

Summary
Bone marrow-derived mesenchymal stem cells (BMSCs) are proposed as the cells of origin of several subtypes of osteosarcoma (OS). However, signals that direct BMSCs to form different subtypes of OS are unclear. Here we show that the default tumor type from spontaneously transformed p53 knockout (p53_KO) BMSCs is osteoblastic OS. The development of this default tumor type caused by p53 loss can be overridden by various oncogenic signals: RAS reprograms p53_KO BMSCs into undifferentiated sarcoma, AKT enhances osteoblastic OS, while cFOS promotes chondroblastic OS formation. We focus on studying the mechanism of cFOS-induced chondroblastic OS formation. Integrated genome-wide studies reveal a regulatory mechanism whereby cFOS binds to the promoter of a key chondroblastic transcription factor, Sox9, and induces its transcription in BMSCs. Importantly, SOX9 mediates cFOS-induced cartilage formation in chondroblastic OS. In summary, oncogenes determine tumor types derived from BMSCs, and the cFOS-SOX9 axis is critical for chondroblastic OS formation.
Keywords: mesenchymal stem cells, p53, osteosarcoma, cFOS, SOX9, RAS, AKT, stem cells, cancer, cancer stem cells
Graphical Abstract
Highlights
-
•
The default tumor type from p53_KO BMSCs is osteoblastic OS
-
•
Oncogenes reprogram p53_KO BMSCs into different sarcomas
-
•
cFOS promotes chondroblastic OS from p53_KO BMSCs
-
•
SOX9 is a mediator of cFOS in promoting chondroblastic OS
In this article, Huang and colleagues show that the default tumor type arising from p53 knockout mouse bone marrow-derived mesenchymal stem cells is osteoblastic osteosarcoma (OS), and oncogenic signals can override this default tumor type choice. They focus on studying the mechanisms underlying cFOS-driven chondroblastic OS and demonstrate that SOX9 is one of the mediators of cFOS.
Introduction
Bone marrow-derived mesenchymal stem cells (BMSCs) are multipotent fibroblast-like cells that have the capacity to differentiate into adipocytes, osteocytes, chondrocytes, and fibroblasts. They also maintain bone marrow homeostasis by generating osteocytes and providing a niche for hematopoietic stem cells (Bianco et al., 2008, Kfoury and Scadden, 2015). The true in vivo identity of BMSCs has not been firmly established (Bianco et al., 2008, Sacchetti et al., 2016, Worthley et al., 2015, Zhou et al., 2014). BMSCs, in vitro, are loosely defined as a group of cells that have the ability of differentiating into adipocytes, osteoblasts, and chondrocytes. Because of their multipotency, BMSCs attract a lot of attention in the tissue regeneration field. Several key transcription factors govern the development of BMSCs into different cell types. RUNX2 is critical for osteogenic differentiation, SOX9 for chondrogenic differentiation, and peroxisome proliferator activated receptor γ (PPARγ) for adipogenic differentiation (Bi et al., 1999). In tumor biology, BMSCs are closely relevant to sarcomagenesis since they are proposed as the cells of origin of several types of sarcomas, such as osteosarcoma (OS), chondrosarcoma, liposarcoma, and Ewing's sarcoma (Tirode et al., 2007). For example, both mouse and human BMSCs can give rise to OS through spontaneous transformation or genetic modifications (Calo et al., 2010, Lin et al., 2009, Xiao et al., 2013).
OS is the most common cancer arising from bone and mainly affects children and adolescents. The knowledge of the genetic and epigenetic causes of OS remains incomplete, although TP53 and RB1 losses are commonly involved (Velletri et al., 2016). Around 25% of mice with heterozygous deletion of trp53 (also called p53) develop OS after a long latency, suggesting that a second genetic or epigenetic event is required for the full development of OS (Donehower et al., 1992, Lang et al., 2004, Olive et al., 2004). A recent genome-wide sequencing study of human OS showed that almost all the human OS have point mutations, deletions, amplifications, and/or translocations in the TP53 gene or the genes encoding other components (e.g., HDM2) in the p53 signaling pathway, demonstrating the importance of p53 loss in osteosarcomagenesis (Chen et al., 2014). In human OS and patient-derived OS cell lines, TP53 deletions and point mutations are common (Chen et al., 2014, He et al., 2015). Recent whole-genome sequencing studies revealed that the PI3K (phosphoinositide 3-kinase)-AKT (v-akt murine thymoma viral oncogene)-mTOR (mammalian target of rapamycin) pathway is also involved in osteosarcomagenesis (Chen et al., 2014, Perry et al., 2014). However, the dysregulation of this pathway explains only a small portion of OS, suggesting that other oncogenic pathways are involved. Another possible oncogene in OS is the cFos (FBJ osteosarcoma oncogene) gene, which was identified in mouse OS (Van Beveren et al., 1983). In human OS, the genetic alterations of cFOS were rare although cFOS upregulation was found (Chen et al., 2014, Perry et al., 2014), raising the possibility that epigenetic or transcriptional upregulation of cFOS is involved.
OS is divided into several subtypes: osteoblastic (about 70%), chondroblastic (about 10%), fibroblastic (about 10%), and others (10%) (Fletcher et al., 2013, Mirabello et al., 2009). Although the prognoses of these subtypes are clinically similar, it is largely unknown how they are generated. Since BMSCs are multipotent and may be the cells of origin of OS, an attractive hypothesis is that oncogenes and/or tumor suppressors regulate the lineage choices of BMSCs and, thus, the subtypes of OS.
We have previously isolated and characterized BMSCs from the bone marrow of p53 knockout (p53_KO) mice (He et al., 2015). In this study, we aim to use these p53_KO BMSCs as a model to study the contribution of oncogenic signals in osteosarcomagenesis. We find that the default tumor type arising from p53_KO BMSCs is osteoblastic OS, and that oncogenic signals can override this default lineage choice. We concentrate on studying the signaling that induces chondroblastic OS and identify an axis of cFOS-SOX9, which enhances cartilage formation during osteosarcomagenesis. Results from human OS tissue microarray support that the cFOS-SOX9 connection is conserved in human chondroblastic OS.
Results
BMSCs Carrying p53 Deletion Spontaneously Form Osteoblastic OS
In mouse models, p53 loss has been shown to be a dominant factor for osteosarcomagenesis (Lin et al., 2009, Walkley et al., 2008). To test whether this can be recapitulated in a cellular model, we assessed the tumorigenicity of p53_KO BMSCs, which were isolated from the bone marrow of 6- to 10-week-old adult p53_KO mice. These p53_KO BMSCs are immortalized and multipotent with the ability of differentiating into three lineages: osteocytes, chondrocytes, and adipocytes (He et al., 2015). We transplanted these cells into immunocompromised NOD, scid, gamma (NSG) mice. Five out of nine mice transplanted with p53_KO BMSCs generated tumors with an average latency (time between transplantation and tumor harvesting) of 44 days; the other four mice did not develop tumors within a 4-month period (Table S1). Pathological analyses revealed that 100% of generated tumors were osteoblastic OS containing woven bones and osteoid (Figure 1). This finding is consistent with our previous in vitro observation showing that p53_KO BMSCs are predisposed to osteoblastic differentiation (He et al., 2015). Thus, the default subtype of OS formed from p53_KO BMSCs is osteoblastic OS.
