

Abstract
The phenomenon of functional selectivity, whereby a ligand preferentially directs the information output of a G-protein coupled receptor (GPCR) along (a) particular effector pathway(s) and away from others, has redefined traditional GPCR signaling paradigms to provide a new approach to structure-based drug design. The two principal cannabinoid receptors (CBRs) 1 and 2 belong to the class-A GPCR subfamily and are considered tenable therapeutic targets for several indications. Yet conventional orthosteric ligands (agonists, antagonists/inverse agonists) for these receptors have had very limited clinical utility due to their propensity to incite on-target adverse events. Chemically distinct classes of cannabinergic ligands exhibit signaling bias at CBRs toward individual subsets of signal transduction pathways. In this review, we discuss the known signaling pathways regulated by CBRs and examine the current evidence for functional selectivity at CBRs in response to endogenous and exogenous cannabinergic ligands as biased agonists. We further discuss the receptor and ligand structural features allowing for selective activation of CBR-dependent functional responses. The design and development of biased ligands may offer a pathway to therapeutic success for novel CBR-targeted drugs.
Keywords: GPCR, Biased agonism, Drug discovery, Effector Pathways
Graphical abstract
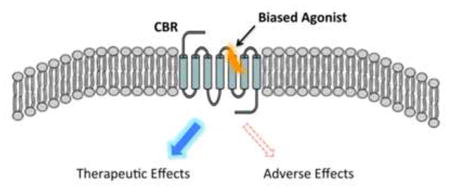
1. Pharmacological and drug-discovery implications of functional selectivity
According to the tenets of the traditional “two-state” model of G protein-coupled receptor (GPCR) function, a GPCR acts as a ligand-controlled “on-off” switch, eliciting, when activated by an agonist, a cascade of cellular signals and effects through specific transducers such as G proteins without any particular directionality to the ensuing information output [1]. Based upon data from an array of biophysical and biochemical studies, this outmoded paradigm has been elaborated to a so-called “multi-state” model that encompasses a variety of GPCR conformations along an activity continuum influenced by bound ligands, effector proteins, and other cellular molecules (Na+, lipids, etc.) [2,3]. Furthermore, it is now well-recognized that GPCRs can mediate cellular signaling through both G protein-dependent (through four major G-protein sub-classes: Gs, Gi/o, Gq/11, and G12/13) [4] and -independent pathways involving, for example, arrestins [5], G-protein receptor kinases (GRKs) [6], ion channels [7], and Src kinases [8]) (Fig. 1).
Fig. 1.
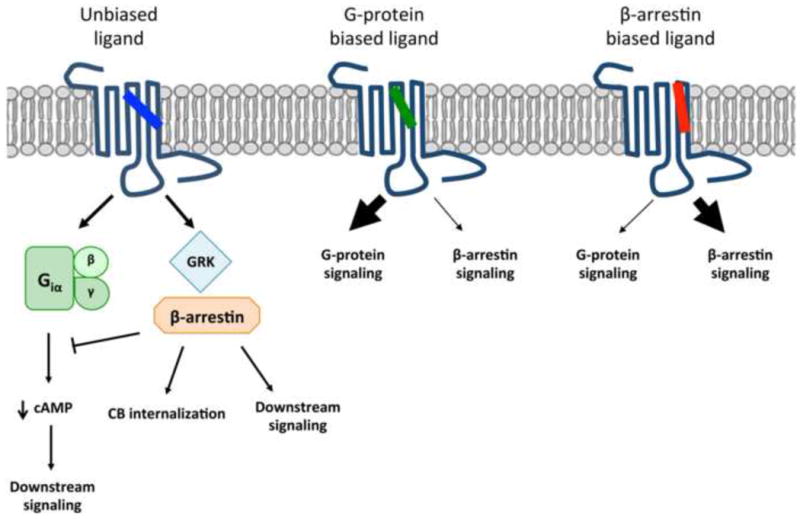
Diagrammatic representation of biased signaling at a G-protein coupled receptor (GPCR). In this example, an unbiased agonist is depicted that engages the GPCR and activates both G protein-dependent [i.e., adenylyl cyclase/cyclic adenosine monophosphate (cAMP)] and -independent (i.e., β-arrestin) signaling pathways. A biased agonist engages the receptor to generate a GPCR-ligand conformational ensemble that preferentially activates one or the other signaling cascade. If the preferentially activated signaling pathway were associated with a therapeutic effect vs. a harmful response from the non-preferred signaling pathway, the resulting signal bias could generate a pharmacologically improved therapeutic with less risk of on-target adverse events as compared to the unbiased ligand.
Traditional GPCR agonists bind to the receptor at the site that engages endogenous ligands, the so-called orthosteric site. Although orthosteric agonists may activate multiple, G protein-dependent and -independent downstream signaling networks, some preferentially activate one (or a few select) effector pathways. This phenomenon, termed “biased agonism” or “functional selectivity,” has been demonstrated for an ever increasing number of GPCRs including 5-HT2A, 5-HT2C, and 5-HT1A serotonin receptors [9,10]; the mu-opioid receptor (MOR) [11]; dopamine D1, D2, and D3 receptors [12-14]; chemokine receptor 7 [15]; melanocortin 4 receptor [16]; α-1a adrenergic receptor [17]; angiotensin type 1 receptor [18]; gonadotrophin-releasing hormone receptor [19]; and type 1 parathyroid hormone receptor (PTH1R) [20]. Comprehensive review articles may be consulted for a global appreciation of the large number and variety of GPCRs to which biased signaling has been attributed [21-23]. Regardless of the specific GPCR involved, current receptor theory holds that biased agonists preferentially promote certain intracellular signaling circuits by selectively stabilizing a subset of possible receptor conformations in different proportions than would an unbiased ligand, leading to pathway-selective biological responses [4,5,11,21]. Biased agonism is typically recognized as an alteration in the balance between GPCR G-protein dependent and -independent signaling mechanisms, the latter often involving recruitment of the multifunctional scaffold protein, β-arrestin2 [5,22].
As detailed elsewhere [21,23,24], functional selectivity carries important implications for drug discovery, especially with respect to populating the developed chemical space around therapeutic GPCRs lacking novel candidate drugs. Biased agonism offers an opportunity for the development of signaling pathway-selective (rather than just receptor-selective) therapies biased toward salutary effector pathways and away from those associated with on-target adverse events. This is particularly important for therapeutic GPCRs whose pharmacologically active ligands carry adverse effects that have restricted, if not obviated, the medical exploitation of those GPCRs as drug targets. The principal cannabinoid GPCRs are two such receptors.
2. Cannabinoid receptors
2.1. Receptor-mediated cannabinoid physiology
The endogenous cannabinoid (“endocannabinoid”) system includes two principal class-A GPCRs: cannabinoid receptor 1 (CB1R), predominantly expressed in the brain [25] and to a lesser extent in the periphery [26], and cannabinoid receptor 2 (CB2R), expressed mainly in immune cells and during inflammatory injury in the central nervous system (CNS) [27]. CB2R has 44% overall sequence identity with CB1R and shows comparatively greater interspecies heterogeneity. Some 25 years ago, identification and initial molecular characterization of CB1R and CB2R were promoted by attempts to understand the mechanism by which the plant-derived cannabinoid (phytocannabinoid) and principal psychoactive constituent of marijuana, Δ9-tetrahydrocannabinol (THC) (1, Fig. 2), exerts its (patho)physiological effects [28]. Other receptors, including GPR55 and GPR119 and members of the transient receptor potential (TRP) family, also bind select endogenous and pharmacologically active synthetic cannabinergic ligands [29,30].
Fig. 2.
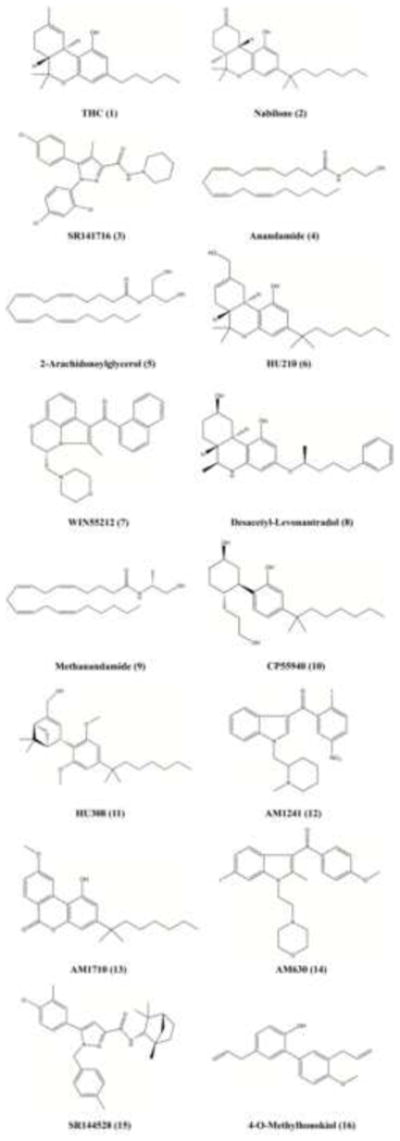
Chemical structures of cannabinoid receptor ligands discussed in the text. A summary of the molecular pharmacology of those ligands that display functional selectivity at CB1R and/or CB2R may be found in Tables 1 and 2, with details in references [47,56-63,65,67].
