

Abstract
Limb-girdle muscular dystrophies (LGMDs) are common in India. Information on LGMDs has been gradually evolving in the recent years. This information is scattered in case series and case studies. The aim of this study is to collate available Indian information on LGMDs and put it in perspective. PubMed search using keywords such as limb-girdle muscular dystrophies in India, sarcoglycanopathies, dysferlinopathy, calpainopathy, and GNE myopathy was carried out. The published information on LGMDs in Indian context suggests that dysferlinopathy, calpainopathy, sarcoglycanopathies, and other myopathies such as GNE myopathy are frequently seen in India. Besides these, anecdotal reports of many other forms are available, some with genetic support and others showing immunocytochemical defects. The genotypic information on LGMDs is gradually evolving and founder mutations have been detected in selected populations. Further multicenter studies are necessary to document the incidence and prevalence of these common conditions in India.
Keywords: Calpainopathy, Dysferlinopathy, GNE myopathy, India, Limb-girdle muscular dystrophy, Sarcoglycanopathies
INTRODUCTION
The term limb-girdle muscular dystrophy (LGMD) is a broad term encompassing many conditions which present with weakness of the girdle musculature. Initially, patients who did not have known dystrophies such as Duchenne and Becker muscular dystrophies, facioscapulohumeral muscular dystrophies, and myotonic muscular dystrophies were clubbed together under this umbrella diagnosis, and for a while, it was unclear as to what this group represents. Early descriptions of LGMDs go back to the late 18th century, when Erb and Leyden-Mobius described patients having weakness which primarily involved the shoulder and the hip girdles, respectively, sparing the facial muscles. In 1951, Levison[1] documented for the first time, the autosomal recessive transmission in patients with LGMDs. In 1953, Stevenson[2] introduced the term “autosomal limb-girdle muscular dystrophy” for his patients with autosomal recessive inheritance. In 1954, Walton and Nattrass[3] defined LGMD as a distinct nosological entity in their classification system. With the development of diagnostic methods, it soon became evident that LGMD, as originally defined by Walton and Nattrass, comprised a wide variety of neuromuscular disorders, for example, Kugelberg Welander disease, polymyositis, endocrine myopathies, and some congenital and metabolic myopathies, and hence, the utility of the entity was limited and questionable. Gardner-Medwin omitted the term from the classification. In 1981, it found place as a major category of dystrophies in the Walton classification[4] and was divided into four subtypes: (1) autosomal recessive or sporadic (Erb, Leyden-Mobius), (2) myopathy limited to quadriceps, (3) autosomal recessive muscular dystrophy in childhood, and (4) late-onset autosomal dominant type. Over the ensuing years, with enormous progress in immunocytochemical evaluation of structural sarcolemmal proteins and molecular characterization with newer methods of testing, clear subgroups have evolved and continue to evolve rapidly. Autosomal dominant LGMDs are termed LGMD 1 and recessive LGMD 2; the numbering goes sequentially as per the time of discovery.
In India, Srinivas et al.[5] presented their initial observations on 35 patients with LGMDs. In the following four decades, studies have accumulated which describe clinical phenotypes of LGMDs. In the early 2000s, immunohistochemistry became available to some tertiary care centers in India and was utilized for diagnosis. Initial literature is about sarcoglycanopathies as these were the only stains available for use then. Studies reported the presence of alpha-sarcoglycanopathy followed by beta- and gamma-sarcoglycanopathy diagnosed by immunohistochemistry as isolated case reports and small cohort studies.[6,7,8,9,10,11,12,13,14,15,16] The second LGMD phenotype diagnosed by immunohistochemistry in India happens to be dysferlinopathy,[17,18,19] followed by calpainopathy. A few other subtypes are also now known to exist in India.
In recent years, genetic analysis of these LGMD phenotypes is becoming increasingly available in the service sector and the research units and studies regarding genetic details of these various types of LGMD are being undertaken in various part of India. Studies describing the genotypes of sarcoglycanopathies, dysferlinopathy, and calpainopathy are now published from India. These efforts are very limited in the context of the population of India, and we have not yet reached the point of understanding the prevalence patterns of these diseases in our large country with diverse population. While hospital-based data regarding prevalence of particular LGMD phenotypes cannot be applied to whole country, it seems that dysferlinopathy, calpainopathy, and other myopathies GNE myopathy are among common LGMDs in India.
