage (SAH) rat. (A) Representation of coronal brain section,21 the black box indicates the selected area for immunofluorescence study in the right cortex. Representative immunofluorescence images of 4′,6′-diamidino-2-phénylindole (DAPI; blue channel, B-D), Glial Fibrillary Acidic Protein (GFAP; red channel, B), Ionized calcium binding adapter molecule 1 (Iba1; red channel, C), and CD11b (green channel, D) of a Sham rat (upper) and a high-grade SAH rat (lower). Scale bars represent 150 µm.
Correlations Between SAH Grades and In Vivo and In Vitro Parameters
There was a high positive correlation between the SAH grade and [18F]DPA-714 SUVr in the cortex and striatum (r = .93, P = .017 in both brain regions, Spearman test). Similarly, a positive correlation was observed between the SAH grade and [3H]PK-11195 binding in the cortex (r = .93, P = .017) and a weak correlation was observed in the striatum (r = .84, P = .058). In addition, in vivo [18F]DPA-714 SUVr and in vitro [3H]PK-11195 binding values were also highly positively correlated in the cortex (r = .98, P = .003) but not correlated in the striatum (r = .66, P = .17).
Discussion
The DCI consecutive to SAH significantly increases both morbidity and mortality.5 These complications were often attributed to vasospasm, but increasing evidence implicates various pathophysiological mechanisms including oxidative stress and neuronal apoptosis.9 In addition, neuroflammation, microglial activation in particular, plays a crucial role in brain damage by contributing to the neural injury and the induction of brain edema,10,22,23 the reduction in vascular reactivity, and the enhancement of blood–brain barrier permeability.22,24,25
Several SAH rat models have been described including the filament model which we used in this study.26,27 This model avoids the need for opening the skull and closely reproduces many features of human SAH. We sought to study the intensity of neuroinflammation as a function of SAH grade, and this required quantifying the degree of hemorrhage.20 In our study, the mortality occurrence before the end or after the surgical procedure was lower than that reported in the literature (1 death 48 hours postsurgery in the SAH group, corresponding to 14%). Indeed, using similar techniques, some authors reported about 33% of death27,28 and about 50% in the “double hemorrhage” model.29,30 The autopsy of the dead rat in our experiment indicated a very high SAH grade (grade 11). Consequently, we tried to reduce the risk of premature death associated with surgery and as a result we obtained few high SAH grade (2 rats of 6). In 2 Sham-operated rats, the ligation of the CCA was required for satisfactory hemostasis. However, this did not affect the results because clamping the CCA does not lead to ipsilateral ischemic brain injury.31 The duration of the procedure for the SAH group was significantly longer than that for the Sham group, largely because the technique was more complex and time consuming.27 It is well established that the grading of the degree of bleeding is based on a personal interpretation of rat brain pictures. In this study, the pictures were analyzed blindly by 2 neurosurgeons, and the correlation between both was almost perfect. The interpretation of the pictures was therefore not considered as a source of significant bias.
We evaluated the density of the neuroinflammation biomarker TSPO,12,13 at 48 hours postsurgery, using a noninvasive PET method with the specific tracer [18F]DPA-714 and observed the highest accumulation of the tracer in the right hemisphere (cortex and striatum) of SAH rats. By contrast, a low accumulation of [18F]DPA-714 was measured in the left hemisphere of these rats or in both hemispheres of Sham rats. Quantitative autoradiography with [3H]PK-11195 in brain sections of same animals showed a greater level of the radio ligand in the cortex and striatum on the lesioned side, but statistical significance was not reached. This could be partly explained by the small number of animals included in the study, and the fact that it was not possible to predict the subsequent SAH grade during surgery, leading to substantial standard deviation values in the SAH group. Indeed, our study included only a small number of high-grade lesions in the SAH group, and severity was determinant to provide microglial activation associated with neuroinflammation. In addition, although both PET imaging with [18F]DPA-714 and autoradiography with [3H]PK-11195 explore the same target, that is, TSPO, difference in methods should be considered. Indeed, whole volumes of the ROIs (cortex and striatum) were analyzed using PET imaging, whereas only some brain sections were analyzed with autoradiography, leading to higher mean standard error values and to a bias resulting in a possible underestimation of the measurement of TSPO binding values by autoradiography.