Figure 1.

p53_KO BMSCs Generate Osteoblastic OS
(A) H&E staining of two representative tumors formed from p53_KO BMSCs transplanted into NSG mice. Woven bone and osteoid structures are shown.
(B) Masson's trichrome staining of two representative tumors formed from p53_KO BMSCs transplanted into NSG mice. Blue color indicates osteoid. Blue indicates collagen in bone.
(C) Alcian blue staining of two representative tumors formed from p53_KO BMSCs transplanted into NSG mice. Blue indicates glycosaminoglycans in cartilages.
Scale bars represent 2 mm for inlets and 200 μm for large microscopic views. See also Table S1.
Different Oncogenes Induce Different Types of Sarcoma from BMSCs
Because not all the transplants of p53_KO BMSCs generated tumors, we tested whether oncogenes can increase the tumor incidence. We examined the effect of three oncogenes, Akt, cFos, and Ras (rat sarcoma viral oncogene), on promoting the tumor formation in p53_KO BMSCs. These oncogenes were chosen because Akt and cFos have been linked to OS and Ras to sarcoma in general (Chen et al., 2014, Perry et al., 2014, Shih et al., 1980, Van Beveren et al., 1983).
We transduced p53_KO BMSCs using murine retroviruses expressing Akt2, cFos, k-Ras (Kirsten Ras) (V12), or h-Ras (Harvey Ras) (V12). Anchorage-independent growth assays were performed to determine the in vitro transformation ability of these cells (Figures S1A and S1B). About 20% of p53_KO BMSCs generated colonies (Figure S1A). cFOS, k-RAS (V12), and AKT2 further increased both colony number (Figure S1A) and size (Figure S1B). These results suggest that p53_KO BMSCs contain some spontaneous transformed cells but the majority of the cells are not transformed. Since these cells are primary cells with less than eight passages, it is likely that the small portion of transformed cells pre-existed before the isolation from bone marrow. However, our results cannot completely rule out the possibility that short-term in vitro culturing enables some cells to be spontaneously transformed. Nonetheless, oncogenes, such as cFos, k-Ras (V12), and Akt2, can greatly enhance the transformation of p53_KO BMSCs.
After determining the anchorage-independent growth capacity of these cells, we transplanted them into NSG mice to test their ability to form tumor. Tumor formation frequency was 100% for p53_KO BMSCs transduced with each of the oncogenes, and tumor formation latency was decreased by oncogenes (Table S1). One prominent observation was that tumor types were drastically different between cells transduced with the different oncogenes. cFOS induced chondroblastic OS, which contained mixed osteoid (stained by Masson’s trichrome) and cartilage (stained by Alcian blue) (Figures 2A–2D), while both k-RAS and h-RAS induced undifferentiated sarcomas, which were negative for Masson’s trichrome and Alcian blue staining (Figures 2E–2H). Tumors from k-RAS (V12)- or h-RAS (V12)-transformed p53_KO BMSCs had telangiectatic change as displayed by abundant blood cells within the tumors (Figures 2 and S1C–S1E). AKT2 induced osteoblastic OS with the characteristic osteoid and woven bone and the absence of cartilage (Figures 2I–2L). Real-time PCR analyses and immunohistochemistry showed that cFOS-driven tumors expressed high levels of Acan (also called Aggrecan), a marker for chondrocytes, while AKT-driven tumors expressed high levels of Bglap (also called Osteocalcin), a marker for osteocytes (Figures 2M and S1F). Ras-driven tumors expressed low levels of these two markers (Figures 2M and S1F).
Figure 2.

Different Oncogenes Direct p53_KO BMSCs to Form Different Types of Sarcomas
(A) Western blot analyses (left) of overexpressing cFos in p53_KO BMSCs with cFOS and β-ACTIN antibodies. Macroscopic view (right) of a representative tumor generated from p53_KO BMSCs with cFos overexpression.
(B) H&E staining.
(C) Masson's trichrome staining. Blue indicates collagen in bone.
(D) Alcian blue staining. Blue indicates glycosaminoglycans in cartilages.
(E) Western blot analyses (left) of overexpressing k-RAS (V12) in p53_KO BMSCs with k-RAS (V12) and β-ACTIN antibodies. Macroscopic view (right) of a representative tumor generated from p53_KO BMSCs with k-RAS (V12) overexpression.
(F) H&E staining.
(G) Masson's trichrome staining.
(H) Alcian blue staining.
(I) Western blot analyses (left) of overexpressing Akt2 in p53_KO BMSCs with AKT2 and β-ACTIN antibodies. Macroscopic view (right) of a representative tumor generated from p53_KO BMSCs with Akt2 overexpression.
(J) H&E staining.
(K) Masson's trichrome staining.
(L) Alcian blue staining.
(M) Real-time PCR to measure the relative normalized RNA levels of Bglap (an osteogenic marker) and Acan (a chondrogenic marker) in cFOS, k-RAS (V12), or AKT2 driven tumors. n = 3 independent experiments; error bar denotes SEM.
Scale bars represent 2 mm for inlets and 200 μm for large microscopic views. See also Figures S1 and S2; Table S2.
To test whether the transplantation sites affect the tumor types, we performed intrabone marrow injection of p53_KO BMSCs transformed with different oncogenes (Figure S2 and Table S2). Similar to the results from intramuscular injection, k-RAS (V12) induced undifferentiated sarcoma while cFOS transformed these cells into chondroblastic OS. However, we did not observe tumor formation from p53_KO BMSCs transformed by AKT2 in the intrabone marrow injection up to 5 months, presumably due to fewer cells injected (1 million cells) and the potential long latency of tumor formation in the bone marrow environment. Therefore, the injection sites do not appear to affect the tumor types, at least for k-RAS (V12)- and cFOS-driven tumors originating from p53_KO BMSCs.
In summary, different oncogenes transform p53_KO BMSCs into different types of sarcomas.
cFOS-Transformed p53_KO BMSCs Are Multipotent
Because the etiology of chondroblastic OS is unknown and this subtype of OS is refractory to chemotherapy, we decided to further study cFOS-induced chondroblastic OS generated from BMSCs. We set out to test whether the enhanced chondrogenesis in tumor can be recapitulated in vitro. To this end, we picked single clones from cFOS-transduced p53_KO BMSCs and performed in vitro chondrogenesis, osteogenesis, and adipogenesis assays. Single clones of BMSCs transformed with cFOS had an enhanced chondrogenesis and decreased osteogenesis compared with control BMSCs (Figures 3A, 3B, S3A, and S3B), while adipogenesis was not affected (Figures 3C and S3C). Similar to polyclonal cFOS-transduced p53_KO BMSCs, these single clones formed chondroblastic OS with spicules of woven bone, osteoid, and chondrocyte-like cells within a cartilaginous matrix (Figures 3D–3F and S3D–S3F). These results indicate that a single cFOS-transformed p53_KO BMSC is multipotent and has the capacity to generate tumors with features of chondroblastic OS. In addition, cFOS-induced chondrogenesis of p53_KO BMSCs does not require host cells and is mainly cell autonomous.