Cannabinoid receptors have been implicated in the regulation of a variety of central and peripheral physiological processes including neurogenesis [31], neuromodulation [32], energy balance and metabolism [33,34], immune-system activity [35], thermoregulation [36], and reproduction [37]. It is thus unremarkable that aberrant CB1R- or CB2R-dependent signaling is implicated in a number of disease states that represent major unsolved medical problems for which CBRs are considered key drug targets. CB1R-selective ligands are currently of interest as potential treatments for overweight/obesity, cardiometabolic and substance-use disorders, and neuropathic pain [38,39]. CB2R-selective agents hold promise for treating neuro-inflammatory diseases such as multiple sclerosis and amyotrophic lateral sclerosis [38,40]. Nonetheless, typical orthosetric CBR ligands (especially CB1R agonists) have a propensity to induce adverse psychobehavioral responses that have limited their therapeutic utility and circumscribed the indications accessible to salutary pharmacotherapeutic CB1R/CB2R modulation [39,41]. At present, although some synthetic CB1R/CB2R orthosteric ligands have utility as pharmacologically-active tool compounds for the laboratory, only two have received regulatory approval. Cesamet® (nabilone) (2, Fig. 2) is a synthetic cannabinoid and potent CB1R and CB2R agonist approved to treat chemotherapy-induced nausea and emesis [42]. Rimonabant® (SR141716) (3, Fig. 2), a CB1R antagonist/inverse agonist approved in 2006 the European Union for weight loss, was withdrawn by the manufacturer in 2009 due to its unacceptable risk:benefit ratio [43]. This situation has promoted the quest for efficacious small molecules with novel molecular-pharmacology phenotypes that express the benefits of CB1R/CB2R modulation with less side-effect risk than conventional CBR (ant)agonists [39,44].
2.2. CBR-dependent signaling
Both CB1R and CB2R preferentially couple to Gi/o -type G proteins, thereby inhibiting the activity of adenylyl cyclase (AC) and decreasing cellular cyclic adenosine monophosphate (cAMP) accumulation (Fig. 3) [45]. Under certain conditions, some cannabinergic agonists activate CBRs coupled to Gs [46] or Gq [47] G-proteins. Through a G-protein dependent mechanism, both CB1R and CB2R can activate different members of the mitogen-activated protein kinase (MAPK) family, including extracellular kinase-1 and -2 (ERK1/2), p38 and p42/p44 MAPKs, and c-Jun N-terminal kinase (JNK) [45,48-50]. CB1Rs can also negatively couple to N- and P/Q-type voltage-gated Ca2+ channels (VGCCs) [51] and positively couple to A-type and inward-rectifying K+ channels [52]. CB1Rs can also activate phospholipase C-b (PLC-b) to elicit an increase in intracellular Ca2+ as well as phosphatidylinositol 3-kinase/protein kinase B (PI3K/PKB) to stimulate glycolysis and modulate cell proliferation [53] (Fig. 3). In contrast, CB2Rs are associated with a more circumscribed repertoire of downstream effector pathways and do not modulate PI3K/PKB signaling, inward rectifying K+ channels, or Ca2+ channels [54]. Both CB1R and CB2R increase ceramide levels either by increasing sphingomyelin hydrolysis or de novo ceramide synthesis and can modulate gene transcription [55].
Fig. 3.
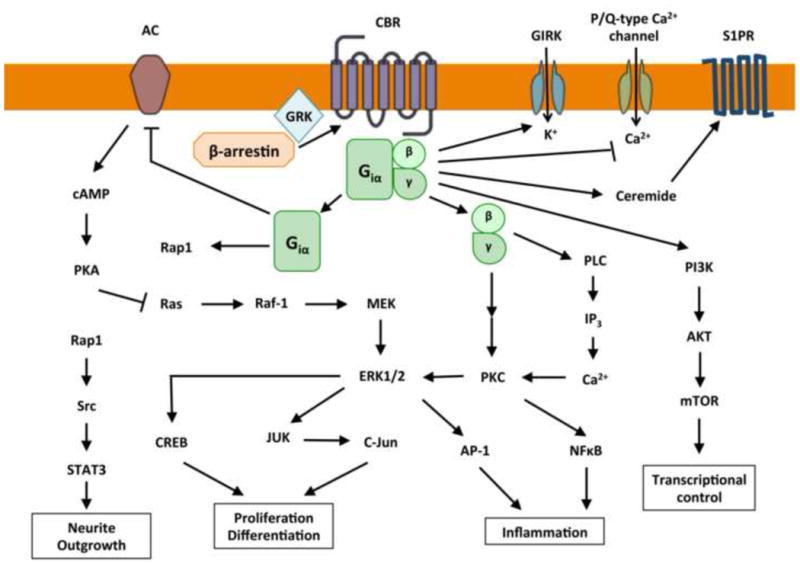
Schematic representation of canonical cannabinoid receptor (CBR)-mediated, G protein-dependent signaling pathways. Upon activation, CBRs preferentially couple to Gi/o-type G proteins and activate a series of downstream signaling cascades through which adenylyl cyclase (AC) is inhibited and mitogen-activated kinase (MAK) and extracellular kinase-1 and -2 (ERK1/2) are activated. Through the G-protein Gβγ subunit, CB1R activation can stimulate phospholipase C (PLC), leading to an increase in intracellular Ca2+ and activation of protein kinase C (PKC). Through Gi/o αβγ, the phosphoinositide 3-kinase (PI3K)/protein kinase B (Akt)/mammalian target of rapamycin (mTOR) intracellular signaling pathway can be stimulated. In addition, CB1R also modulates N- and P/Q-type voltage-gated Ca2+ channels, A-type and inward-rectifying K+ channels (GIRKs), and the sphingosine 1 receptor (S1PR) through which ceramide levels are regulated. These cell signaling circuits can modulate such diverse functions as neurite growth, cell proliferation and differentiation and inflammation and can also control gene transcription.
Certain ligands that engage CBRs are functionally biased in that they can activate preferentially one (or more) downstream signaling pathways (Tables 1 and 2; Fig. 2). Multiple lines of evidence supporting this proposition come from both cellular overexpression models and biological systems that express endogenous CB1R or CB2R, the latter suggesting the physiological significance of signaling bias at these receptors and inviting the opportunity for developing targeted, pathway-selective ligands that modulate GPCR-mediated cannabinergic signaling as pharmacotherapeutics. Those data will now be discussed with respect to key structure-function correlates of biased cannabinergic ligands. Particular emphasis will be placed upon findings that inform the design of functionally biased ligands as potential drugs targeted to these two endocannabinoid-system GPCRs at their respective orthosteric sites, at which naturally-occurring endocannabinoid lipid mediators bind.
Table 1. Evidence of CB1R functional selectivity.
| Ligand | Functional selectivity | Experimental approach | Reference |
|---|---|---|---|
| Endocannabinoids | |||
| 2-AG | No bias towards cAMP inhibition over pERK 1/2 | Alphascreen cAMP and SureFire assay with CB1R-expressing CHO cells | [60] |
| Increases CB1R surface expression through Gβγ signaling | cAMP accumulation assay in CB1R-expressing CHO cells. | [58] | |
| Anandamide | Biased towards Gi over Go | CB1R urea-extracted from sf9 membranes and reconstituted with varying Ga and Gβγ subunits. | [56] |
| Biased towards Gi over Gs and doesnot signal through Gq | cAMP accumulation assay in CB1R-expressing CHO cells. | [58] | |
| Biased towards cAMP inhibition over pERK 1/2 | Alphascreen cAMP and SureFire assay with CB1R-expressing CHO cells | [60] | |
| Eicosanoids | |||
| Methanandamide | Inverse agonist at Gi1 and Gi2; Agonist at Gi3 | CB1R-G protein complex were CHAPS-extracted from N18TG2 cell membrane and quantified by coimmunoprecipitation | [57] |
| Biased towards cAMP inhibition over pERK 1/2 | Alphascreen cAMP and SureFire assay with CB1R-expressing CHO cells | [60] | |
| Classical Cannabinoids | |||
| THC | Biased towards cAMP inhibition over pERK 1/2 | Alphascreen cAMP and SureFire assay with CB1R-expressing CHO cells | [60] |
| Induces expression of Tyrosine Hydrooxylase | mRNA and Protein analysis in mouse neuroblastoma cells N1E-115 | [61] | |
| Induced receptor internalization through β-arrestin recruitment | CREB phosphorylation in mouse striatal medium spiny projection neurons (STHdhQ7/Q7) | [59] | |
| Desacetyl-Levonantradol | Agonist at Gi1 and Gi2; Inverse agonist at Gi3 | CB1R-G protein complex were CHAPS-extracted from N18TG2 cell membrane and quantified by coimmunoprecipitation | [57] |
| HU210 | Non-selective between Gi, Go and Gs pathways | cAMP accumulation assay in CB1R-expressing CHO cells. | [58] |
| Biased towards cAMP inhibition over pERK 1/2 | Alphascreen cAMP and SureFire assay with CB1R-expressing CHO cells | [60] | |
| Decreases Tyrosine Hydroxylase expression unlike CP55940 | Tyrosine hydroxylase expression in N1E-115 Neuroblastoma cells | [61] | |
| Non-classical Cannabinoids | |||
| CP55940 | Activates Gs pathway | CREB phosphorylation in mouse striatal medium spiny projection neurons (STHdhQ7/Q7) | [59] |
| Biased towards Gi over Gs and does not signal through Go and Gq | cAMP accumulation assay in CB1R-expressing CHO cells. | [58] | |
| Induced receptor internalization through β-arrestin recruitment | CREB phosphorylation in mouse striatal medium spiny projection neurons (STHdhQ7/Q7) | [59] | |
| Aminoalkylindole | |||
| WIN55212 | Activates Gq pathway | Intracellular calcium accumulation in CB1R-expressing HEK293 cells and cultured hippocampal neurons. | [47] |
| Non-selective between Gi1, 2 and 3 subtypes | CB1R-G protein complex were CHAPS-extracted from N18TG2 cell membrane and quantified by coimmunoprecipitation | [57] |
Table 2. Evidence of CB2R functional selectivity.