One interesting finding is that the grade of the lesion was strongly and positively correlated with in vivo imaging and in vitro autoradiography in the cortex, although this correlation was not significant in the striatum. It was previously reported that the clinical neurological grade of SAH rats was correlated with moderate and severe SAH but that the correlation did not extend to cases of low-grade SAH.20 In the striatum, which is distant from the lesion site, TSPO imaging with [18F]DPA-714 was highly correlated with the SAH grade, indicating that neuroinflammation distant from the lesion site can be detected by PET imaging.
There is currently no consensus regarding the precise type of glial cells responding to a proinflammatory insult through TSPO upregulation. Some studies have reported the increase of TSPO expression in both microglia and astrocytes,32,33 whereas other studies pointed to a selective microglia expression.34 Our qualitative immunofluorescence study on the adjacent sections to those used in the autoradiographic study showed that the astrocytic marker Glial Fibrillary Acidic protein (GFAP) as well as the activated microglial markers CD11b (including infiltrated macrophages) and Iba1 were upregulated. Such upregulations have recently been observed in experimental models of SAH.35,36 Therefore, this indicated that the increase in TSPO density detected both by PET and autoradiographic studies reflected astrocyte and/or microglial activation. Of note, the antibodies we used do not distinguish between the local microglia and the circulating macrophages that could be recruited to the injured brain.
We report herein the first in vivo use of [18F]DPA-714 in a rat model of SAH. Our findings support the hypothesis that the intensity of the neuroinflammation assessed both by PET and by autoradiographic approaches using TSPO quantification is correlated with the severity of SAH. This suggests that in vivo exploration of neuroinflammation by PET imaging in humans may reflect the initial severity of SAH and the risks of unfavorable secondary evolution (DCI). This imaging approach could help evaluate the efficacy of various therapies that are currently being developed for such lesions.4,5,8 The mechanisms underlying the activation of the inflammatory cascade remain unclear. Nevertheless, in vivo studies in humans may make it possible to predict complications and assist with the understanding of SAH pathophysiology.
Acknowledgments
We thank the Laboratoires Cyclopharma for providing fluor-18, and Sylvie Bodard for technical assistance.
Footnotes
Declaration of Conflicting Interests: The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The author(s) received no financial support for the research, authorship, and/or publication of this article: The research leading to these results has received funding from the European Union’s Seventh Framework Programme (FP7/2007-2013) under grant agreement 278850 (INMiND), from Labex IRON (ANR-11-LABX-18-01) and from the Région Centre (2014 00094049 – AP 2014 – 850).