Figure 3.

cFOS Enhances Chondrogenic Differentiation of p53_KO BMSCs In Vitro and In Vivo
(A) Alcian blue staining of in vitro chondroblastic differentiation of p53_KO BMSCs single clone #1 with or without cFos overexpression. Scale bars, 200 μm.
(B) Alizarin red staining of in vitro osteogenic differentiation of p53_KO BMSCs single clone #1 with or without cFos overexpression. Scale bars, 1 mm.
(C) Oil red O staining of in vitro adipogenic differentiation of p53_KO BMSCs single clone #1 with or without cFos overexpression. Scale bars, 100 μm.
(D) H&E section staining of a tumor generated from p53_KO BMSCs single cell-derived clone #1 with cFos overexpression.
(E) Masson's trichrome staining of the sections of tumor generated from p53_KO BMSCs single cell-derived clone #1 with cFos overexpression.
(F) Alcian blue staining of the sections of tumor generated from p53_KO BMSCs single cell-derived clone #1 with cFos overexpression.
Scale bars in (D) to (F) represent 2 mm for inlets and 200 μm for large microscopic views. See also Figure S3.
Pathways Regulated by cFOS in BMSCs
To identify pathways that are regulated by cFOS in p53_KO BMSCs, we performed RNA sequencing (RNA-seq) using p53_KO BMSCs transduced with retroviruses expressing luciferase (Luc) or cFos. In total, 449 transcripts were determined to be cFos-regulated transcripts (a p value less than 0.01 and fold change larger than 1.5) (Table S3 and Figure 4A). To investigate whether cFOS directly regulates these transcripts, we also performed chromatin immunoprecipitation sequencing (ChIP-seq) and assigned cFos ChIP-seq peaks to a transcript if the peak located within 25 kb away from a transcript (He et al., 2015, Li et al., 2012, Shin et al., 2016). Among the 31,099 annotated transcripts in the mouse genome (build mm9), 8,635 transcripts had at least one associated cFOS peak, and these transcripts were defined as cFOS bound. After overlapping cFos-regulated transcripts (449) and cFOS-bound transcripts (8,635), we found that 275 transcripts were directly regulated by cFOS (Table S3 and Figure 4B). We then focused on these 275 cFOS direct targets and subjected them to pathway analysis, which identified several cancer-related pathways, such as the focal adhesion pathway and the MAPK signaling pathway (Figure 4C).
Figure 4.

Direct Targets of cFOS
(A) Hierarchy clustering of differentially expressed transcripts (by RNA-seq, fold change >1.5, p < 0.01) in p53_KO BMSCs transduced with retroviruses expressing luciferase (LUC) or cFOS.
(B) Venn diagram of cFOS-regulated transcripts and cFOS-bound transcripts to show direct targets of cFOS.
(C) Pathways enriched in the 275 transcripts that are directly regulated by cFOS in p53_KO BMSCs.
cFOS Induces the Expression of Sox9
These identified cFOS-regulated pathways explained the tumorigenic ability of cFOS. However, they did not shed light on the chondrogenic induction by cFOS. This role of cFOS in chondrogenesis does not appear to be associated with cell proliferation because cFOS does not affect the cell proliferation of p53_KO BMSCs (Figure S4A). To gain insight into cFOS-regulated lineage choice, we inspected the 275 direct targets of cFOS and found that Sox9 was within the list (Table S3 and Figure 5A). Sox9 is a master regulator of cartilage formation (Bi et al., 1999). The Sox9 mRNA was induced 2- to 3-fold by cFOS based on RNA-seq and real-time PCR, respectively (Figures 5B and 5C). The SOX9 protein was also induced (Figure 5D). To further validate that Sox9 is a cFOS target, we examined the promoter of Sox9 and detected two putative consensus binding motifs of cFOS (Figure 5E; Fleming et al., 2013). We cloned this promoter and performed luciferase assay. cFOS induced the luciferase activity driven by this promoter, further demonstrating that Sox9 is a direct target of cFOS (Figure 5F). Using site-directed mutagenesis, we modified these two putative motifs of cFOS binding (Figure 5E). Both sites were involved in the regulation of Sox9 by cFOS, since mutation of either or both sites decreased the induction of luciferase activity (Figure 5G), although disruption of both sites had a more profound effect than either single site.
Figure 5.

Sox9 Is a Direct Target of cFOS
(A) ChIP-seq (upper) shows the binding of cFOS in the promoter of the Sox9 locus. The black box shows the identified binding site for cFOS. RNA-seq shows the induction of Sox9 by cFOS. Gene structure of Sox9 is shown. Arrow indicates the direction of transcription. RNA-seq (lower) shows the expression of Sox9.
(B) RNA-seq showing the expression of Sox9. Fragments per kilobase per million reads (FPKM) of Sox9 in p53_KO BMSCs transduced with retroviruses expressing luciferase and cFOS. Error bars denote SEM, n = 3 independent experiments. ∗∗p < 0.01.
(C) Real-time PCR to measure mRNA levels of Sox9, Runx2, and Pparg in p53_KO BMSCs with or without cFOS overexpression. Error bars denote SEM, n = 3 independent experiments. ∗0.01 < p < 0.05, ∗∗p < 0.01.
(D) Western blot analyses using cFOS, SOX9, RUNX2, PPARγ, and β-ACTIN antibodies.
(E) Schematics of the wild-type and mutated Sox9 promoter. Putative cFOS binding sites are shown in bold. Red color shows the mutated nucleotides of the promoter of Sox9 using the QuikChange (QC) method. The numbers after “QC” indicate the mutated position of putative cFos targeting sites.
(F) Fold induction of luciferase activity of Sox9 promoter co-transfected with vector expressing cFOS versus empty vector control. Value of empty vector was set to 1. Error bars denote SEM, n = 3 independent experiments. ∗∗p < 0.01.
(G) Fold induction of luciferase activity using wild-type and various mutated versions of promoter (pr) of Sox9. Error bars denote SEM, n = 3 independent experiments. ∗∗p < 0.01, ∗p < 0.05.
See also Figure S4.
Sox9 Mediates cFOS-Driven Chondrogenesis in Chondroblastic OS
To test whether Sox9 mediates cFOS-driven chondrogenesis, we used two lentiviral short hairpin RNAs (shRNAs) to reduce the levels of Sox9 in cFOS-transduced p53_KO BMSCs (Figure 6A). In vitro chondrogenesis assay showed that these two shRNAs decreased the chondrogenesis of cFOS-transduced p53_KO BMSCs (Figure 6B). We then transplanted control cells (shRNA against luciferase) and knockdown cells into NSG mice to form tumors. Knockdown of Sox9 in cFOS-transduced p53_KO BMSCs decreased the percentage of cartilage in tumors (Figures 6C and 6D). All the control cells (p53_KO BMSCs + cFos + shLuc) formed chondroblastic OS while Sox9 knockdown cells (p53_KO BMSCs + cFos + shSox9) had decreased cartilage formation (Figures 6C and 6D). Thus, these results showed that Sox9 is one of the downstream mediators of cFOS in the induction of chondroblastic OS. Of note, Sox9 knockdown did not change the incidence of tumor formation and tumor growth of cFOS-transduced p53_KO BMSCs, suggesting that cFOS has two roles in osteosarcomagenesis: a cartilage induction role mediated by Sox9 and a tumor-promoting role.