LOCAL ISSUES PERTAINING TO PATTERNS OF INHERITANCE OF LIMB-GIRDLE MUSCULAR DYSTROPHIES IN INDIA
In India, consanguineous marriages are customary, leading to increased prevalence of autosomal recessive LGMDs. The large population of India, from a matrimonial aspect, is divided into many small segments, based on the castes, subcastes, etc. People tend to marry within these small genetic pools and hence combinations within the genetic stock take place. Within a community, there are multiple “gotras” and care is taken to see that the bride and bridegroom do not belong to the same “gotra” to avoid inbreeding. However, this becomes insufficient as the current custom only looks at the paternal “gotra” and not the maternal. Conventionally, Manusmriti, the ancient Indian treatise, advises to ensure that there is no repetition among the four gotras, two paternal and two maternal. Currently, in communities with a small number of gotras, it becomes difficult to follow this ancient wisdom and hence the modern short cuts are being applied. Some communities advocate marriage of first cousins, increasing autosomal recessive diseases. In India, genetic diseases are shunned and hence obtaining family history can be met with difficulties. Furthermore, people have to be made aware that information of the maternal family is required as well; surprisingly, a lot of families consider maternal section as a different family.
Dysferlinopathy, GNE myopathy, calpainopathy, and sarcoglycanopathies are common and have been documented in various investigative studies. GNE myopathy is included in this discussion as the clinical presentation is of limb weakness and is a common disease in the Indian context. We shall now describe the clinical, immunocytochemical, and genetic characterization of these diseases with particular references to our local settings. An algorithm has been presented to help the clinical evaluation [Algorithm 1].
Algorithm 1.
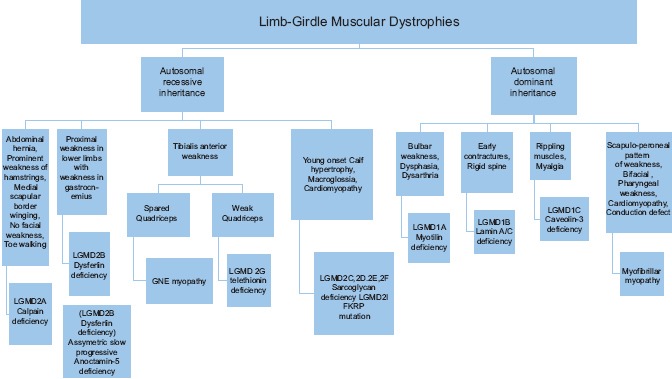
Clinical evaluation of limb-girdle muscular dystrophies
DYSFERLINOPATHY
Clinical characteristics
Dysferlinopathy is an autosomal recessive LGMD resulting from the deficiency of the sarcolemmal protein dysferlin secondary to DYSF gene mutation on chromosome 2p. Two common clinical entities are described under the label of dysferlinopathy: Miyoshi myopathy (distal onset) and LGMD 2B (proximal onset). Initially, Miyoshi and colleagues described the distal form with burnt on the gastrocnemius muscles and labeled it as Miyoshi distal myopathy. The genetic identification of Miyoshi myopathy coincided with the same genetic defect being identified in a set of proximal myopathies termed LGMD 2B. Hence, both these presentations are clubbed together as dysferlinopathy. While the condition was originally discovered in Japan, it is now recognized to be a common LGMD in most parts of the world.
Patients present in the second decade of life, earlier presentations are uncommon. The initial weakness is in the gastrocnemius muscles, which is usually discovered while standing on toes for sporting activities or exercise programs. Gradually, patients are unable to stand on toes and calf muscles are wasted [Figure 1a]. Hamstrings and hip flexors become progressively weakened and patients develop difficulties in climbing stairs and rising from the ground. As people in India customarily squat to defecate, proximal muscle weakness comes to early attention. Upper limbs are affected later and biceps may show a “lump.” This lump is not unique to this condition but is frequently seen. Similarly, the quadriceps muscle is known to show a diamond-like configuration of hypertrophy and atrophy [Figure 1b]. Ambulation is maintained for many years and the progress is gradual. A proportion of patients begins with proximal weakness and then goes on to have distal involvement. Clinically, the proximodistal weakness is of most frequent occurrence. Uncommonly, the brunt of the disease could be on tibialis anterior muscles (DMAT) Distal Myopathy with Anterior Tibial onset and minority of patients goes through a phase of intense calf pains with some swelling and tenderness before they waste and weaken. Other less common clinical patterns are asymptomatic elevation of serum creatine phosphokinase and bent spine syndrome or camptocormia.
Figure 1.