| Ligand | Functional selectivity | Experimental approach | Reference |
|---|---|---|---|
| Endocannabinoids | |||
| 2-AG | Actiavted ERK-MAPK pathway with good potency but required higher concentrations for adenylyl cyclase inhibition and intracellular calcium release | Western Blotting analysis of ERK-MAPK phosphorylation and fluorescence-based calcium assay in human CB2R-expressing CHO cells | [62] |
| Anandamide | Did not induce receptor internalization even in the presence of FAAH inhibitors | Antibody-based analysis of cell-surface CB2R in HEK293 cells | [63] |
| Classical Cannabinoids | |||
| THC | Did not induce receptor internalization unlike CP55940 | Antibody-based analysis of cell-surface CB2R in HEK293 cells | [63] |
| Cannabilactone | |||
| AM1710 | Robustly internalized CB2R and recruited β-arrestin2, but weakly activated MAPK and did not affect VGCCs | Antibody-based analysis of cell-surface CB2R in HEK293 cells | [63] |
| Aminoalkylindole | |||
| WIN55212 | Induced cAMP and MAPK pathways but not receptor internalization and VGCC activation | Antibody-based analysis of cell-surface CB2R in HEK293 cells | [63] |
| AM1241 | Agonist at human CB2R, but an inverse agonist at rat and mouse CB2R | HitHunter cAMP XS assay with CB2R-expressing CHO-K1 cells | [65] |
| AM630 | Inverse agonist at Gi/o pathway but neutral antagonist at CB2R internalization and β-arrestin2 recruitment | Antibody-based analysis of cell-surface CB2R in HEK293 cells | [63] |
| Inverse agonist at Gi/o but no effect on 2-AG induced release of intracellular calcium | cAMP assay and FLIPR calcium assay in CB2R-expressing CHO-K1 cells | [67] | |
| Natural Product | |||
| 4′-O-methylhonokiol | Inverse agonist at Gi/o but induces CB2R-mediated release of intracellular calcium | cAMP assay and FLIPR calcium assay in CB2R-expressing CHO-K1 cells | [67] |
3. Functional selectivity at CB1R
All endocannabinoid signaling lipids, including the principal, arachidonic acid-derived mediators found in mammals, anandamide (AEA) (4, Fig. 2) and 2-arachidonoylglycerol (2-AG) (5, Fig. 2), preferentially activate Gi/o G proteins at CB1R (Table 1). Certain structurally distinct cannabinergic ligands, upon binding to CB1R, can elicit the receptor's differential interaction with Gi and/or Go G-protein subtypes. HU210 (6, Fig. 2) activates both Gi and Go proteins to maximal efficacy, whereas WIN55212 (7, Fig. 2) and AEA are biased towards Gi [56]. WIN55212 and SR141716 behave as an agonist and inverse agonist, respectively, at all Gi subtypes (Gi1, 2 and 3). Desacetyl-levonantradol (8, Fig. 2) is an agonist for Gi1 and Gi2 and an inverse agonist for Gi3, whereas methanandamide (9, Fig. 2) behaves as an agonist at only Gi3 and as an inverse agonist at Gi1 and Gi2 [57].
Although CB1R preferentially couples to Gi, under certain circumstances cannabinoid agonists such as HU210, WIN55212-A, and CP55940 (10, Fig. 2) can activate adenylyl cyclase through Gs-linked G-protein pathways [46,58,59]. Similarly, WIN55212 couples to Gq/11 G protein and activates Ca2+ channels to increase intracellular calcium [47]. Other agonists such as HU210, THC, and CP55940 do not signal through Gq/11 [47]. Taken together, these data demonstrate that CB1R exhibits complex, ligand-dependent signaling effects at the G-protein level that potentially govern the overall in vivo efficacy of these agents. At CB1R, preferential ligand bias through β-arrestin has also been observed, albeit less frequently than at different G proteins: In a murine cell-culture model of striatal medium spiny projection neurons endogenously expressing CB1R, THC and CP55940 promoted CB1R internalization through biased β-arrestin2 recruitment [59].
Endogenous cannabinoids may also show distinct signaling biases at CB1R. Unlike 2-AG, which displays no preference between adenylyl cyclase- and pERK1/2-dependent signaling pathways, AEA exhibits a 7-fold bias towards the former. Similar to 2-AG, WIN55212 does not exhibit any bias between adenylyl cyclase inhibition and pERK1/2 activation. However, THC and synthetic cannabinoids such as CP55940, HU210, and methanandamide all have significant bias towards cAMP inhibition over pERK1/2 activation [60]. Signaling bias has also been observed in further downstream events such as regulation of tyrosine hydroxylase (TH) transcription: in a murine neuroblastoma cell line, CB1R agonists (including THC and WIN55212) stimulate, whereas CP55940 inhibits, TH transcription [61].
4. Functional selectivity at CB2R
Reminiscent of CB1R, CB2R can be activated by cananbinergic agents that signal differentially through various intracellular information pathways in a ligand-dependent manner (Table 2). CB2R preferably interacts with Gi over Go, the latter not widely expressed in the peripheral tissues where CB2R expression is high. Furthermore, at CB2R, HU210 produces a maximal Gi response unlike AEA, which produces a partial response [56]. At CB2R, low concentrations of CP55940 inhibit adenylyl cyclase and stimulate ERK1/2 phosphorylation, whereas low concentrations of 2-AG behave similarly to CP55940 and stimulate ERK1/2 activity. Significantly greater 2-AG concentrations are required to inhibit CB2R-dependent adenylyl cyclase activity [62].
The specificity for downstream signaling pathways from CB2R may be generalized based on different classes of cannabinergic ligands [63]. In experiments with rat CB2R, non-classical cannabinoid agonists such as HU308 (11, Fig. 2) and CP55940 induced β-arrestin recruitment followed by receptor internalization. In contrast, aminoalkylindoles such as the nonselective agonist WIN55212 and the CB2R-selective agonist AM1241 (12, Fig. 2) [64] did not themselves elicit CB2R internalization, but rather antagonized CP55940-induced receptor internalization in a concentration-dependent manner. However, WIN55212 still promoted ERK1/2 activation and β-arrestin recruitment to the plasma membrane. In cAMP inhibition assays, racemic (R)- and (S)-AM1241 was an agonist at human CB2R, but an inverse agonist at rat and mouse CB2R [65]. Cannabilactones such as AM1710 (13, Fig. 2) were found to cause β-arrestin2 recruitment and CB2R internalization, but only weakly activated MAPK, and did not affect voltage-gated calcium channel function. The endocannabinoid AEA, a weak CB2R partial agonist, did not elicit CB2R internalization [66].
Signaling bias was also observed with antagonists/inverse agonists at CB2R [67]. The CB2R-selective inverse agonists AM630 (14, Fig. 2) and SR144528 (15, Fig. 2) reversed the inhibition of cAMP accumulation caused by cannabinoid agonists, but only SR144528 antagonized 2-AG-induced Ca2+ accumulation. AM630 did not influence CB2R-dependent, 2-AG-induced Ca2+ accumulation. In contrast, 4-O-methylhonokiol (16, Fig. 2), a synthetic CB2R-specific ligand that acts as an inverse agonist on 2-AG-induced cAMP inhibition, further potentiated agonist-induced Ca2+ ion flux. Similarly, SR144528 reversed agonist-induced CB2R internalization, leading to augmented CB2R levels on the cell surface, whereas AM630 acted as a neutral antagonist, displaying no detectable inverse-agonist efficacy.
5. Structural basis for signaling bias at cannabinoid GPCRs
Exponential progress in atomic-level structure determination of membrane proteins in the last few years has enabled the crystal structures of some 30 unique GPCRs, and over 119 different GPCR structures have been solved [68]. These structures have improved our understanding of the GPCR conformational changes associated with (especially orthosteric) ligand engagement as they pertain to various activity states. The large outward displacement of transmembrane helix (TMH) 6 and inward movement of TMH7 upon agonist binding appear characteristic of GPCR activation. Other hallmark structural accommodations associated with GPCR activation include rearrangements in the side chains of residues in conserved D(E)RY and NPxxY motifs in TMH3 and TMH7, respectively [68]. These activation-associated changes in GPCR TMH topology modulate interactions between the GPCR and intracellular effector molecules such as G-proteins, GRKs, and β-arrestin.
Diverse techniques have been used to interrogate the structure-function correlates of GPCR functional selectivity, as detailed elsewhere [69-71]. In this regard, β-adrenergic and serotonin (5-HT2) receptor subtypes have been used most extensively as model systems, especially in X-ray crystallographic studies. Emerging data indicate that conformational changes in TMHs 3, 5, and 6 are associated with G-protein activation [72], whereas TMH7, carboxy-terminal cytoplasmic helix 8, and intracellular loop (ICL) 2 play particularly critical roles in β-arrestin-mediated signaling [73]. Crystal structures of the liganded β1-adrenergic receptor suggest specific interactions between β-arrestin biased ligands and receptor residues in TMH7 and extracellular loop (ECL) 2 not associated with unbiased ligands [74]. Crystals of the 5-HT1B receptor bound to the strongly biased ligand ergotamine exhibited a classical, agonist-induced active-state conformation, whereas those of the ergotamine-bound serotonin 2B (5-HT2B) receptor showed characteristics of both active- or inactive-state conformations: the NPxxY motif and TMH7 exhibited pronounced active-state features, while the D(E)RY motif and TMH6 exhibited inactive-state features, indicating a β-arrestin bias consistent with biochemical data [75]. Similarly, a series of polar amino-acid interactions extending from its extracellular loops to the transmembrane helical bundle has been implicated in the signaling bias of the glucagon-like peptide 1 receptor (GLP-1R) [76].