Figure 6.

SOX9 Mediates cFOS-Driven Chondrogenesis In Vitro and In Vivo
(A) Western blot analyses of Sox9 knockdown in p53_KO BMSCs with cFOS overexpression.
(B) In vitro chondrogenic differentiation of p53_KO BMSCs expressing cFOS transduced with lentiviruses expressing shRNA against luciferase (shLuc) or Sox9 (shSox9_#1 and shSox9_#3). Scale bars represent 300 μm for inlets and 200 μm for large microscopic views.
(C) H&E staining and Alcian blue staining showing the reduction of cartilage in tumors generated from p53_KO BMSCs + cFos transduced with shSox9_#1 or shSox9_#3 in comparison with shLuc control. Scale bars represent 2 mm for inlets and 50 μm for large microscopic views.
(D) Quantitative analyses of the percentage of cartilage area of tumors. Upper panel: a representative of pseudo-color mark-up of cartilage area within a tumor, as acquired using quantitative pattern recognition image analysis. Cart, cartilage. Lower panel: statistical analyses of percentage of cartilage area. n = 8 mice; ∗∗p < 0.01. Scale bars, 50 μm.
To test whether Sox9 is an oncogene, we overexpressed Sox9 in p53_KO BMSCs (Figure S4B). The levels of Sox9 were higher than cFOS-induced Sox9 (Figure S4B). However, anchorage-independent growth assay showed that Sox9 did not transform p53_KO BMSCs in vitro (Figures S4C and S4D). These results together demonstrate that Sox9, by itself, has no capacity to transform p53_KO BMSCs. Thus, Sox9 is unlikely a bona fide oncogene.
cFOS and SOX9 Are Co-expressed in Human Chondroblastic OS Cells
To explore the relevance of the cFOS-SOX9 connection in human OS tumor samples, we performed Alcian blue staining, and cFOS and SOX9 immunohistochemistry (IHC) using a human OS tumor microarray (TMA). We found that cFOS and SOX9 were both located in the nuclei of human chondroblastic OS (Figures 7A, S5A, and S5B). We also detected signal of cFOS and SOX9 in normal human chondrocytes, but the signals of both proteins were much lower than those in chondroblastic OS. Although we detected SOX9 signal in other subtypes of OS, cFOS staining was either negative or cytoplasmic (Figure S5C). These results suggest that the regulation of SOX9 by cFOS may be conserved in human chondroblastic OS. Of note, our result does not exclude the possibility that other factors also regulate the expression of SOX9 in human OS.
Figure 7.

cFOS and SOX9 Are Co-expressed in Human Chondroblastic Osteosarcomas
(A) Alcian blue staining of two representative tumors (tumor 1 and tumor 2) from human OS TMA and normal human chondrocytes.
(B) cFOS IHC staining of two representative chondroblastic OS from human OS TMA and normal human chondrocytes.
(C) SOX9 IHC staining of two representative chondroblastic OS from human OS TMA and normal human chondrocytes.
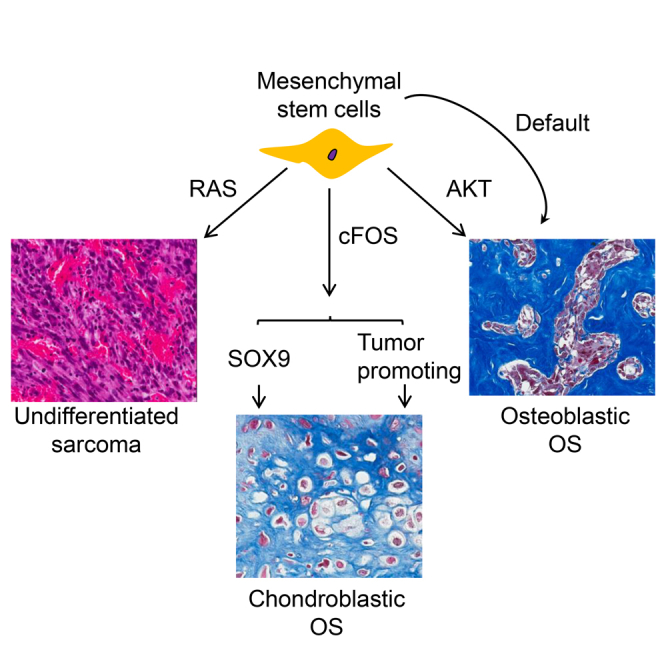
(D) A model of oncogene-driven lineage determination of p53_KO BMSCs.
Scale bars represent 200 μm for both the insets and larger microscopic views. See also Figure S5.
Discussion
The Roles of p53 in BMSCs and Osteosarcomagenesis
The effect of p53 loss in MSCs on osteosarcomagenesis has been reported (Berman et al., 2008, Lin et al., 2009, Rodriguez et al., 2009, Rubio et al., 2010, Tataria et al., 2006, Walkley et al., 2008). In the ex vivo studies, the tissues from which MSCs are derived affect the tumor types. For example, p53 loss in MSCs derived from adipose tissue generated fibrosarcoma or leiomyosarcoma in immunocompromised mice (Rodriguez et al., 2009, Rubio et al., 2010). This is probably due to the epigenetic “memory” of MSCs. In the in vivo studies, the p53 gene was conditionally deleted using Prx1-driven Cre recombinase. Mice developed osteosarcoma and fibrosarcoma, as well as other types of sarcomas (Berman et al., 2008, Lin et al., 2009, Walkley et al., 2008). Because Prx1 is expressed in the mesenchymal tissue during embryonic stage, the cell of origin of these sarcomas is still unknown.
Within these previously reported models, the latency of osteosarcoma formation ranges from 4 to 12 months. Our sarcoma models are based on exogenously expressed oncogenes in isolated BMSCs, which give rise to tumors within 1–2 months, depending on site of injection and the number of transplanted cells (Tables S1 and S2). In addition, our models use p53_KO MSCs derived from mouse bone marrow, and therefore are more relevant to osteosarcomagenesis than MSCs isolated from other tissues. Overall, our models serve as useful complementary approaches to previously reported models.
The Role of cFOS in Chondrocyte Development and Chondroblastic Osteosarcomagenesis
cFOS was discovered at an insertion site for a murine retrovirus that promotes OS formation (Van Beveren et al., 1983). In transgenic and knockout mouse models, cFOS has been shown to play an important role in cartilage formation (Wang et al., 1991, Wang et al., 1992). In human OS, upregulation of cFOS has been documented (Wu et al., 1990). However, it is not due to cFOS mutation or amplification, suggesting that transcriptional or epigenetic upregulation may be involved (Chen et al., 2014, Perry et al., 2014). Thus, the role of cFOS in OS is conserved between human and mouse, although the mechanisms may differ. Similar to the observations in mouse models, we observed a role of cFOS in inducing chondrogenesis using BMSCs as a cellular model. Furthermore, we demonstrated that the cFos proto-oncogene connects to the chondrogenic differentiation program of BMSCs by inducing the transcription of Sox9 and that the cFOS-SOX9 axis is critical for the role of cFOS in the induction of chondroblastic OS (Figure 7D). Of note, it is very likely that cFOS is not the only transcription factor inducing the expression of Sox9, and other factors may also play a role in Sox9 expression.