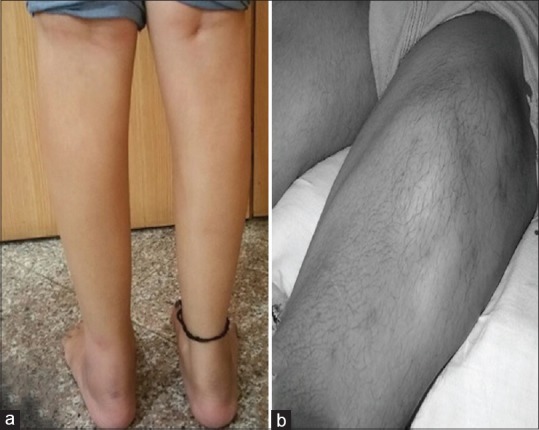
Dysferlinopathy. (a) Wasting of calf muscles. (b) Diamond sign
In India, three studies depicting clinical features of dysferlinopathy are available.[17,18,19] Khadilkar et al.[17] in 2004 investigated 14 patients with dysferlinopathy. In their series, the initial symptoms in nine patients were of distal weakness and wasting while remaining five had proximal weakness. Six patients had asymmetrical onset of disease. Three patients had initial calf pains with transient hypertrophy, followed by atrophy. Gastrocnemius and tibialis were affected almost equally. In proximal weakness, muscles involved were iliopsoas, hip adductors, hamstrings, and quadriceps. Upper limbs were slightly affected. Overall, the phenotype was mild and patients were ambulatory till late in disease. In the study undertaken by Nalini et al. in 2008,[19] out of 28 patients, 12 patients had distal onset (gastrocnemius weakness), 12 patients had proximal onset, 2 had proximodistal onset, and 2 had tibial onset distal weakness. Muscle pain was the prominent symptom (47%). In upper limb, deltoid and biceps were weak and wasted. Ambulation was maintained till late in the disease course.
Pradhan[18] noted two interesting signs in dysferlinopathy; calf head on trophy sign and diamond on quadriceps. Calf head on trophy sign is seen in Miyoshi myopathy. It is observed from posterior side with patient's shoulders abducted and elbows flexed at 90°. It is due to selective atrophy and sparing of certain muscles in periscapular region. Diamond on quadriceps is the result of selective wasting of vastus lateralis and rectus femoris in their upper and lower part with sparing of middle part. Whenever the muscle is in action, demarcation between wasted and preserved part becomes prominent giving rise to diamond on quadriceps sign.
Histopathology
The histological findings in dysferlinopathy are suggestive of a dystrophic process evidenced by de- and re-generation of muscle fibers, increase in connective tissue, and myophagocytosis. One interesting aspect of the histology is the presence of inflammatory cell infiltrates. In some biopsies, these can be very prominent and lead to a confusion with inflammatory myopathy. In fact, from time to time, we come across patients in India who have received corticosteroids following biopsy, with modest or no results and some cases, deterioration of muscle strength. On immunostaining and immunoblotting, dysferlin staining is reduced or is absent in the muscle fibers.
Dysferlin is probably the only muscle protein to be expressed in the peripheral blood monocytes. Western blotting for dysferlin can be utilized in the diagnosis of dysferlinopathy as the concordance between the finding of Western blotting of peripheral monocytes and genetic confirmation is favorable. However, the blotting process can be expensive, offsetting the benefits.
Genetics
Dysferlinopathy results from mutations in dysferlin gene on chromosome 2p13.3-p13.1, which contains 55 codon exons.[20] Missense, nonsense, small deletions, small insertions, and splice mutation have all been described. Most mutations introduce a stop codon or result in premature truncation of dysferlin protein.[21] While there is no known mutational hot spot in gene, there are seven recorded founder mutations in various populations worldwide. These have been detected in Portuguese (c.1180_1180 + 7delAGTGCGTG, c.5492G>A),[22,23] native Canadian (c.2745C>G),[24] Caucasian Jewish (c.2779delG),[25] Italian (c.2875C>T),[26] Lebanese Jewish (c.4872delG.fsX9),[27] and Spanish (c.6086C>T)[28] populations.
In India, a center for genetic diagnosis of dysferlinopathy is set up at Mumbai with the efforts and aids from the Jain Foundation, United States of America. The center has been active and receives samples from multiple places in India and recently also from Pakistan and Sri Lanka. These samples are currently processed free of cost. Over a hundred cases have been genetically characterized. No founder mutation has been identified and missense, frameshift, and splice site mutations have been detected. Uncommonly, intronic mutations are also described in this cohort.
CALPAINOPATHY
Clinical characteristics
The first autosomal recessive LGMD to be described is calpainopathy (LGMD 2A) caused by mutations in CAPN 3 gene encoding calpain 3 protein resulting in calpain deficiency. The age of onset is usually in the second decade of life and both sexes are affected. Rarely, early and severe presentations are encountered. Three phenotypes have been identified based on the distribution of muscle weakness.