References
- 1. Becker KJ. Epidemiology and clinical presentation of aneurysmal subarachnoid hemorrhage. Neurosurg Clin N Am. 1998;9(3):435-444. [PubMed] [Google Scholar]
- 2. Haug T, Sorteberg A, Sorteberg W, Lindegaard KF, Lundar T, Finset A. Cognitive outcome after aneurysmal subarachnoid hemorrhage: time course of recovery and relationship to clinical, radiological, and management parameters. Neurosurgery. 2007;60(4):649-656. [DOI] [PubMed] [Google Scholar]
- 3. Alaraj A, Charbel FT, Amin-Hanjani S. Peri-operative measures for treatment and prevention of cerebral vasospasm following subarachnoid hemorrhage. Neurol Res. 2009;31(6):651-659. [DOI] [PubMed] [Google Scholar]
- 4. Eddleman CS, Hurley MC, Naidech AM, Batjer HH, Bendok BR. Endovascular options in the treatment of delayed ischemic neurological deficits due to cerebral vasospasm. Neurosurg Focus. 2009;26(3):E6. [DOI] [PubMed] [Google Scholar]
- 5. Dorsch NW. Therapeutic approaches to vasospasm in subarachnoid hemorrhage. Curr Opin Crit Care. 2002;8(2):128-133. [DOI] [PubMed] [Google Scholar]
- 6. Dankbaar JW, de Rooij NK, Velthuis BK, et al. Diagnosing delayed cerebral ischemia with different CT modalities in patients with subarachnoid hemorrhage with clinical deterioration. Stroke. 2009;40(11):3493-3498. [DOI] [PubMed] [Google Scholar]
- 7. Dankbaar JW, Rijsdijk M, van der Schaaf IC, Velthuis BK, Wermer MJ, Rinkel GJ. Relationship between vasospasm, cerebral perfusion, and delayed cerebral ischemia after aneurysmal subarachnoid hemorrhage. Neuroradiology. 2009;51(12):813-819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Pluta RM, Hansen-Schwartz J, Dreier J, et al. Cerebral vasospasm following subarachnoid hemorrhage: time for a new world of thought. Neurol Res. 2009;31(2):151-158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sehba FA, Hou J, Pluta RM, Zhang JH. The importance of early brain injury after subarachnoid hemorrhage. Prog Neurobiol. 2012;97(1):14-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Schneider UC, Davids A-M, Brandenburg S, et al. Microglia inflict delayed brain injury after subarachnoid hemorrhage. Acta Neuropathol. 2015;130(2):215-231. [DOI] [PubMed] [Google Scholar]
- 11. Heneka MT, Rodríguez JJ, Verkhratsky A. Neuroglia in neurodegeneration. Brain Res Rev 2010;63(1-2):189-211. [DOI] [PubMed] [Google Scholar]
- 12. Papadopoulos V, Baraldi M, Guilarte TR, et al. Translocator protein (18kDa): new nomenclature for the peripheral-type benzodiazepine receptor based on its structure and molecular function. Trends Pharmacol Sci. 2006;27(8):402-409. [DOI] [PubMed] [Google Scholar]
- 13. Chen MK, Guilarte TR. Translocator protein 18 kDa (TSPO): molecular sensor of brain injury and repair. Pharmacol Ther. 2008;118(1):1-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Venneti S, Lopresti BJ, Wiley CA. The peripheral benzodiazepine receptor (Translocator protein 18kDa) in microglia: from pathology to imaging. Prog Neurobiol. 2006;80(6):308-322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chauveau F, Boutin H, Van Camp N, Dollé F, Tavitian B. Nuclear imaging of neuroinflammation: a comprehensive review of [11C]PK11195 challengers. Eur J Nucl Med Mol Imaging. 2008;35(12):2304-2319. [DOI] [PubMed] [Google Scholar]
- 16. James ML, Fulton RR, Vercoullie J, et al. DPA-714, a new translocator protein-specific ligand: synthesis, radiofluorination, and pharmacologic characterization. J Nucl Med. 2008;49(5):814-822. [DOI] [PubMed] [Google Scholar]
- 17. Corcia P, Tauber C, Vercouillie J, et al. Molecular imaging of microglial activation in amyotrophic lateral sclerosis. PLoS One. 2012;7(12):e52941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Boutin H, Prenant C, Maroy R, et al. [18F]DPA-714: direct comparison with [11C]PK11195 in a model of cerebral ischemia in rats. PLoS One. 2013;8(2):e56441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Schiffer WK, Mirrione MM, Biegon A, Alexoff DL, Patel V, Dewey SL. Serial microPET measures of the metabolic reaction to a microdialysis probe implant. J Neurosci Methods. 2006;155(2):272-284. [DOI] [PubMed] [Google Scholar]