Because Sox9 knockdown changes only the subtype but not the tumorigenicity of cFOS-transduced p53_KO BMSCs, our results reveal that cFOS has at least two roles in osteosarcomagenesis: an oncogenic role in promoting tumor formation and a role in the induction of cartilaginous tumor mediated by SOX9. The dual roles of cFOS in OS are reminiscent of the dual roles of p53 in BMSCs, in which p53 not only prevents tumorigenesis but also affects the lineage choice of these cells (He et al., 2015). What, then, is the relationship between the roles of cFOS in development and cancer? During normal chondrocyte development the action of cFOS may be transient, which may avoid the tumorigenic potential of cFOS. However, when cFOS is consistently upregulated, as in our models and some tumors, cartilaginous tumors form because of the consistent upregulation of cancer-related signaling regulated by cFOS (Figure 4C).
It is worth noting that osteoblastic OS, chondroblastic OS, and fibroblastic OS have a similar prognosis. Although chondroblastic OS is resistant to chemotherapy, this negative effect is offset by its good outcome. Nonetheless, our study provides an explanation to BMSCs' capacity of being the cells of origin of many subtypes of OS and undifferentiated sarcoma, and merits future studies to improve the clinical outcomes of OS.
In our chondroblastic OS model, p53 was deleted in BMSCs prior to the introduction of exogenous cFos. However, in the human setting it is still elusive whether these p53/cFOS/SOX9 sequential events are conserved.
The Oncogenic Signal Determines the Subtypes of OS
One of the long-standing questions is how BMSCs can be the cells of origin of different types of sarcomas. One possibility is that BMSCs are a mixture of committed cell types and that each of these cell types can develop into different subtypes of sarcoma. Using single clones of p53_KO BMSCs, our study supports an alternative explanation: p53_KO BMSCs are multipotent in the generation of different types of sarcomas and oncogenic signals dictate the tumor types. Although the default tumor type generated from p53_KO BMSCs is osteoblastic OS, oncogenic signals can override this default program and direct these cells to develop into various lineage tumors. The plasticity of generating different tumor types of BMSCs probably is tightly associated with the multipotency of these cells during normal development. Therefore, these results provide an explanation for how BMSCs can serve as the cells of origin for many different types of sarcomas.
The PI3K-AKT and RAS signaling pathways are two of the most common oncogenic signals in human cancers (Shaw and Cantley, 2006). In particular, the AKT pathway was aberrantly upregulated in OS through PTEN (Phosphatase and tensin homolog) deletions or PI3K mutations (Chen et al., 2014, Perry et al., 2014). Interestingly, the RAS signaling pathway regulates the PI3K pathway as well as the RALGDS pathway, RAF pathway, and PLC-PKC pathway (Downward, 2003). Thus, it is intriguing to speculate that the RALGDS pathway, RAF pathway, and PLC-PKC pathway mediate the role of RAS in the induction of undifferentiated sarcomas.
RAS is an oncogene in sarcomas but its point mutations or amplifications in OS are rare (Chen et al., 2014, Perry et al., 2014). Since both k-RAS and h-RAS reprogram p53_KO BMSCs into undifferentiated sarcomas, our results are consistent with the relative infrequency of Ras mutations in human osteoblastic OS.
Cells of Origin of OS
Understanding the cell of origin could facilitate the development of novel therapeutic strategies for OS. In various mouse models, it has been shown that committed pre-osteoblasts, osteoblasts, and uncommitted embryonic mesenchymal progenitors are the cells of origin of OS (Calo et al., 2010, Lin et al., 2009, Walkley et al., 2008). All of these mouse models use a tissue- or cell-type-specific Cre recombinase to conditionally delete p53 and/or RB1. Because of the intrinsic heterogeneous expression of the Cre transgene, the “true” cell of origin of OS is still elusive. Using previously characterized BMSCs isolated from the bone marrow from adult mice (He et al., 2015), our results demonstrate that adult BMSCs indeed have the ability of developing into OS in the transplantation assay. Of note, our results do not exclude the possibility that other cell types, such as osteoblasts, can also serve as cells of origin of OS. Since oncogenes can determine the tumor types, it is likely that OS may have several cells of origin depending on the oncogenic signals. Nonetheless, in the presence of persistent cFos upregulation, BMSCs become the origin of chondroblastic OS.
Experimental Procedures
Mouse Strains
p53+/+ and p53−/− mice were described previously (Donehower et al., 1992) and housed in the mouse facility of the National Cancer Institute. The immunocompromised NSG (also known as NOD-scid-gamma, strain #005557) mice were purchased from the Jackson Laboratory. Mice were maintained under the guidelines of the Institutional Animal Care and Use Committee-approved protocols of the National Cancer Institute and National Heart, Lung, and Blood Institute.
Intramuscular Injection and Intrabone Marrow Injection for Tumorigenesis
Ten million p53_KO, p53_KO+ cFos BMSCs or 1 million human OS cells were resuspended in medium + 25 mM HEPES, mixed with same volume of Matrigel (BD Biosciences, catalog #356231), and injected into hindlimb muscle of immunocompromised NSG mice. Tumors were harvested when they reached about 1.5 cm in diameter or the mice started to show signs of pain. The tumors were dissected, cut into 5-mm thick portions, fixed in 10% formalin overnight, and paraffin embedded, and 5-μm sections were placed onto positively charged slides.
For intrabone marrow injection, the right tibia was gently drilled with a 26-gauge needle through the patellar tendon. One million p53_KO BMSCs transduced with an oncogene was injected into the drilled tibia through the patellar hole using a Hamilton microsyringe.
Isolating and Culturing of Mouse BMSCs
The procedures of isolating and characterizing mouse BMSCs were mostly described previously (Anjos-Afonso and Bonnet, 2008, He et al., 2015). In brief, mouse BMSCs were isolated from 6- to 10-week-old adult mice using the attachment approach followed by double-negative cell sorting of CD11b and CD45. Sorted mouse BMSCs were cultured in a proprietary BMSC medium (STEMCELL Technologies, #05512). Because BMSCs are prone to spontaneous transformation, we only used BMSCs with passages less than or equal to 8 at a split ratio of 1:10.
In Vitro Osteogenic Differentiation and Adipogenic Differentiation
Osteogenic differentiation and adipogenic differentiation of mouse BMSCs were performed as we previously described (He et al., 2015).