Leyden-Mobius LGMD phenotype in which muscle weakness is first evident in the pelvic girdle and later in the shoulder girdle with onset before age 12 years or after age 30 years
Erb LGMD phenotype in which muscle weakness is first evident in the shoulder girdle and later in the pelvic girdle. This is milder variant with early age of onset
HyperCKemia in which asymptomatic individuals have only high serum creatine kinase (CK) concentrations. Usually, it occurs in young children.
There is proximal muscle weakness (pelvic and/or shoulder girdle) with atrophy and sparing of facial and neck muscles. Scapular winging, scoliosis, Achilles tendon, and other joint contractures are common. There is no evidence of cardiomyopathy. Scapular winging [Figure 2a] is prominent in calpainopathy as compared to other LGMDs and often is evident early in the course of the disease. Similarly, tendon Achilles contractures tend to be early and prominent and patients tend to walk on their toes. Patients in whom the disease begins in the shoulder girdle, a differential diagnosis of facioscapulohumeral muscular dystrophy needs to be considered. Patients with calpainopathy do not have facial weakness. Within the hip girdle musculature, hip adductors are severely affected in calpainopathy. Some patients develop severe weakness of the external oblique muscles and abdominal hernia [Figure 2b]. In the author's experience, one such patient presented initially to a surgeon and then was directed to the neurology service. He/she required hernia repair. Children presenting with hyperCKemia are often investigated extensively without a definitive diagnosis, till the clinical features start evolving, pointing to the diagnosis.
Figure 2.
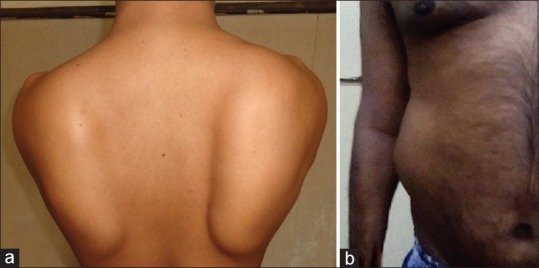
Calpainopathy. (a) Scapular winging. (b) Abdominal hernia due to weakness of external oblique muscle
While the prevalence of calpainopathy is unclear, it seems to be a common form of LGMD in India. In a histologically oriented study, Pathak et al.[29] documented abnormalities of the calpain protein in almost 40% of their LGMD biopsies. This study did not include genetic information and hence secondary protein changes, particularly with dysferlinopathy, dilute the inferences.
Immunoblotting
This muscle protein cannot be immunostained but has to be western blotted. All the three (94, 60, and 30 Kd) fractions of the protein should be checked as only some may be reduced. Approximately 80% of individuals with mutation of CAPN3 show absence or a severe reduction of calpain-3 bands on Western blot. While these quantitative measurements are helpful, they lack specificity as secondary reductions of the calpain protein have been reported in other muscular dystrophies such as dysferlinopathy, dystrophinopathy, and Udd muscular dystrophy (titinopathy).
Genetics
Leiden muscular dystrophy mutation database documents 490 CAPN3 mutations, which are mostly of the missense type (about 60%). c.550delA mutation is the most common allele in Caucasian populations.[30] Founder effect for the c.550delA mutation has been demonstrated in the Eastern Mediterranean region (Russia, Croatia, Bulgaria, and Northern Italy)[31,32,33,34,35,36,37,38,39,40,41,42] Other rare mutations which have a founder effect in small populations due to cultural inbreeding associated with historical, demographic, or religious factors have been reported in the population of La Reunion Island in the Indian Ocean (c.946-1G>A),[43] in the Old Order Amish community in Northern Indiana in the USA (c.2306G>A),[44,45] in the Guipuzcoa region of the Basque Country of Spain and in Brazil (c.2362_2363delAGinsTCATCT),[46,47,48] Japan (c.1795_1796insA),[49,50,51] the Chioggia village in the Venetian lagoon (p.R490Q),[35] the Mocheni population in the Italian Alps (c.1193 + 6T>A),[52] and the Agarwal community in Northern India (p.D780H and c.2099-1G>T).[53] The last one is relevant to India and hence is discussed further.
Founder mutations in Agarwal
Clinical observations that a rather uniform phenotype results in scapular winging and toe walking lead to long-term in-depth studies of this Agarwal cohort of individuals in Mumbai. Investigations detected two ancestral founder mutations in calpain gene, a missense (c.2338G>C; p.D780H) and a splice-site (c.2099-1G>T) mutation.[53] Patients can be either heterozygous for both or homozygous for either of these mutations. These mutations are believed to have arisen due to the practice of intra-communal exogamy in the community of Agarwal. Recently, Khadilkar et al. have published the utility of founder mutations in CAPN3 gene in Agarwal community. This founder allele analysis can be utilized as the initial noninvasive diagnostic step for index cases, carrier detection, and counseling, with almost 90% yield.[54]
SARCOGLYCANOPATHIES
While sarcoglycanopathies were the initially identified group of LGMDs in India, they are uncommon and form a differential diagnosis of Duchene phenotype in both males and females.