Although a crystallized receptor construct of human CB1R (hCB1R) in complex with a stabilizing antagonist has recently been described [77], the data for that inactive liganded receptor state cannot provide structural details as to the features of ligand docking poses or CB1R interactions with G-proteins or β-arrestin that determine biased agonism at hGPCR. An X-ray crystal structure of CB2R has not yet been reported. Mutational studies, often in conjunction with in-silico modeling, have shed some light on the structural features of activation at these cannabinoid receptors. Most of the residues critical for CBR activation are within highly conserved GPCR functional motifs. These amino acid residues include TMH2 D2.50 (of the SLAxAD motif); TMH3 R3.50 (of the D(E)RY motif); TMH6 D6.30 (of the TMH3-7 salt bridge) and P6.50 (of the CWxP motif); and TMH7 P7.50 (of the NPxxY motif). As with other class-A GPCRs, disruption of the ionic interactions between D2.50 of the SLAxAD motif and N7.49 of the NPxxY motif, and between D3.49 of the DRY motif and D6.30 of TMH6, is critical for CBR activation [78].
A number of structural features unique to CBRs have been associated with receptor activation. CB1R sequence analysis revealed the presence of a relatively lengthy extracellular N-terminal end and the absence of the highly conserved proline residue in TMH5. It has been speculated that the long N-terminal tail may play a role in chaperone-mediated regulation of CB1R synthesis, folding, maturation and trafficking [79]. ECL2 connecting TMH4 and TMH5 is considered to be important to the stability of and ligand engagement by most GPCRs. In CB1R, C256ECL2 and C264ECL2 form an intra-loop disulfide bond critical for receptor stabilization and activation, but not CP55940 binding [80]. Furthermore, CBRs lack a cysteine at the N-terminal end of TMH3, which prevents formation of a disulfide bond with the distal cysteine of the ECL2 that is present in most other GPCRs [81]. Similarly, most class-A GPCRs feature a CWxP motif (including residue W6.48) in their binding pocket that serves as a “toggle switch” for receptor activation upon agonist binding. However, both CB1R and CB2R lack an analogous, critical TMH6 aromatic residue at 6.52 position. Mutation studies suggest that W2586.48 pairs with the F1173.36 residue to form the toggle switch in CBR1, with consequent loss of aromatic stacking, leading to receptor activation [82]. In contrast to other GPCRs that engage hydrophilic ligands, lipophilic cannabinergic ligands may access CBR binding pockets through the membrane bilayer via an intramembranous portal between TMH6 and TMH7 [83].
Overall, CBRs exhibit high conformational flexibility, as suggested by their high basal activity [84], making them interesting candidates for studying the structural basis of GPCR signaling bias. Yet few experimental studies have interrogated the structural basis of functional selectivity in CBRs. Biochemical/mutation data with cannabinergic ligands and information on the structures of other class-A GPCRs have been used to identify candidate residues potentially involved in ligand functional selectivity at CBRs. Similar to other GPCRs, double mutation of D2133.49 and R2143.50 in the highly conserved aspartic acid-arginine-tyrosine (“DRY”) motif that plays a pivotal role in regulating GPCR conformational/activity states caused CB1R to bias towards β-arrestin signaling and away from G-protein activation without significant loss in binding affinity of cannabinoid agonists [85]. It was also observed that C3556.47 of the CWxP motif was critical for binding of CP55940 and other classical cannabinoids that are known to induce receptor internalization through β-arrestin-mediated pathways [85]. However, WIN55212, an aminoalkylindole derivative that does not induce receptor internalization, maintained high affinity for CB1R, even when C3556.47 had been mutated. These data invited speculation that ligands interacting with W3566.48 by aromatic stacking do not induce receptor internalization [82]. Unlike classical agonists, inverse agonists such as the biarylpyrazole SR141716 bind very poorly to CB1R when C3867.41 is mutated to a bulky group [81]. The reduced affinity can be explained by the loss of critical aromatic interactions involving W3566.48 and F2003.36 within the binding pocket. Thus, the unique biochemical characteristics of WIN55212 can be explained by the presence of both inactive- and active-state structural features, reminiscent of the situation with ergotamine in 5-HT2B receptor [75]. In-depth experimental studies are required to understand and confirm the structural features associated with the functional selectivity of specific cannabinoid ligands at CB1R and CB2R.
6. Pharmacotherapeutic implications of biased signaling at CBRs
As a component of its overall pharmacological profile, the ability of a GPCR-targeted therapeutic candidate to activate differentially specific intracellular effector pathways carries critical implications for drug discovery. For example, niacin acts therapeutically as an antilipolytic agent by activating GPR109A receptor-mediated G-protein signaling. However, it also activates the G protein-independent β-arrestin1 pathway responsible for cutaneous flushing and other adverse effects [86]. Similarly, in kappa opioid receptors, activation of ERK signaling while retaining the G-protein signaling bias over β-arrestin2 pathway is effective in relieving pain without inducing adverse effects such as dysphoria, sedation, and diuresis associated with β-arrestin signaling [87,88]. The pharmacological profiles of specific ligands for other GPCRs, including dopamine [89], glucagon-like peptide [90], opioid [91], and angiotensin II [92] receptors, show selective activation of particular signaling pathway(s) that could potentially support a clinically relevant therapeutic effect vs. inciting other, deleterious signaling cascades.
Knockout studies have shown that the anti-nociceptive effect of CB1R cannabinergic agonists is not significantly affected by β-arrestin2. However, in β-arrestin2 knockout animals, development of tolerance to antinociception was attenuated following repeated THC administration, an effect attributed to a decrease in receptor desensitization for G-protein-dependent signaling. In contrast, β-arrestin2 knockout resulted in an increased tolerance with respect to cannabinoid dependence characteristics such as catalepsy and also potentiated the hypothermia response to cannabinoid agonists [93]. Therefore, developing CB1R-targeted biased agonists with attenuated β-arrestin2 recruitment may not only improve their anti-nociceptive efficacy, but also reduce the potential for unwanted catalepsy and drug tolerance. This view is supported by demonstration that β-arrestin2-null mice evidence enhanced sensitivity to the principal psychoactive phytocannabinoid in marijuana, THC [94]. In contrast, CB2R-selective antagonists/inverse agonists that preferentially activate the β-arrestin2 pathway incite cytoskeletal rearrangements in immune cells [95,96] and in lung airway [97] to modulate chemotaxis and invasion of immune and cancerous cells. Although these studies illustrate the important role of β-arrestin in normal, G-protein-dependent GPCR desensitization mechanisms and do not exemplify β-arrestin-biased signaling per se, the results suggest that the degree of β-arrestin bias in vivo is an important characteristic of potential, CBR-targeted therapeutics.
7. Future research directions
Although it is well established that most GPCR ligands may exhibit pluridimensional efficacies with respect to different signaling pathways, challenges remain in the study of CBR functional selectivity. Experimental nuances such as kinetics of response, response read-out bias, cell/tissue-specific variations, and the system-dependency of the observed pharmacological effects may influence the qualitative nature of biased signaling and its quantification [98]. Stereochemical [65] and species-dependent [99] pharmacological effects among orthosteric cannabinergic ligands and differences in their ability to elicit receptor oligomers that actively signal [100,101] may further extend the pharmacological scope of biased signaling at CBRs.
Over the past decade, novel biased ligands have been profiled preclinically for some GPCRs including the β2-adrenergic [102], kappa-opioid [103], mu-opioid [104], angiotensin II (Type 1) [105], parathyroid hormone [106], and serotonin [107] receptors. A few such ligands are in various stages of clinical development. TRV130, a G protein-biased ligand that targets the mu opioid receptor, provides significantly better pain relief than either placebo or morphine in a phase-II clinical study for treating severe acute pain [108]. Furthermore, TRV130 did not elicit hypercapnia-induced respiratory drive or severe nausea at equi- (or greater) analgesic doses than morphine in healthy volunteers [109]. However, TRV027, a β-arrestin-biased ligand targeting the angiotensin II type 1 receptor, failed to meet both primary and secondary endpoints for treating acute heart failure in a phase-II clinical study [110].
Optimal therapeutic exploitation of functionally selective CB1R and CB2R ligands requires several aspects of their pharmacology to be detailed. Primarily, for a thorough analysis of the molecular mechanisms of signaling bias within the endocannabinoid system, novel biased molecules need to be designed, synthesized, and profiled in both over-expressed and native cell lines under similar experimental conditions to avoid system/context-specific pharmacological activity that could reflect, for example, the relative prevalence of G protein-related and -independent signaling partners across cell-based assays rather than true physiological ligand bias [111,112]. The extent to which a biased phenotype for any given GPCR ligand may directly translate into the systems physiology of a living organism in vivo remains of great concern, given that GPCR ligand bias has been most consistently demonstrated at heterologously expressed receptors in functional assays employing cultured-cell or isolated-tissue backgrounds (e.g., Tables 1 and 2 for CBRs)-- not in complex biological systems [113,114]. This concern is exacerbated by the fact that delineation of the relationships between apparent ligand bias defined by cell- or tissue-based screening criteria in vitro and therapeutically-relevant biological responses in vivo is crucial to drug discovery. More generally, the mechanism and biological/therapeutic significance of cannabinergic signaling pathways need to be better characterized in both in vitro studies and in vivo animal models exhibiting normal physiology and relevant disease phenotypes. In this regard, studies of functional selectivity using cells or tissues from patients with clearly defined disease conditions could provide valuable insight into the biological and pathological significance of CBR signaling bias. Ultimately, these efforts may result in cannabinergic drugs with improved therapeutic profiles and less risk of clinically significant adverse events. Such knowledge could also help inform the application of cannabis extracts as well as THC and other plant-derived cannabinoids in medical practice so as to leverage any potential clinical benefit and militate against undesirable adverse events, a subject of much current controversy and speculation for public health [115].