Chondrogenic Differentiation and Alcian Blue Staining
Alginate solution (1.5% [w/v]) in 150 mM NaCl was made 2 hr before cells were ready. Ten million BMSCs were resuspended in 900 μL of alginate solution and the alginate/cell suspension was rapidly dispensed into a 100-mM CaCl2 solution while stirring with a 3-mL syringe fitted with a 27-gauge needle to form chondrogenic microbeads. The chondrogenic microbeads were washed with PBS and grown in four wells of a 24-well plate under hypoxia condition (5% oxygen) in chondrogenic induction medium, which contains DMEM high glucose plus 6.25 μg/mL insulin, 6.25 μg/mL transferrin, 6.25 ng/mL sodium selenite, 5.33 μg/mL linoleic acid, 1.25 mg/mL BSA, 100 nM dexamethasone, 10 ng/mL transforming growth factor β3, and 50 μM ascorbic acid. The medium was changed every 3 days for up to 4 weeks. After differentiation, the beads were fixed for 24 hr in 4% paraformaldehyde, 100 mM sodium cacodylate, and 50 mM BaCl2. The fixed alginate beads were further processed for paraffin embedding and sectioning using standard methods (HistoServ). The deparaffinized and hydrated slides were stained with 0.5% Alcian blue in 0.1 N HCl for 30 min, washed with water, stained with nuclear fast red for 10 min, and rinsed with water. After staining, these slides were dehydrated through graded alcohols and coverslipped with resinous mounting medium.
Human OS Cell Lines
SAOS2 cells were originally purchased from ATCC. These cells were grown in DMEM with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin (100 units/mL). Hu09-M112 was generously provided by Dr. Jun Yokota (Biology Division, National Cancer Center Research Institute, Japan) and grown in RPMI-1640 with 10% FBS and 1% penicillin-streptomycin (100 units/mL). Puromycin (2 μg/mL) was added to the medium 24 hr after virus infection for selecting positive clones. The derived cells were maintained in medium with 1 μg/mL puromycin after selection.
cFos Overexpression in BMSCs
Mouse cFos cDNA was cloned into the pENTR/D-TOPO vector using sense primer 5′-CAC CAA GCT TAT GAT GTT CTC GGG TTT CAA CGC-3′ and antisense primer 5′-CCT TGC GGC CGC TCA CAG GGC CAG CAG CGT GGG-3′, and confirmed by sequencing. The cDNA was then subcloned into retrovirus expressing vector pMXs_puro using gateway recombination. Two micrograms of pMXs_puro_cFos(m) plasmid was transfected into Plat-E cells for making retrovirus. The medium containing retrovirus was collected 48 hr and 72 hr after transfection. One milliliter of supernatant containing virus from each collection was used to infect 0.1 million BMSCs in each well of a 6-well plate at same time of virus collection. Puromycin (4 μg/mL) was added into the medium 24 hr after second infection for selection. After selection, the cells were maintained in medium with 2 μg/mL puromycin.
Western Blot
Procedures of western blotting were described previously (He et al., 2015, Li et al., 2012, Li et al., 2015, Shin et al., 2016, Zhang et al., 2013). Antibodies were 1:1,000 cFOS (Santa Cruz Biotechnology, #sc-7202), 1:1,000 AKT (Cell Signaling Technology, #4691P), 1:1,000 RAS (Cell Signaling, #3339S), 1:1,000 SOX9 (Millipore, #AB5535), 1:1,000 RUNX2 (Cell Signaling, #8486), 1:500 PPARγ (Cell Signaling, #2443), 1:2,000 α-tubulin (Sigma, #T9026).
Reporter Assay
The putative promoter region was amplified (see Table S4 file for details) and cloned into pGL4.20-luc2-MCS2, which expressed Firefly luciferase (Promega). The reporter containing Sox9 promoter was co-expressed together with a reporter expressing Renilla luciferase (internal control) plus an empty vector or a vector expressing cFos in p53_KO BMSCs using Turbo DNAfectin 3000 (EGFIE). Cells were lysed and luciferase activity was measured as previously described (He et al., 2015, Li et al., 2012).
RNA-Seq, ChIP-Seq, and Pathway Analyses
For RNA-seq, 1 μg of total RNA for each sample was sent to the Next Generation Sequencing Facility at the Center for Cancer Research at the National Cancer Institute followed by rRNA removal, size selection, reverse transcription, cluster generation, and high-throughput sequencing on the HiSeq 2500 or NextSeq 500 platforms. For ChIP-seq, we followed our previous procedures without major modifications (He et al., 2015, Li et al., 2012, Shin et al., 2016). Pathway analyses using DAVID Bioinformatics Resources were previously described (Huang da et al., 2009, Lee et al., 2010).
shRNA Knockdown of Sox9
The sequences of shRNAs for knocking down mouse Sox9 were: shSox9(m)_#1: 5′-CCG GGC GAC GTC ATC TCC AAC ATT GCT CGA GCA ATG TTG GAG ATG ACG TCG CTT TTT-3′; shSox9(m)_#3: 5′-CCG GAG ACT CAC ATC TCT CCT AAT GCT CGA GCA TTA GGA GAG ATG TGA GTC TTT TTT-3′. shSox9(h) were cloned into pLKO.1_puro and shSox9(m) were cloned into pLKO.1_hygro lentivirus expressing vectors. Puromycin (2 μg/mL) and hygromycin B (300 μg/mL) were added into medium for selection of the positive clones after virus infection. Puromycin (1 μg/mL) and hygromycin B (100 μg/mL) were used in medium for maintaining the knockdown cells.
Quantitative Analysis of Cartilaginous Differentiation in Paraffin-Embedded, Sectioned Xenografted Tumors
Digital image data files were created for each tumor specimen by optically scanning individual H&E-stained tissue section slides using the Aperio AT2 digital slide scanner (Leica Biosystems) at a resolution of 0.49 μm/pixel. The whole slide images were imported into HALO imaging software (Indica Labs) where tumor areas were manually segmented using SnapTo and flood tools. In addition, non-tumor tissue features were excluded from regions of interest (ROI) on a case-by-case basis from each tissue section to avoid analysis of non-tumor tissue. To identify tumor regions having cartilaginous differentiation, we created a pattern-recognition feature classification image analysis algorithm using the HALO pattern classifier tool to delineate cartilage, in contrast to osteoid and other soft tissue within H&E-stained tissue sections. This algorithm was applied to all segmented ROI within all the H&E-stained specimens to quantify evidence of cartilaginous differentiation. Acceptance of algorithm parameters, post image-processing output, and quality assurance steps conformed to that described previously (Webster et al., 2012).
Masson's Trichrome Staining
NovaUltra Masson’s trichrome stain kit from IHC World was used to stain the collagen in OS sections. In brief, after deparaffinizing and hydrating, the tumor sections were stained in Weigert's iron hematoxylin solution for 10 min, rinsed in tap water for 10 min, and washed in distilled water. The sections were then stained in Biebrich Scarlet-Acid Fuchsin Solution for 10 min, washed in distilled water, differentiated in phosphotungstic-phosphomolybdic acid solution for 10 min, and directly stained in aniline blue solution for 5 min. Afterward, the sections were briefly washed in distilled water, differentiated in acetic acid solution for 5 min, washed again in distilled water, dehydrated very quickly through 95% alcohol and two changes of 100% alcohol for 1 min each, cleared in two changes of xylene for 5 min, and coverslipped with resinous mounting medium.