Clinical characteristics
Sarcoglycanopathies tend to present in the first decade of life with a phenotype similar to the common Duchenne muscular dystrophy (DMD). Milder presentations are known but uncommon. As this is an autosomal recessive disease, both males and females present with equal severity, which is exceptional for manifesting carrier females of DMD. Children have easy falls and difficulty in climbing stairs and getting up from ground. Calves are enlarged, sometimes very much so, and weakness pattern remains largely proximal. Children walk with waddle and exhibit lordosis. Cardiac involvement is known but not as common or prominent as in dystrophinopathies.
Available studies[11,16,55,56] on sarcoglycanopathies from India have been largely based on immunocytochemical characterization and hence need to be put in that context. Only one small study of genetic characterization is available.[57] Mean age of onset varies in all 4 studies ranging from 6 to 21 years. Initial symptoms started in hip girdle muscles followed by shoulder girdle involvement.
Histopathology
Skin biopsy has been utilized for immunocytochemical studies in few LGMDs. One study described role of skin biopsy in diagnosis of sarcoglycanopathy in two cases.[58] Studies have also evaluated role of skin biopsy in diagnosing dystrophinopathy[59,60] and few congenital muscular dystrophies such as merosin-deficient congenital muscular dystrophy,[61,62] X-linked Emery–Dreifuss muscular dystrophy,[63] and Ullrich muscular dystrophy.[64] Larger studies are required for validation of skin biopsy in various LGMDs and other dystrophies. At present, it can be used as adjunct to the muscle biopsy.
Sarcoglycans (SG) are components of the dystrophin-associated glycoproteins on the muscle membrane. Four subunits (alpha, beta, gamma, and delta) can be individually immunostained on the fresh frozen muscle. As they are functionally interlinked, loss of more than one subunit on the immunostaining is common and immunostaining is often proves to be inadequate to know the primary defect. Worldwide alpha-SG deficiency is most common. This is followed by gamma- and beta-SG deficiency. Delta-SG deficiency is least common. However, the prevalence of subtypes varies with the population under evaluation; for example, gamma-SG opathy is most common in Tunisia.[65] Secondary dystrophin deficiency is well known in SG opathies, a fact that needs to be borne in mind, when evaluating children with the DMD phenotype.
Genetics
Mutations in one of the four genes for SG complex in skeletal muscle can cause LGMD 2C-F. LGMD 2C (gamma-sarcoglycanopathy), 2D (alpha-sarcoglycanopathy), 2E (beta-sarcoglycanopathy), and 2F (delta-sarcoglycanopathy) are caused by mutations in SGCG gene (13q12), SGCA gene (17q12-q21.3), SGCB gene (4q12), and SGCD (5q33), respectively. Founder mutations in beta-sarcoglycanopathy are found in Amish population (p.T151R)[66,67] and Northern Italy (c.377–384dupl).[68] In gamma-sarcoglycanopathy, founder mutations found in North Africans (c.525delT),[69] Roma Gypsies (p.C283Y),[70] and Northern Italy (c.87insT),[69] In delta-sarcoglycanopathy, founder mutation has been found in Brazilian population (c.656delC).[71] Most common mutation in alpha-sarcoglycanopathy is p.R77C[72] with founder effect in Finland[73] and Magdalen.[74] Other founder mutations are p.R34 L in Taiwan[75] and p.R192X in Egypt.[76]
In India, genetically confirmed patients are few and one small series and some case reports are available. Khadilkar et al.[57] analyzed genetic aspects of sarcoglycanopathies. In this study, a total of 18 patients showed mutation in SG genes. Among them, gamma-SG gene mutation (44.4%) was most common followed by delta (27.77%), alpha (22.22%), and beta (5.55%). Among gamma-SG mutation, 525del T was encountered in this study, again a curious fact as this abnormality is seen in the Maghrebian population.[77] Haplotype analysis however has not been undertaken in these patients.