Aside from biased orthosteric modulators, other ligands with pharmacological properties distinct from conventional, unbiased agonists and antagonists/inverse agonists are being studied for their ability to modulate therapeutic CBR signaling. The potential for the inverse-agonist property of CB1R antagonists/inverse agonists to elicit adverse events by influencing physiologically important CB1R constitutive (i.e., ligand-independent) signaling has raised interest in so-called “neutral” or “silent” CB1R antagonists that display little, if any, intrinsic inverse-agonist efficacy [116]. CBR ligands that engage sites functionally and topographically distinct from the binding pockets of endogenous ligands (“allosteric ligands”) are gaining considerable pharmacological and therapeutic attention, since allosteric GPCR modulation is associated with several theoretical advantages that could make for safer, more efficacious pharmacotherpaeutics, especially at GPCRs such as CB1R where orthosteric agonists carry significant adverse-event risk [117,118]. Perhaps most intriguingly, CB1R-selective allosteric modulators may themselves bias receptor-dependent information output along particular signaling circuits [119,120]. As with functionally-selective orthosteric CB1R and CB2R ligands, the underlying molecular mechanisms biasing allosteric effector pathways will hopefully emerge in sufficient detail to help realize the potential of CBR-targeted drug discovery in the clinic. Indeed, this cutting-edge proposition is already driving the discovery and profiling of proprietary biased CB1R allosteric modulators with demonstrable preclinical therapeutic efficacy while free of endogenous inverse-agonist activity [121,122].
Abbreviations
- THC
Δ9-tetrahydrocannabinol
- 5-HT2B
serotonin 2B receptor
- AC
adenylyl cyclase
- AEA
anandamide
- Akt
protein kinase B
- cAMP
cyclic adenosine monophosphate
- CB1R
cannabinoid receptor 1
- CB2R
cannabinoid receptor 2
- CNS
central nervous system
- ECL
extracellular loop
- ERK1/2
extracellular kinase-1 and -2
- GIRK
G protein-coupled inwardly-rectifying potassium channel
- GLP-1R
glucagon-like peptide 1 receptor
- GPCR
G-protein coupled receptors
- GRKs
G-protein receptor kinases
- hCB1R
human cannabinoid receptor 1
- JNK
c-Jun N-terminal kinase
- MAK
mitogen activated kinase
- MAPK
mitogen activated protein kinase
- MOR
mu-opioid receptors
- mTor
mammalian target of rapamycin
- PI3K/PKB
phosphatidylinositol 3-kinase/protein kinase B
- PKC
protein kinase C
- S1PR
sphingosine 1 receptor
- TH
tyrosine hydroxylase
- TMH
transmembrane helix
- TRP
transient receptor potential
Footnotes
Conflict of interest: The authors declare no conflicts of interest regarding the subject of this paper.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.De Lean A, Stadel JM, Lefkowitz RJ. A ternary complex model explains the agonist-specific binding properties of the adenylate cyclase-coupled beta-adrenergic receptor. J Biol Chem. 1980;255:7108–7117. [PubMed] [Google Scholar]
- 2.Kobilka BK, Deupi X. Conformational complexity of G-protein-coupled receptors. Trends Pharmacol Sci. 2007;28:397–406. doi: 10.1016/j.tips.2007.06.003. [DOI] [PubMed] [Google Scholar]
- 3.Manglik A, Kobilka B. The role of protein dynamics in GPCR function: insights from the β2AR and rhodopsin. Curr Opin Cell Biol. 2014;27:136–143. doi: 10.1016/j.ceb.2014.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hamm HE. The many faces of G protein signaling. J Biol Chem. 1998;273:669–672. doi: 10.1074/jbc.273.2.669. [DOI] [PubMed] [Google Scholar]
- 5.Luttrell LM, Lefkowitz RJ. The role of beta-arrestins in the termination and transduction of G-protein-coupled receptor signals. J Cell Sci. 2002;115:455–465. doi: 10.1242/jcs.115.3.455. [DOI] [PubMed] [Google Scholar]
- 6.Ribas C, Penela P, Murga C, Salcedo A, García-Hoz C, Jurado-Pueyo M, et al. The G protein-coupled receptor kinase (GRK) interactome: Role of GRKs in GPCR regulation and signaling. Biochim Biophys Acta BBA - Biomembr. 2007;1768:913–922. doi: 10.1016/j.bbamem.2006.09.019. [DOI] [PubMed] [Google Scholar]
- 7.Dascal N. Ion-channel regulation by G proteins. Trends Endocrinol Metab. 2001;12:391–398. doi: 10.1016/S1043-2760(01)00475-1. [DOI] [PubMed] [Google Scholar]
- 8.Luttrell DK, Luttrell LM. Not so strange bedfellows: G-protein-coupled receptors and Src family kinases. Oncogene. 2004;23:7969–7978. doi: 10.1038/sj.onc.1208162. [DOI] [PubMed] [Google Scholar]
- 9.Berg KA, Maayani S, Goldfarb J, Scaramellini C, Leff P, Clarke WP. Effector pathway-dependent relative efficacy at serotonin type 2A and 2C receptors: evidence for agonist-directed trafficking of receptor stimulus. Mol Pharmacol. 1998;54:94–104. [PubMed] [Google Scholar]
- 10.Newman-Tancredi A, Martel JC, Assié MB, Buritova J, Lauressergues E, Cosi C, et al. Signal transduction and functional selectivity of F15599, a preferential post-synaptic 5-HT1A receptor agonist. Br J Pharmacol. 2009;156:338–353. doi: 10.1111/j.1476-5381.2008.00001.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Raehal KM, Schmid CL, Groer CE, Bohn LM. Functional selectivity at the μ-opioid receptor: implications for understanding opioid analgesia and tolerance. Pharmacol Rev. 2011;63:1001–1019. doi: 10.1124/pr.111.004598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mottola DM, Kilts JD, Lewis MM, Connery HS, Walker QD, Jones SR, et al. Functional selectivity of dopamine receptor agonists. I. Selective activation of postsynaptic dopamine D2 receptors linked to adenylate cyclase. J Pharmacol Exp Ther. 2002;301:1166–1178. doi: 10.1124/jpet.301.3.1166. [DOI] [PubMed] [Google Scholar]
- 13.Möller D, Kling RC, Skultety M, Leuner K, Hübner H, Gmeiner P. Functionally selective dopamine D2, D3 receptor partial agonists. J Med Chem. 2014 doi: 10.1021/jm5004039. [DOI] [PubMed] [Google Scholar]
- 14.Ryman-Rasmussen JP, Griffith A, Oloff S, Vaidehi N, Brown JT, Goddard WA, et al. Functional selectivity of dopamine D1 receptor agonists in regulating the fate of internalized receptors. Neuropharmacology. 2007;52:562–575. doi: 10.1016/j.neuropharm.2006.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kohout TA, Nicholas SL, Perry SJ, Reinhart G, Junger S, Struthers RS. Differential desensitization, receptor phosphorylation, beta-arrestin recruitment, and ERK1/2 activation by the two endogenous ligands for the CC chemokine receptor 7. J Biol Chem. 2004;279:23214–23222. doi: 10.1074/jbc.M402125200. [DOI] [PubMed] [Google Scholar]
- 16.Nickolls SA, Fleck B, Hoare SRJ, Maki RA. Functional selectivity of melanocortin 4 receptor peptide and nonpeptide agonists: evidence for ligand-specific conformational states. J Pharmacol Exp Ther. 2005;313:1281–1288. doi: 10.1124/jpet.105.083337. [DOI] [PubMed] [Google Scholar]
- 17.Evans BA, Broxton N, Merlin J, Sato M, Hutchinson DS, Christopoulos A, et al. Quantification of functional selectivity at the human α1A-adrenoceptor. Mol Pharmacol. 2011;79:298–307. doi: 10.1124/mol.110.067454. [DOI] [PubMed] [Google Scholar]
- 18.Balakumar P, Jagadeesh G. Structural determinants for binding, activation, and functional selectivity of the angiotensin AT1 receptor. J Mol Endocrinol. 2014;53:R71–R92. doi: 10.1530/JME-14-0125. [DOI] [PubMed] [Google Scholar]
- 19.McArdle CA. Gonadotropin-releasing hormone receptor signaling: biased and unbiased. Mini Rev Med Chem. 2012;12:841–850. doi: 10.2174/138955712800959080. [DOI] [PubMed] [Google Scholar]
- 20.Bohinc BN, Gesty-Palmer D. β-arrestin-biased agonism at the parathyroid hormone receptor uncouples bone formation from bone resorption. Endocr Metab Immune Disord Drug Targets. 2011;11:112–119. doi: 10.2174/187153011795564151. [DOI] [PubMed] [Google Scholar]
- 21.Whalen EJ, Rajagopal S, Lefkowitz RJ. Therapeutic potential of β-arrestin- and G-protein-biased agonists. Trends Mol Med. 2011;17:126–139. doi: 10.1016/j.molmed.2010.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Reiter E, Ahn S, Shukla AK, Lefkowitz RJ. Molecular michanism of β-arrestin-biased agonism at seven-transmembrane receptors. Annu Rev Pharmacol Toxicol. 2012;52:179–197. doi: 10.1146/annurev.pharmtox.010909.105800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Luttrell LM. Minireview: more than just a hammer: ligand ‘bias’ and pharmaceutical discovery. Mol Endocrinol. 2014;28:281–294. doi: 10.1210/me.2013-1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shonberg J, Lopez L, Scammells PJ, Christopoulos A, Capuano B, Lane JR. Biased agonism at G protein-coupled receptors: the promise and the challenges—A medicinal chemistry perspective. Med Res Rev. 2014;34:1280–1330. doi: 10.1002/med.21318. [DOI] [PubMed] [Google Scholar]