Tumor Microarray of OS
The TMA, as described previously (Shin et al., 2016), was generated using 88 tumors collected from 88 patients with OS under an Institutional Review Board (IRB)-approved protocol of the Montefiore Medical Center. For Immunohistochemistry (IHC), formalin-fixed paraffin-embedded slides were deparaffinized in xylene, 100% ethanol, and 95% ethanol. Antigens were retrieved by boiling slides in 10 mM sodium citrate for 10 min. After cooling, slides were treated with 3% H2O2 for 10 min followed by washing with PBS + 0.1% Tween 20 and blocked with serum. Slides were then incubated with 1:200 cFOS (Santa Cruz) or 1:200 SOX9 antibody (Millipore) overnight at 4°, washed three times with PBS, and incubated with biotinylated goat anti-rabbit immunoglobulin G secondary antibody (Vectastain ABC Kit) for 1 hr at room temperature. After washing with biotin-avidin solution for 30 min at room temperature, slides were rinsed with PBS three times, and DAB solution was added to allow color development for 2–5 min. The procedures of using this TMA for IHC were approved under an exemption of IRB (#12806) by the Office of Human Subjects of Research at the NIH. Normal human chondrocyte tissue was purchased from US Biomax.
Oligo Sequences
Oligo sequences are shown in Table S4.
Author Contributions
B.M., R.M.S., and J.H. designed the experiments; Y.H., W.Z., M.H.S., J.G., C. L., W.D., S.B.H., S.J., and E.M. performed experiments and analyses of the data. Y.H. and J.H. wrote the manuscript, which everyone read and commented on.
Acknowledgments
This work was supported by the National Cancer Institute, USA, intramural grant 1ZIABC011504-02 to J.H. We thank Stuart Yuspa for useful suggestions, Bao Tran's Next Generation Sequencing Facility at the Center for Cancer Research (CCR) for RNA-seq and ChIP-seq, and the Office of Science and Technology Resources (OSTR) at CCR, NCI, for partial funding support.
Published: May 25, 2017
Footnotes
Supplemental Information includes five figures and four tables and can be found with this article online at http://dx.doi.org/10.1016/j.stemcr.2017.04.029.
Accession Numbers
Sequencing data are available from the GEO database with accession number GEO: GSE89015.
Supplemental Information
References
- Anjos-Afonso F., Bonnet D. Isolation, culture, and differentiation potential of mouse marrow stromal cells. Curr. Protoc. Stem Cell Biol. 2008;Chapter 2:Unit 2B 3. doi: 10.1002/9780470151808.sc02b03s7. [DOI] [PubMed] [Google Scholar]
- Berman S.D., Calo E., Landman A.S., Danielian P.S., Miller E.S., West J.C., Fonhoue B.D., Caron A., Bronson R., Bouxsein M.L. Metastatic osteosarcoma induced by inactivation of Rb and p53 in the osteoblast lineage. Proc. Natl. Acad. Sci. USA. 2008;105:11851–11856. doi: 10.1073/pnas.0805462105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bi W., Deng J.M., Zhang Z., Behringer R.R., de Crombrugghe B. Sox9 is required for cartilage formation. Nat. Genet. 1999;22:85–89. doi: 10.1038/8792. [DOI] [PubMed] [Google Scholar]
- Bianco P., Robey P.G., Simmons P.J. Mesenchymal stem cells: revisiting history, concepts, and assays. Cell Stem Cell. 2008;2:313–319. doi: 10.1016/j.stem.2008.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calo E., Quintero-Estades J.A., Danielian P.S., Nedelcu S., Berman S.D., Lees J.A. Rb regulates fate choice and lineage commitment in vivo. Nature. 2010;466:1110–1114. doi: 10.1038/nature09264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X., Bahrami A., Pappo A., Easton J., Dalton J., Hedlund E., Ellison D., Shurtleff S., Wu G., Wei L. Recurrent somatic structural variations contribute to tumorigenesis in pediatric osteosarcoma. Cell Rep. 2014;7:104–112. doi: 10.1016/j.celrep.2014.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donehower L.A., Harvey M., Slagle B.L., McArthur M.J., Montgomery C.A., Jr., Butel J.S., Bradley A. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature. 1992;356:215–221. doi: 10.1038/356215a0. [DOI] [PubMed] [Google Scholar]
- Downward J. Targeting RAS signalling pathways in cancer therapy. Nat. Rev. Cancer. 2003;3:11–22. doi: 10.1038/nrc969. [DOI] [PubMed] [Google Scholar]
- Fleming J.D., Pavesi G., Benatti P., Imbriano C., Mantovani R., Struhl K. NF-Y coassociates with FOS at promoters, enhancers, repetitive elements, and inactive chromatin regions, and is stereo-positioned with growth-controlling transcription factors. Genome Res. 2013;23:1195–1209. doi: 10.1101/gr.148080.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fletcher C.D.M., Bridge J.A., Hogendoorn P., Mertens F. Fourth Edition. IARC WHO Classification of Tumours; 2013. WHO Classification of Tumours of Soft Tissue and Bone. [Google Scholar]
- He Y., de Castro L.F., Shin M.H., Dubois W., Yang H.H., Jiang S., Mishra P.J., Ren L., Gou H., Lal A. p53 loss increases the osteogenic differentiation of bone marrow stromal cells. Stem Cells. 2015;33:1304–1319. doi: 10.1002/stem.1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang da W., Sherman B.T., Lempicki R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- Kfoury Y., Scadden D.T. Mesenchymal cell contributions to the stem cell niche. Cell Stem Cell. 2015;16:239–253. doi: 10.1016/j.stem.2015.02.019. [DOI] [PubMed] [Google Scholar]
- Lang G.A., Iwakuma T., Suh Y.A., Liu G., Rao V.A., Parant J.M., Valentin-Vega Y.A., Terzian T., Caldwell L.C., Strong L.C. Gain of function of a p53 hot spot mutation in a mouse model of Li-Fraumeni syndrome. Cell. 2004;119:861–872. doi: 10.1016/j.cell.2004.11.006. [DOI] [PubMed] [Google Scholar]
- Lee K.H., Li M., Michalowski A.M., Zhang X., Liao H., Chen L., Xu Y., Wu X., Huang J. A genomewide study identifies the Wnt signaling pathway as a major target of p53 in murine embryonic stem cells. Proc. Natl. Acad. Sci. USA. 2010;107:69–74. doi: 10.1073/pnas.0909734107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M., He Y., Dubois W., Wu X., Shi J., Huang J. Distinct regulatory mechanisms and functions for p53-activated and p53-repressed DNA damage response genes in embryonic stem cells. Mol. Cell. 2012;46:30–42. doi: 10.1016/j.molcel.2012.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M., Gou H., Tripathi B.K., Huang J., Jiang S., Dubois W., Waybright T., Lei M., Shi J., Zhou M. An apela RNA-containing negative feedback loop regulates p53-mediated apoptosis in embryonic stem Cells. Cell Stem Cell. 2015;16:669–683. doi: 10.1016/j.stem.2015.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin P.P., Pandey M.K., Jin F., Raymond A.K., Akiyama H., Lozano G. Targeted mutation of p53 and Rb in mesenchymal cells of the limb bud produces sarcomas in mice. Carcinogenesis. 2009;30:1789–1795. doi: 10.1093/carcin/bgp180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirabello L., Troisi R.J., Savage S.A. Osteosarcoma incidence and survival rates from 1973 to 2004: data from the surveillance, epidemiology, and end results program. Cancer. 2009;115:1531–1543. doi: 10.1002/cncr.24121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olive K.P., Tuveson D.A., Ruhe Z.C., Yin B., Willis N.A., Bronson R.T., Crowley D., Jacks T. Mutant p53 gain of function in two mouse models of Li-Fraumeni syndrome. Cell. 2004;119:847–860. doi: 10.1016/j.cell.2004.11.004. [DOI] [PubMed] [Google Scholar]