- 20. Sugawara T, Ayer R, Jadhav V, Zhang JH. A new grading system evaluating bleeding scale in filament perforation subarachnoid hemorrhage rat model. J Neurosci Methods. 2008;167(2):327-334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Paxinos G, Watson C. The Rat Brain in Stereotaxic Coordinates. 6th ed San Diego, CA: Elsevier Academic Press; 2007. [Google Scholar]
- 22. Schneider UC, Schiffler J, Hakiy N, Horn P, Vajkoczy P. Functional analysis of Pro-inflammatory properties within the cerebrospinal fluid after subarachnoid hemorrhage in vivo and in vitro. J Neuroinflammation. 2012;9:28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chaichana KL, Pradilla G, Huang J, Tamargo RJ. Role of inflammation (leukocyte-endothelial cell interactions) in vasospasm after subarachnoid hemorrhage. World Neurosurg. 2010;73(1):22-41. [DOI] [PubMed] [Google Scholar]
- 24. Fassbender K, Hodapp B, Rossol S, et al. Inflammatory cytokines in subarachnoid haemorrhage: association with abnormal blood flow velocities in basal cerebral arteries. J Neurol Neurosurg Psychiatry. 2001;70(4):534-537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Macdonald RL. Delayed neurological deterioration after subarachnoid haemorrhage. Nat Rev Neurol. 2014;10(1):44-58. [DOI] [PubMed] [Google Scholar]
- 26. Bederson JB, Germano IM, Guarino L. Cortical blood flow and cerebral perfusion pressure in a new noncraniotomy model of subarachnoid hemorrhage in the rat. Stroke. 1995;26(6):1086-1091. [DOI] [PubMed] [Google Scholar]
- 27. Park IS, Meno JR, Witt CE, et al. Subarachnoid hemorrhage model in the rat: modification of the endovascular filament model. J Neurosci Methods. 2008;172(2):195-200. [DOI] [PubMed] [Google Scholar]
- 28. Cahill J, Calvert JW, Zhang JH. Mechanisms of early brain injury after subarachnoid hemorrhage. J Cereb Blood Flow Metab. 2006;26(11):1341-1353. [DOI] [PubMed] [Google Scholar]
- 29. Lee JY, Huang DL, Keep R, Sagher O. Characterization of an improved double hemorrhage rat model for the study of delayed cerebral vasospasm. J Neurosci Methods. 2008;168(2):358-366. [DOI] [PubMed] [Google Scholar]
- 30. Guresir E, Raabe A, Jaiimsin A, et al. Histological evidence of delayed ischemic brain tissue damage in the rat double-hemorrhage model. J Neurol Sci. 2010;293(1-2):18-22. [DOI] [PubMed] [Google Scholar]
- 31. Guo H, Itoh Y, Toriumi H, et al. Capillary remodeling and collateral growth without angiogenesis after unilateral common carotid artery occlusion in mice. Microcirculation. 2011;18(3):221-227. [DOI] [PubMed] [Google Scholar]
- 32. Cosenza-Nashat M, Zhao ML, Suh HS, et al. Expression of the translocator protein of 18 kDa by microglia, macrophages and astrocytes based on immunohistochemical localization in abnormal human brain. Neuropathol Appl Neurobiol. 2009;35(3):306-328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lavisse S, Guillermier M, Hérard AS, et al. Reactive astrocytes overexpress TSPO and are detected by TSPO positron emission tomography imaging. J Neurosci. 2012;32(32):10809-10818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wang Y, Yue X, Kieseetter DO, Niu G, Teng G, Chen X. PET imaging of neuroinflammation in a rat traumatic brain injury model with radiolabeled TSPO ligand DPA-714. Eur J Nucl Med Mol Imaging. 2014;41(7):1440-1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Murakami K, Koide M, Dumont TM, Russell SR, Tranmer BI, Wellman GC. Subarachnoid hemorrhage induces gliosis and increased expression of the pro-inflammatory cytokine high mobility group box 1 protein. Transl Stroke Res. 2011;2(1):72-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kooijman E, Nijboer CH, van Velthoven CT, Kavelaars A, Kesecioglu J, Heijnen CJ. The rodent endovascular puncture model of subarachnoid hemorrhage: mechanisms of brain damage and therapeutic strategies. J Neuroinflammation. 2014;11:2. [DOI] [PMC free article] [PubMed] [Google Scholar]