- 25.Herkenham M, Lynn AB, Little MD, Johnson MR, Melvin LS, de Costa BR, et al. Cannabinoid receptor localization in brain. Proc Natl Acad Sci U S A. 1990;87:1932–1936. doi: 10.1073/pnas.87.5.1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sañudo-Peña MC, Strangman NM, Mackie K, Walker JM, Tsou K. CB1 receptor localization in rat spinal cord and roots, dorsal root ganglion, and peripheral nerve. Zhongguo Yao Li Xue Bao. 1999;20:1115–1120. [PubMed] [Google Scholar]
- 27.Munro S, Thomas KL, Abu-Shaar M. Molecular characterization of a peripheral receptor for cannabinoids. Nature. 1993;365:61–65. doi: 10.1038/365061a0. [DOI] [PubMed] [Google Scholar]
- 28.Ligresti A, De Petrocellis L, Di Marzo V. From phytocannabinoids to cannabinoid receptors and endocannabinoids: pleiotropic physiological and pathological roles through complex pharmacology. Physiol Rev. 2016;96:1593–1659. doi: 10.1152/physrev.00002.2016. [DOI] [PubMed] [Google Scholar]
- 29.Brown AJ. Novel cannabinoid receptors. Br J Pharmacol. 2007;152:567–575. doi: 10.1038/sj.bjp.0707481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Janero DR, Makriyannis A. Terpenes and lipids of the endocannabinoid and transient-receptor-potential-channel biosignaling systems. ACS Chem Neurosci. 2014;5:1097–1106. doi: 10.1021/cn5000875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jin K, Xie L, Kim SH, Parmentier-Batteur S, Sun Y, Mao XO, et al. Defective adult neurogenesis in CB1 cannabinoid receptor knockout mice. Mol Pharmacol. 2004;66:204–208. doi: 10.1124/mol.66.2.204. [DOI] [PubMed] [Google Scholar]
- 32.Di Marzo V, Melck D, Bisogno T, De Petrocellis L. Endocannabinoids: endogenous cannabinoid receptor ligands with neuromodulatory action. Trends Neurosci. 1998;21:521–528. doi: 10.1016/s0166-2236(98)01283-1. [DOI] [PubMed] [Google Scholar]
- 33.Cardinal P, Bellocchio L, Clark S, Cannich A, Klugmann M, Lutz B, et al. Hypothalamic CB1 cannabinoid receptors regulate energy balance in mice. Endocrinology. 2012;153:4136–4143. doi: 10.1210/en.2012-1405. [DOI] [PubMed] [Google Scholar]
- 34.Guzmán M, Sánchez C. Effects of cannabinoids on energy metabolism. Life Sci. 1999;65:657–664. doi: 10.1016/s0024-3205(99)00288-x. [DOI] [PubMed] [Google Scholar]
- 35.Kaminski NE. Evidence for a cannabinoid receptor in immunomodulation by cannabinoid compounds. Adv Exp Med Biol. 1993;335:115–120. doi: 10.1007/978-1-4615-2980-4_16. [DOI] [PubMed] [Google Scholar]
- 36.Wenger T, Moldrich G. The role of endocannabinoids in the hypothalamic regulation of visceral function. Prostaglandins Leukot Essent Fatty Acids. 2002;66:301–307. doi: 10.1054/plef.2001.0353. [DOI] [PubMed] [Google Scholar]
- 37.Maccarrone M. CB2 receptors in reproduction. Br J Pharmacol. 2008;153:189–198. doi: 10.1038/sj.bjp.0707444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Singh J, Budhiraja S. Therapeutic potential of cannabinoid receptor ligands: current status. Methods Find Exp Clin Pharmacol. 2006;28:177–183. doi: 10.1358/mf.2006.28.3.985231. [DOI] [PubMed] [Google Scholar]
- 39.Pertwee RG. Targeting the endocannabinoid system with cannabinoid receptor agonists: pharmacological strategies and therapeutic possibilities. Philos Trans R Soc B Biol Sci. 2012;367:3353–3363. doi: 10.1098/rstb.2011.0381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kim K, Moore DH, Makriyannis A, Abood ME. AM1241, a cannabinoid CB2 receptor selective compound, delays disease progression in a mouse model of amyotrophic lateral sclerosis. Eur J Pharmacol. 2006;542:100–105. doi: 10.1016/j.ejphar.2006.05.025. [DOI] [PubMed] [Google Scholar]
- 41.Pertwee RG. Endocannabinoids and their pharmacological actions. Handb Exp Pharmacol. 2015;231:1–37. doi: 10.1007/978-3-319-20825-1_1. [DOI] [PubMed] [Google Scholar]
- 42.Davis MP. Oral nabilone capsules in the treatment of chemotherapy-induced nausea and vomiting and pain. Expert Opin Investig Drugs. 2008;17:85–95. doi: 10.1517/13543784.17.1.85. [DOI] [PubMed] [Google Scholar]
- 43.Krentz AJ, Fujioka K, Hompesch M. Evolution of pharmacological obesity treatments: focus on adverse side-effect profiles. Diabetes Obes Metab. 2016;18:558–570. doi: 10.1111/dom.12657. [DOI] [PubMed] [Google Scholar]
- 44.Janero DR, Lindsley L, Vemuri VK, Makriyannis A. Cannabinoid 1 G protein-coupled receptor (periphero-)neutral antagonists: emerging therapeutics for treating obesity-driven metabolic disease and reducing cardiovascular risk. Expert Opin Drug Discov. 2011;6:995–1025. doi: 10.1517/17460441.2011.608063. [DOI] [PubMed] [Google Scholar]
- 45.Howlett AC. In: Cannabinoid Receptor Signaling. Pertwee RG, editor. Cannabinoids: Springer Berlin Heidelberg; 2005. pp. 53–79. [DOI] [PubMed] [Google Scholar]
- 46.Glass M, Felder CC. Concurrent stimulation of cannabinoid CB1 and dopamine D2 receptors augments cAMP accumulation in striatal neurons: evidence for a Gs linkage to the CB1 receptor. J Neurosci. 1997;17:5327–5333. doi: 10.1523/JNEUROSCI.17-14-05327.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lauckner JE, Hille B, Mackie K. The cannabinoid agonist WIN55,212-2 increases intracellular calcium via CB1 receptor coupling to Gq/11 G proteins. Proc Natl Acad Sci U S A. 2005;102:19144–19149. doi: 10.1073/pnas.0509588102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rueda D, Galve-Roperh I, Haro A, Guzmán M. The CB1 cannabinoid receptor is coupled to the activation of c-Jun N-terminal kinase. Mol Pharmacol. 2000;58:814–820. doi: 10.1124/mol.58.4.814. [DOI] [PubMed] [Google Scholar]
- 49.Bouaboula M, Poinot-Chazel C, Bourrié B, Canat X, Calandra B, Rinaldi-Carmona M, et al. Activation of mitogen-activated protein kinases by stimulation of the central cannabinoid receptor CB1. Biochem J. 1995;312:637–641. doi: 10.1042/bj3120637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liu J, Gao B, Mirshahi F, Sanyal AJ, Khanolkar AD, Makriyannis A, et al. Functional CB1 cannabinoid receptors in human vascular endothelial cells. Biochem J. 2000;346:835–840. doi: 10.1042/bj3460835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Twitchell W, Brown S, Mackie K. Cannabinoids inhibit N- and P/Q-type calcium channels in cultured rat hippocampal neurons. J Neurophysiol. 1997;78:43–50. doi: 10.1152/jn.1997.78.1.43. [DOI] [PubMed] [Google Scholar]
- 52.Ho BY, Uezono Y, Takada S, Takase I, Izumi F. Coupling of the expressed cannabinoid CB1 and CB2 receptors to phospholipase C and G protein-coupled inwardly rectifying K+ channels. Receptors Channels. 1999;6:363–374. [PubMed] [Google Scholar]
- 53.Gómez del Pulgar T, Velasco G, Guzmán M. The CB1 cannabinoid receptor is coupled to the activation of protein kinase B/Akt. Biochem J. 2000;347:369–373. doi: 10.1042/0264-6021:3470369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Felder CC, Joyce KE, Briley EM, Mansouri J, Mackie K, Blond O, et al. Comparison of the pharmacology and signal transduction of the human cannabinoid CB1 and CB2 receptors. Mol Pharmacol. 1995;48:443–450. [PubMed] [Google Scholar]
- 55.Velasco G, Galve-Roperh I, Sánchez C, Blázquez C, Haro A, Guzmán M. Cannabinoids and ceramide: two lipids acting hand-by-hand. Life Sci. 2005;19:1723–1731. doi: 10.1016/j.lfs.2005.05.015. [DOI] [PubMed] [Google Scholar]
- 56.Glass M, Northup JK. Agonist selective regulation of G proteins by cannabinoid CB1 and CB2 receptors. Mol Pharmacol. 1999;56:1362–1369. doi: 10.1124/mol.56.6.1362. [DOI] [PubMed] [Google Scholar]
- 57.Mukhopadhyay S, Howlett AC. Chemically distinct ligands promote differential CB1 cannabinoid receptor-Gi protein interactions. Mol Pharmacol. 2005;67:2016–2024. doi: 10.1124/mol.104.003558. [DOI] [PubMed] [Google Scholar]
- 58.Bonhaus DW, Chang LK, Kwan J, Martin GR. Dual activation and inhibition of adenylyl cyclase by cannabinoid receptor agonists: evidence for agonist-specific trafficking of intracellular responses. J Pharmacol Exp Ther. 1998;287:884–888. [PubMed] [Google Scholar]
- 59.Laprairie RB, Bagher AM, Kelly MEM, Dupré DJ, Denovan-Wright EM. Type 1 cannabinoid receptor ligands display functional selectivity in a cell culture model of striatal medium spiny projection neurons. J Biol Chem. 2014;289:24845–24862. doi: 10.1074/jbc.M114.557025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Khajehali E, Malone DT, Glass M, Sexton PM, Christopoulos A, Leach K. Biased agonism and biased allosteric modulation at the CB1 cannabinoid receptor. Mol Pharmacol. 2015;88:368–379. doi: 10.1124/mol.115.099192. mol.115.099192. [DOI] [PubMed] [Google Scholar]