OTHER LIMB-GIRDLE MUSCULAR DYSTROPHIES IN INDIA
Other LGMDs have been infrequently reported from India,[78] and these are discussed below in brief. LGMD 2G known as telethoninopathy is caused by mutation in TCAP gene on chromosome 17q11-12. Age of onset is in the second decade and presented with pelvic girdle weakness with foot drop. LGMD 2H is caused by mutation in TRIM32 (cytosolic enzyme) on chromosome 9q33.1., characterized by proximal weakness, atrophy, and moderately raised levels of CK. D487N mutation of TRIM32 causes the more severe sarcotubular myopathy. LGMD 2I and 2K are caused by mutation in proteins FKRP and POMT1, respectively, which is required for glycosylation of transmembrane protein named dystroglycan. Among this, LGMD 2I is fairly common[79] and cardiac and respiratory functions get involved frequently. LGMD 2I clinically resembles severe Duchenne-like or milder late-onset Becker-like phenotypes. In large series by Nalini et al.,[78] 8 patients of LGMD 2G, 16 patients of LGMD 2I, 10 patients of LGMD 2K, 2 patients of LGMD 2H and 2J each have been detected, based on immunohistochemistry and Western blotting. LGMD 2 L is caused by mutations in the anoctamin-5 (ANO5) gene at 11p14.3. Clinically, it is characterized by asymmetric quadriceps, hamstring, biceps, brachioradialis, or calf weakness and atrophy. Muscle pain is frequent symptom. Anecdotal occurrence of anoctaminopathy in India is known (Khadilkar et al., unpublished data). LGMD 2J is caused by mutation in the titin gene on chromosome 2q31.2. Clinically, patients present in 10–40 years of age group with proximal limb-girdle weakness and tibialis anterior weakness. Nalini et al.[78] identified two such patients who had lower-limb proximal muscle weakness with foot drop, severe wasting of tibialis anterior.
GNE (UDP-N-ACETYLGLUCOSAMINE 2-EPIMERASE/N-ACETYLMANNOSAMINE KINASE) MYOPATHY
While GNE myopathy is classified under distal myopathies, it is included in the review as it is frequent, and as the disease progresses, it affects the hip and shoulder girdle musculatures. This disorder has been named variously as hereditary inclusion body myopathy, distal myopathy with rimmed vacuoles, Nonaka myopathy, and quadriceps sparing myopathy. It is an autosomal recessive or sporadic distal myopathy due to mutation in gene encoding bifunctional enzyme UDP-N-acetylglucosamine 2-epimerase and N-acetylmannosamine kinase. Uncommon autosomal dominant transmissions are known. GNE myopathy has been encountered in most parts of India. Initial studies have been from NIMHANS, Bengaluru, which documented the existence of GNE myopathy in India and gave the early clinical and histological characteristics of the condition.[80] A case series[81] from the north of India has recently been available. Data from Mumbai also document GNE myopathy, with genetic peculiarities in the people with Rajasthan ancestry.[82]
Clinical characteristics
GNE myopathy is adult-onset distal myopathy. Patients present in the second or third decade, characteristically with a foot drop. Wasting of tibialis anterior becomes evident [Figure 3a]. This makes them vulnerable to falls and they cannot run well. This is then followed by weakness of the posterior thigh muscles, iliopsoas, and biceps muscles. Distal upper limb muscles are also affected, and first dorsal interosseous muscle may be wasted [Figure 3b]. Quadriceps muscle remains strong in most patients till late in the illness. In fact, the sparing of the quadriceps is so striking in the face of extreme weakness of some other groups of muscles that the disease also bore the name of quadriceps sparing myopathy. The initial distal weakness and wasting of the lower and sometimes upper limb muscles lead the clinician to believe that the nature of the illness could be neurogenic and initial misdiagnosis as inherited neuropathy or spinal muscular atrophy is not uncommon. Moreover, the serum CK is only modestly elevated in this disease, adding to difficulties.
Figure 3.
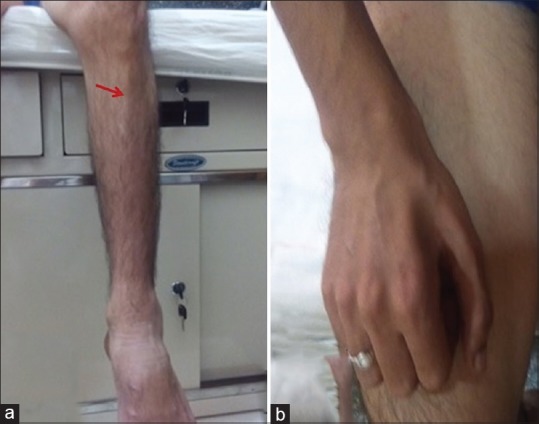
GNE myopathy. (a) Wasting of tibialis anterior muscle resulting in prominence of shin of tibia. (b) Wasting of first dorsal interosseous muscle
In studies available from India, mean age of onset is 27–28 years. An initial symptom was foot drop in large number of patients.[80] Symptoms of proximal weakness are also encountered in India, first as patients come to attention late into their illness and second due to the custom of squatting and sitting on the floor. Thus, proximal as well as distal weakness is often seen at the time of examination.