- Perry J.A., Kiezun A., Tonzi P., Van Allen E.M., Carter S.L., Baca S.C., Cowley G.S., Bhatt A.S., Rheinbay E., Pedamallu C.S. Complementary genomic approaches highlight the PI3K/mTOR pathway as a common vulnerability in osteosarcoma. Proc. Natl. Acad. Sci. USA. 2014;111:E5564–E5573. doi: 10.1073/pnas.1419260111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez R., Rubio R., Masip M., Catalina P., Nieto A., de la Cueva T., Arriero M., San Martin N., de la Cueva E., Balomenos D. Loss of p53 induces tumorigenesis in p21-deficient mesenchymal stem cells. Neoplasia. 2009;11:397–407. doi: 10.1593/neo.81620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubio R., Garcia-Castro J., Gutierrez-Aranda I., Paramio J., Santos M., Catalina P., Leone P.E., Menendez P., Rodriguez R. Deficiency in p53 but not retinoblastoma induces the transformation of mesenchymal stem cells in vitro and initiates leiomyosarcoma in vivo. Cancer Res. 2010;70:4185–4194. doi: 10.1158/0008-5472.CAN-09-4640. [DOI] [PubMed] [Google Scholar]
- Sacchetti B., Funari A., Remoli C., Giannicola G., Kogler G., Liedtke S., Cossu G., Serafini M., Sampaolesi M., Tagliafico E. No identical “mesenchymal stem cells” at different times and sites: human committed progenitors of distinct origin and differentiation potential are incorporated as adventitial cells in microvessels. Stem Cell Reports. 2016;6:897–913. doi: 10.1016/j.stemcr.2016.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw R.J., Cantley L.C. Ras, PI(3)K and mTOR signalling controls tumour cell growth. Nature. 2006;441:424–430. doi: 10.1038/nature04869. [DOI] [PubMed] [Google Scholar]
- Shih T.Y., Papageorge A.G., Stokes P.E., Weeks M.O., Scolnick E.M. Guanine nucleotide-binding and autophosphorylating activities associated with the p21src protein of Harvey murine sarcoma virus. Nature. 1980;287:686–691. doi: 10.1038/287686a0. [DOI] [PubMed] [Google Scholar]
- Shin M.H., He Y., Marrogi E., Piperdi S., Ren L., Khanna C., Gorlick R., Liu C., Huang J. A RUNX2-mediated epigenetic regulation of the survival of p53 defective cancer cells. PLoS Genet. 2016;12:e1005884. doi: 10.1371/journal.pgen.1005884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tataria M., Quarto N., Longaker M.T., Sylvester K.G. Absence of the p53 tumor suppressor gene promotes osteogenesis in mesenchymal stem cells. J. Pediatr. Surg. 2006;41:624–632. doi: 10.1016/j.jpedsurg.2005.12.001. discussion 624–632. [DOI] [PubMed] [Google Scholar]
- Tirode F., Laud-Duval K., Prieur A., Delorme B., Charbord P., Delattre O. Mesenchymal stem cell features of Ewing tumors. Cancer Cell. 2007;11:421–429. doi: 10.1016/j.ccr.2007.02.027. [DOI] [PubMed] [Google Scholar]
- Van Beveren C., van Straaten F., Curran T., Muller R., Verma I.M. Analysis of FBJ-MuSV provirus and c-fos (mouse) gene reveals that viral and cellular fos gene products have different carboxy termini. Cell. 1983;32:1241–1255. doi: 10.1016/0092-8674(83)90306-9. [DOI] [PubMed] [Google Scholar]
- Velletri T., Xie N., Wang Y., Huang Y., Yang Q., Chen X., Chen Q., Shou P., Gan Y., Cao G. P53 functional abnormality in mesenchymal stem cells promotes osteosarcoma development. Cell Death Dis. 2016;7:e2015. doi: 10.1038/cddis.2015.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walkley C.R., Qudsi R., Sankaran V.G., Perry J.A., Gostissa M., Roth S.I., Rodda S.J., Snay E., Dunning P., Fahey F.H. Conditional mouse osteosarcoma, dependent on p53 loss and potentiated by loss of Rb, mimics the human disease. Genes Dev. 2008;22:1662–1676. doi: 10.1101/gad.1656808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z.Q., Grigoriadis A.E., Mohle-Steinlein U., Wagner E.F. A novel target cell for c-fos-induced oncogenesis: development of chondrogenic tumours in embryonic stem cell chimeras. EMBO J. 1991;10:2437–2450. doi: 10.1002/j.1460-2075.1991.tb07783.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z.Q., Ovitt C., Grigoriadis A.E., Mohle-Steinlein U., Ruther U., Wagner E.F. Bone and haematopoietic defects in mice lacking c-fos. Nature. 1992;360:741–745. doi: 10.1038/360741a0. [DOI] [PubMed] [Google Scholar]
- Webster J.D., Michalowski A.M., Dwyer J.E., Corps K.N., Wei B.R., Juopperi T., Hoover S.B., Simpson R.M. Investigation into diagnostic agreement using automated computer-assisted histopathology pattern recognition image analysis. J. Pathol. Inform. 2012;3:18. doi: 10.4103/2153-3539.95130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worthley D.L., Churchill M., Compton J.T., Tailor Y., Rao M., Si Y., Levin D., Schwartz M.G., Uygur A., Hayakawa Y. Gremlin 1 identifies a skeletal stem cell with bone, cartilage, and reticular stromal potential. Cell. 2015;160:269–284. doi: 10.1016/j.cell.2014.11.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J.X., Carpenter P.M., Gresens C., Keh R., Niman H., Morris J.W., Mercola D. The proto-oncogene c-fos is over-expressed in the majority of human osteosarcomas. Oncogene. 1990;5:989–1000. [PubMed] [Google Scholar]
- Xiao W., Mohseny A.B., Hogendoorn P.C., Cleton-Jansen A.M. Mesenchymal stem cell transformation and sarcoma genesis. Clin. Sarcoma Res. 2013;3:10. doi: 10.1186/2045-3329-3-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X., He Y., Lee K.H., Dubois W., Li Z., Wu X., Kovalchuk A., Zhang W., Huang J. Rap2b, a novel p53 target, regulates p53-mediated pro-survival function. Cell Cycle. 2013;12:1279–1291. doi: 10.4161/cc.24364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou B.O., Yue R., Murphy M.M., Peyer J.G., Morrison S.J. Leptin-receptor-expressing mesenchymal stromal cells represent the main source of bone formed by adult bone marrow. Cell Stem Cell. 2014;15:154–168. doi: 10.1016/j.stem.2014.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.