- 61.Bosier B, Tilleux S, Najimi M, Lambert DM, Hermans E. Agonist selective modulation of tyrosine hydroxylase expression by cannabinoid ligands in a murine neuroblastoma cell line. J Neurochem. 2007;102:1996–2007. doi: 10.1111/j.1471-4159.2007.04679.x. [DOI] [PubMed] [Google Scholar]
- 62.Shoemaker JL, Ruckle MB, Mayeux PR, Prather PL. Agonist-directed trafficking of response by endocannabinoids acting at CB2 receptors. J Pharmacol Exp Ther. 2005;315:828–838. doi: 10.1124/jpet.105.089474. [DOI] [PubMed] [Google Scholar]
- 63.Atwood BK, Wager-Miller J, Haskins C, Straiker A, Mackie K. Functional selectivity in CB2 cannabinoid receptor signaling and regulation: implications for the therapeutic potential of CB2 ligands. Mol Pharmacol. 2012;81:250–263. doi: 10.1124/mol.111.074013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rahn EJ, Zvonok AM, Makriyannis A, Hohmann AG. Antinociceptive effects of racemic AM1241 and its chirally synthesized enantiomers: lack of dependence upon opioid receptor activation. AAPS J. 2010;12:147–157. doi: 10.1208/s12248-009-9170-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bingham B, Jones PG, Uveges AJ, Kotnis S, Lu P, Smith VA, et al. Species-specific in vitro pharmacological effects of the cannabinoid receptor 2 (CB2) selective ligand AM1241 and its resolved enantiomers. Br J Pharmacol. 2007;151:1061–1070. doi: 10.1038/sj.bjp.0707303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Rahn EJ, Thakur GA, Wood JAT, Zvonok AM, Makriyannis A, Hohmann AG. Pharmacological characterization of AM1710, a putative cannabinoid CB2 agonist from the cannabilactone class: Antinociception without central nervous system side-effects. Pharmacol Biochem Behav. 2011;98:493–502. doi: 10.1016/j.pbb.2011.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Schuehly W, Paredes JMV, Kleyer J, Huefner A, Anavi-Goffer S, Raduner S, et al. Mechanisms of osteoclastogenesis inhibition by a novel class of biphenyl-type cannabinoid CB2 receptor inverse agonists. Chem Biol. 2011;18:1053–1064. doi: 10.1016/j.chembiol.2011.05.012. [DOI] [PubMed] [Google Scholar]
- 68.Katritch V, Cherezov V, Stevens RC. Structure-function of the G protein-coupled receptor superfamily. Annu Rev Pharmacol Toxicol. 2013;53:531–556. doi: 10.1146/annurev-pharmtox-032112-135923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.McNeely PM, Naranjo AN, Robinson AS. Structure-function studies with G protein-coupled receptors as a paradigm for improving drug discovery and therapeutic development. Biotechnol J. 2012;7:1451–1461. doi: 10.1002/biot.201200076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Shukla AK, Singh G, Ghosh E. Emerging structural insights into biased GPCR signaling. Trends Biochem Sci. 2014;39:594–602. doi: 10.1016/j.tibs.2014.10.001. [DOI] [PubMed] [Google Scholar]
- 71.Jaeger W, Armstrong S, Hill S, Pfleger KDG. Biophysical detection of diversity and bias in GPCR function. Mol Struct Endocrinol. 2014;5:26. doi: 10.3389/fendo.2014.00026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Nygaard R, Frimurer TM, Holst B, Rosenkilde MM, Schwartz TW. Ligand binding and micro-switches in 7TM receptor structures. Trends Pharmacol Sci. 2009;30:249–259. doi: 10.1016/j.tips.2009.02.006. [DOI] [PubMed] [Google Scholar]
- 73.Kang Y, Zhou XE, Gao X, He Y, Liu W, Ishchenko A, et al. Melcher K, Xu HE. Crystal structure of rhodopsin bound to arrestin by femtosecond X-ray laser. Nature. 2015;523:561–567. doi: 10.1038/nature14656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Warne T, Edwards PC, Leslie AGW, Tate CG. Crystal structures of a stabilized β1-adrenoceptor bound to the biased agonists bucindolol and carvedilol. Struct Lond Engl 1993. 2012;20:841–849. doi: 10.1016/j.str.2012.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wacker D, Wang C, Katritch V, Han GW, Huang XP, Vardy E, et al. Structural features for functional selectivity at serotonin receptors. Science. 2013;340:615–619. doi: 10.1126/science.1232808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wootten D, Reynolds CA, Smith KJ, Mobarec JC, Koole C, Savage EE, et al. The extracellular surface of the GLP-1 receptor is a molecular trigger for biased agonism. Cell. 2016;165:1632–1643. doi: 10.1016/j.cell.2016.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hua T, Vemuri K, Pu M, Qu L, Han GW, Wu Y, et al. Crystal structure of the human cannabinoid receptor CB1. Cell. 2016;167:750–762. doi: 10.1016/j.cell.2016.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Shim JY. Understanding functional residues of the cannabinoid CB1 receptor for drug discovery. Curr Top Med Chem. 2010;10:779–798. doi: 10.2174/156802610791164210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Andersson H, D'Antona AM, Kendall DA, Von Heijne G, Chin CN. Membrane assembly of the cannabinoid receptor 1: impact of a long N-terminal tail. Mol Pharmacol. 2003;64:570–577. doi: 10.1124/mol.64.3.570. [DOI] [PubMed] [Google Scholar]
- 80.Lu R, Hubbard JR, Martin BR, Kalimi MY. Roles of sulfhydryl and disulfide groups in the binding of CP-55,940 to rat brain cannabinoid receptor. Mol Cell Biochem. 1993;121:119–126. doi: 10.1007/BF00925970. [DOI] [PubMed] [Google Scholar]
- 81.Fay JF, Dunham TD, Farrens DL. Cysteine residues in the human cannabinoid receptor: only C257 and C264 are required for a functional receptor, and steric bulk at C386 impairs antagonist SR141716A binding. Biochemistry (Moscow) 2005;44:8757–8769. doi: 10.1021/bi0472651. [DOI] [PubMed] [Google Scholar]
- 82.McAllister SD, Hurst DP, Barnett-Norris J, Lynch D, Reggio PH, Abood ME. Structural mimicry in class A GpProtein-coupled receptor rotamer toggle switches. The importance of the F3.36(201)/W6.48(357) interaction in cannabinoid CB1 receptor activation. J Biol Chem. 2004;279:48024–48037. doi: 10.1074/jbc.M406648200. [DOI] [PubMed] [Google Scholar]
- 83.Hurst DP, Grossfield A, Lynch DL, Feller S, Romo TD, Gawrisch K, Pitman MC, Reggio PH. A lipid pathway for ligand binding is necessary for a cannabinoid G protein-coupled receptor. J Biol Chem. 2010;285:17954–17964. doi: 10.1074/jbc.M109.041590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Meye FJ, Ramakers GMJ, Adan RAH. The vital role of constitutive GPCR activity in the mesolimbic dopamine system. Transl Psychiatry. 2014;4:e361. doi: 10.1038/tp.2013.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Gyombolai P, Tóth AD, Tímár D, Turu G, Hunyady L. Mutations in the “DRY” motif of the CB1 cannabinoid receptor result in biased receptor variants. J Mol Endocrinol. 2015;54:75–89. doi: 10.1530/JME-14-0219. [DOI] [PubMed] [Google Scholar]
- 86.Walters RW, Shukla AK, Kovacs JJ, Violin JD, DeWire SM, Lam CM, et al. beta-Arrestin1 mediates nicotinic acid-induced flushing, but not its antilipolytic effect, in mice. J Clin Invest. 2009;119:1312–1321. doi: 10.1172/JCI36806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Dogra S, Yadav PN. Biased agonism at kappa opioid receptors: Implication in pain and mood disorders. Eur J Pharmacol. 2015;763(Part B):184–190. doi: 10.1016/j.ejphar.2015.07.018. [DOI] [PubMed] [Google Scholar]
- 88.Lovell KM, Frankowski KJ, Stahl EL, Slauson SR, Yoo E, Prisinzano TE, et al. Structure-activity relationship studies of functionally selective kappa opioid receptor agonists that modulate ERK 1/2 phosphorylation while preserving G protein over (βarrestin2 signaling bias. ACS Chem Neurosci. 2015;6:1411–1419. doi: 10.1021/acschemneuro.5b00092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Allen JA, Yost JM, Setola V, Chen X, Sassano MF, Chen M, et al. Discovery of (β-arrestin-biased dopamine D2 ligands for probing signal transduction pathways essential for antipsychotic efficacy. Proc Natl Acad Sci. 2011;108:18488–18493. doi: 10.1073/pnas.1104807108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Zhang H, Sturchler E, Zhu J, Nieto A, Cistrone PA, Xie J, et al. Lerner RA. Autocrine selection of a GLP-1R G-protein biased agonist with potent antidiabetic effects. Nat Commun. 2015;6:8918. doi: 10.1038/ncomms9918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Chavkin C. The therapeutic potential of κ-opioids for treatment of pain and addiction. Neuropsychopharmacology. 2011;36:369–370. doi: 10.1038/npp.2010.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.DeWire SM, Violin JD. Biased ligands for better cardiovascular drugs, Dissecting G-Protein-coupled receptor pharmacology. Circ Res. 2011;109:205–216. doi: 10.1161/CIRCRESAHA.110.231308. [DOI] [PubMed] [Google Scholar]