Histopathology
An important practical point is the site of muscle biopsy. Conventionally, quadriceps has been chosen for biopsy, which is almost unaffected in this condition. Biopsies from biceps or the upper third of tibialis anterior muscles are suitable to show changes. Rimmed vacuoles are consistently but not invariably seen in histology GNE myopathy patients. Other nonspecific findings of myopathy such as atrophy of muscle fibers and fiber size variation are present in most patients.
Genetics
GNE myopathy results from mutation in GNE gene on chromosome 9. Patients with Middle Eastern origin are found to harbor a GNE gene founder mutation p.M743T.[83] Two other founder mutations have been recognized in Japanese population p.D207V[84,85] and p.V603 L.[85] Recently, European Roma Gypsies has been detected to have founder mutation p.I618T.[86] p.A662V and p.V727M are a frequently found mutation in Southeast Asia.[87,88,89] A potential founder mutation p.A409T was described in a cohort of GNE myopathy patients from the British Islands.[90]
Limited information on the genetic constellation of GNE myopathy patients is available from India. c.2179G>A (p.V727M) is a common mutation 75%[89] and 32.7%[82] found in India. Khadilkar et al. have recently documented an interesting occurrence of homozygous mutation c.1853T>C (p.I618T) (24.5%) in Rajasthani Jain and Maheshwari communities: a founder mutation encountered exclusively in European Roma Gypsies.[82] This observation further strengthens the view that European Roma Gypsies originated from Northwestern India.
AUTOSOMAL DOMINANT LIMB-GIRDLE MUSCULAR DYSTROPHIES 1
Autosomal dominant LGMDs are less common than autosomal recessive forms. Anecdotal case reports are available from India. Jadhav et al. from South India reported one Indian family of EDMD2 with familial dilated cardiomyopathy and cardiac dysrhythmias, in whom LMNA gene sequencing was performed.[91] Recently, Jain et al. from All India Institute of Medical Sciences, Delhi, reported a case of myofibrillar myopathy presenting as neonatal intestinal pseudo-obstruction.[92]
CONCLUDING REMARKS
LGMDs are commonly seen in India and information on the subtypes is slowly accumulating. Regional variations in the prevalence are expected and founder mutations may be detected in populations of India due to the common practice of intracommunal exogamy. At present, dysferlinopathy, calpainopathy, and GNE myopathy seem to be much commoner than others in small hospital-based studies. This information may be of use in selecting the investigative panels for these diseases. With widespread use of genetic tests, genetic counseling, antenatal diagnosis, and preventive measures are expected to be in the forefront in the coming years.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
REFERENCES
- 1.Levison H. Dystrophia musculorum progressiva; clinical and diagnostic criteria, inheritance. Acta Psychiatr Neurol Scand Suppl. 1951;76:1–176. [PubMed] [Google Scholar]
- 2.Stevenson A. Muscular dystrophy in Northern Ireland, I. An account of the condition in fifty - one families. Ann Eugen. 1953;18:50–93. doi: 10.1111/j.1469-1809.1952.tb02497.x. [DOI] [PubMed] [Google Scholar]
- 3.Walton JN, Nattrass FJ. On the classification, natural history and treatment of the myopathies. Brain. 1954;77:169–231. doi: 10.1093/brain/77.2.169. [DOI] [PubMed] [Google Scholar]
- 4.Walton JN, Gardner-Medwin D. Progressive muscular dystrophy and the myotonic disorders. In: Walton J, editor. Disorders of Voluntary Muscle. 4th ed. Edinburgh: Churchill Livingstone; 1981. pp. 481–524. [Google Scholar]
- 5.Srinivas K. The myopathies (1950-1975) Proc Inst Neurol. 1975;5:102–12. [Google Scholar]