- 93.Nguyen PT, Schmid CL, Raehal KM, Selley DE, Bohn LM, Sim-Selley LJ. β-arrestin2 regulates cannabinoid CB1 receptor signaling and adaptation in a central nervous system region-dependent manner. Biol Psychiatry. 2012;71:714–724. doi: 10.1016/j.biopsych.2011.11.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Breivogel CS, Lambert JM, Gerfin S, Huffman JW, Razdan RK. Sensitivity to A9-tetrahydrocannabinol is selectively enhanced in beta-arrestin2-/- mice. Behav Pharmacol. 2008;19:298–307. doi: 10.1097/FBP.0b013e328308f1e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Montecucco F, Burger F, Mach F, Steffens S. CB2 cannabinoid receptor agonist JWH-015 modulates human monocyte migration through defined intracellular signaling pathways. Am J Physiol - Heart Circ Physiol. 2008;294:H1145–H1155. doi: 10.1152/ajpheart.01328.2007. [DOI] [PubMed] [Google Scholar]
- 96.Jordà MA, Verbakel SE, Valk PJM, Vankan-Berkhoudt YV, Maccarrone M, Finazzi-Agrò A, et al. Hematopoietic cells expressing the peripheral cannabinoid receptor migrate in response to the endocannabinoid 2-arachidonoylglycerol. Blood. 2002;99:2786–2793. doi: 10.1182/blood.V99.8.2786. [DOI] [PubMed] [Google Scholar]
- 97.Preet A, Qamri Z, Nasser MW, Prasad A, Shilo K, Zou X, et al. Cannabinoid receptors, CB1 and CB2, as novel targets for inhibition of non-small cell lung cancer growth and metastasis. Cancer Prev Res Phila Pa. 2011;4:65–75. doi: 10.1158/1940-6207.CAPR-10-0181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Kenakin T, Watson C, Muniz-Medina V, Christopoulos A, Novick S. A simple method for quantifying functional selectivity and agonist bias. ACS Chem Neurosci. 2011;3:193–203. doi: 10.1021/cn200111m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Iyer MR, Cinar R, Liu J, Godlewski G, Szanda G, Puhl H, et al. Structural basis of species-dependent differential affinity of 6-alkoxy-5-aryl-3-pyridinecarboxamide cannabinoid-1 receptor antagonists. Mol Pharmacol. 2015;88:238–244. doi: 10.1124/mol.115.098541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Hudson BD, Hébert TE, Kelly MEM. Ligand- and heterodimer-directed signaling of the CB(1) cannabinoid receptor. Mol Pharmacol. 2010;77:1–9. doi: 10.1124/mol.109.060251. [DOI] [PubMed] [Google Scholar]
- 101.Fribourg M, Moreno JL, Holloway T, Provasi D, Baki L, Mahajan R, et al. Decoding the signaling of a GPCR heteromeric complex reveals a unifying mechanism of action of antipsychotic drugs. Cell. 2011;147:1011–1023. doi: 10.1016/j.cell.2011.09.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.van der Westhuizen ET, Breton B, Christopoulos A, Bouvier M. Quantification of ligand bias for clinically relevant β2-adrenergic receptor ligands: implications for drug taxonomy. Mol Pharmacol. 2014;85:492–509. doi: 10.1124/mol.113.088880. [DOI] [PubMed] [Google Scholar]
- 103.White KL, Robinson JE, Zhu H, DiBerto JF, Polepally PR, Zjawiony JK, et al. The G protein-biased κ-opioid receptor agonist RB-64 is analgesic with a unique spectrum of activities in vivo. J Pharmacol Exp Ther. 2015;352:98–109. doi: 10.1124/jpet.114.216820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.DeWire SM, Yamashita DS, Rominger DH, Liu G, Cowan CL, Graczyk TM, et al. A G protein-biased ligand at the μ-opioid receptor is potently analgesic with reduced gastrointestinal and respiratory dysfunction compared with morphine. J Pharmacol Exp Ther. 2013;344:708–717. doi: 10.1124/jpet.112.201616. [DOI] [PubMed] [Google Scholar]
- 105.Violin JD, DeWire SM, Yamashita D, Rominger DH, Nguyen L, Schiller K, et al. Selectively engaging β-arrestins at the angiotensin II type 1 receptor reduces blood pressure and increases cardiac performance. J Pharmacol Exp Ther. 2010;335:572–579. doi: 10.1124/jpet.110.173005. [DOI] [PubMed] [Google Scholar]
- 106.Gesty-Palmer D, Yuan L, Martin B, Wood WH, Lee MH, Janech MG, et al. β-arrestin-selective G protein-coupled receptor agonists engender unique biological efficacy in vivo. Mol Endocrinol Baltim Md. 2013;27:296–314. doi: 10.1210/me.2012-1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Schmid CL, Bohn LM. Serotonin, but not N-methyltryptamines, activates the serotonin 2A receptor via a βarrestin2/Src/Akt signaling complex in vivo. J Neurosci Off J Soc Neurosci. 2010;30:13513–13524. doi: 10.1523/JNEUROSCI.1665-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Viscusi ER, Webster L, Kuss M, Daniels S, Bolognese JA, Zuckerman S, et al. A randomized, phase 2 study investigating TRV130, a biased ligand of the μ-opioid receptor, for the intravenous treatment of acute pain. Pain. 2015:1. doi: 10.1097/j.pain.0000000000000363. [DOI] [PubMed] [Google Scholar]
- 109.Soergel DG, Subach RA, Burnham N, Lark MW, James IE, Sadler BM, et al. Biased agonism of the μ-opioid receptor by TRV130 increases analgesia and reduces on-target adverse effects versus morphine: A randomized, double-blind, placebo-controlled, crossover study in healthy volunteers. Pain. 2014;155:1829–1835. doi: 10.1016/j.pain.2014.06.011. [DOI] [PubMed] [Google Scholar]
- 110.Soergel D, Subach RA, James IE, Cowan CL, Gowen M, Lark M. TRVO27, A beta-arrestin biased ligand at the angiotensin 2 type 1 receptor, produces rapid, reversible changes in hemodynamics in patients with stable systolic heart failure. J Am Coll Cardiol. 2013;61 doi: 10.1016/S0735-10971360683-X. [DOI] [Google Scholar]
- 111.Stott LA, Hall DA, Holliday ND. Unravelling intrinsic efficacy and ligand bias at G protein coupled receptors: a practical guide to assessing functional data. Biochem Pharmacol. 2016;101:1–12. doi: 10.1016/j.bcp.2015.10.011. [DOI] [PubMed] [Google Scholar]
- 112.Thompson GL, Lane JR, Coudrat T, Sexton PM, Christopoulos A, Canals M. Systematic analysis of factors influencing observations of biased agonism at the mu-opioid receptor. Biochem Pharmacol. 2016;113:70–87. doi: 10.1016/j.bcp.2016.05.014. [DOI] [PubMed] [Google Scholar]
- 113.Zhou L, Bohn LM. Functional selectivity of GPCR signaling in animals. Curr Opin Cell Biol. 2014;27:102–108. doi: 10.1016/j.ceb.2013.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Luttrell LM, Maudsley S, Bohn LM. Fulfilling the promise of “biased” G protein-coupled receptor agonism. Mol Pharmacol. 2015;88:579–588. doi: 10.1124/mol.115.099630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Volkow ND, Swanson JM, Evins AE, DeLisi LE, Meier MH, Gonzalez R, et al. Effects of cannabis use on human behavior, including cognition, motivation, and psychosis: a review, JAMA Psychiatry. 2016;73:292–297. doi: 10.1001/jamapsychiatry.2015.3278. [DOI] [PubMed] [Google Scholar]
- 116.Janero DR. Cannabinoid-1 receptor (CB1R) blockers as medicines: beyond obesity and cardiometabolic disorders to substance abuse/drug addiction with CB1R neutral antagonists. Expert Opin Emerg Drugs. 2012;17:17–29. doi: 10.1517/14728214.2012.660916. [DOI] [PubMed] [Google Scholar]
- 117.Bradley SJ, Tobin AB. Design of next-generation G protein-coupled receptor drugs: linking novel pharmacology and in vivo animal models. Annu Rev Pharmacol Toxicol. 2016;56:535–559. doi: 10.1146/annurev-pharmtox-011613-140012. [DOI] [PubMed] [Google Scholar]
- 118.Janero DR, Thakur GA. Leveraging allostery to improve G protein-coupled receptor (GPCR)-directed therapeutics: cannabinoid receptor 1 as discovery target. Expert Opin Drug Discov. 2016;11 doi: 10.1080/17460441.2016.1245289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Baillie GL, Horswill JG, Anavi-Goffer S, Reggio PH, Bolognini D, Abood ME, et al. CB1 receptor allosteric modulators display both agonist and signaling pathway specificity. Mol Pharmacol. 2013;83:322–338. doi: 10.1124/mol.112.080879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Ahn KH, Mahmoud MM, Shim JY, Kendall DA. Distinct roles of β-arrestin 1 and β-arrestin 2 in ORG27569-induced biased signaling and internalization of the cannabinoid receptor 1 (CB1) J Biol Chem. 2013;288:9790–9800. doi: 10.1074/jbc.M112.438804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Kulkarni PM, Kulkarni AR, Korde A, Tichkule RB, Laprairie RB, Denovan-Wright EM, et al. Novel electrophilic and photoaffinity covalent probes for mapping the cannabinoid 1 receptor allosteric site(s) J Med Chem. 2016;59:44–60. doi: 10.1021/acs.jmedchem.5b01303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Laprairie RB, Kulkarni AR, Kulkarni PM, Hurst DP, Lynch D, Reggio PH, et al. Mapping cannabinoid 1 receptor allosteric site(s): critical molecular determinant and signaling profile of GAT100, a novel, potent, and irreversibly binding probe. ACS Chem Neurosci. 2016;7:776–798. doi: 10.1021/acschemneuro.6b00041. [DOI] [PMC free article] [PubMed] [Google Scholar]