- 6.Handa V, Mital A, Gupta M, Goyle S. Deficiency of the 50 kDa dystrophin-associated-glycoprotein (adhalin) in an Indian autosomal recessive limb girdle muscular dystrophy patient: Immunochemical analysis and clinical aspects. Neurol India. 2001;49:19–24. [PubMed] [Google Scholar]
- 7.Dua T, Kalra V, Sharma MC, Kabra M. Adhalin deficiency: An unusual cause of muscular dystrophy. Indian J Pediatr. 2001;68:1083–5. doi: 10.1007/BF02722364. [DOI] [PubMed] [Google Scholar]
- 8.Joshi S. DMD in a female child. Indian Pediatr. 2002;39:98. [PubMed] [Google Scholar]
- 9.Kapoor S, Tatke M, Aggarwal S, Gupta A. Beta-sarcoglycanopathy. Indian J Pediatr. 2005;72:71–4. doi: 10.1007/BF02760585. [DOI] [PubMed] [Google Scholar]
- 10.Gulati S, Leekha S, Sharma MC, Kalra V. Gamma-sarcoglycanopathy. Indian Pediatr. 2003;40:1077–81. [PubMed] [Google Scholar]
- 11.Khadilkar SV, Singh RK, Katrak SM. Sarcoglycanopathies: A report of 25 cases. Neurol India. 2002;50:27–32. [PubMed] [Google Scholar]
- 12.Khadilkar SV, Singh RK. Current concepts in Limb girdle muscular dystrophy. Does hip adductor weakness mark the Indian phenotype? Reviews in Neurology. 2000:34–43. [Google Scholar]
- 13.Khadilkar SV, Singh RK. Hip abduction sign: A new clinical sign in sarcoglycanopathy. J Clin Neuromuscul Dis. 2001;3:13–5. doi: 10.1097/00131402-200109000-00003. [DOI] [PubMed] [Google Scholar]
- 14.Sharma MC, Mannan R, Singh NG, Gulati S, Kalra V, Sarkar C. Sarcoglycanopathies: A clinicopathological study of 13 cases [corrected] Neurol India. 2004;52:446–9. [PubMed] [Google Scholar]
- 15.Meena AK, Sreenivas D, Sundaram C, Rajasekhar R, Sita JS, Borgohain R, et al. Sarcoglycanopathies: A clinico-pathological study. Neurol India. 2007;55:117–21. doi: 10.4103/0028-3886.32781. [DOI] [PubMed] [Google Scholar]
- 16.Nalini A, Gayathri N, Thaha F, Das S, Shylashree S. Sarcoglycanopathy: Clinical and histochemical characteristics in 66 patients. Neurol India. 2010;58:691–6. doi: 10.4103/0028-3886.72164. [DOI] [PubMed] [Google Scholar]
- 17.Khadilkar SV, Singh RK, Kulkarni KS, Chitale AR. A study of clinical and laboratory features of 14 Indian patients with dysferlinopathy. J Clin Neuromuscul Dis. 2004;6:1–8. doi: 10.1097/01.cnd.0000134859.41385.6e. [DOI] [PubMed] [Google Scholar]
- 18.Pradhan S. Diamond on quadriceps: A frequent sign in dysferlinopathy. Neurology. 2008;70:322. doi: 10.1212/01.wnl.0000298091.07609.a0. [DOI] [PubMed] [Google Scholar]
- 19.Nalini A, Gayathri N. Dysferlinopathy: A clinical and histopathological study of 28 patients from India. Neurol India. 2008;56:379–85. doi: 10.4103/0028-3886.40964. [DOI] [PubMed] [Google Scholar]
- 20.Aoki M, Liu J, Richard I, Bashir R, Britton S, Keers SM, et al. Genomic organization of the dysferlin gene and novel mutations in Miyoshi myopathy. Neurology. 2001;57:271–8. doi: 10.1212/wnl.57.2.271. [DOI] [PubMed] [Google Scholar]
- 21.Wenzel K, Carl M, Perrot A, Zabojszcza J, Assadi M, Ebeling M, et al. Novel sequence variants in dysferlin-deficient muscular dystrophy leading to mRNA decay and possible C2-domain misfolding. Hum Mutat. 2006;27:599–600. doi: 10.1002/humu.9424. [DOI] [PubMed] [Google Scholar]
- 22.Vernengo L, Oliveira J, Krahn M, Vieira E, Santos R, Carrasco L, et al. Novel ancestral dysferlin splicing mutation which migrated from the Iberian peninsula to South America. Neuromuscul Disord. 2011;21:328–37. doi: 10.1016/j.nmd.2011.02.003. [DOI] [PubMed] [Google Scholar]
- 23.Santos R, Oliveira J, Vieira E, Coelho T, Carneiro AL, Evangelista T, et al. Private dysferlin exon skipping mutation (c.5492G>A) with a founder effect reveals further alternative splicing involving exons 49-51. J Hum Genet. 2010;55:546–9. doi: 10.1038/jhg.2010.60. [DOI] [PubMed] [Google Scholar]
- 24.Weiler T, Bashir R, Anderson LV, Davison K, Moss JA, Britton S, et al. Identical mutation in patients with limb girdle muscular dystrophy type 2B or Miyoshi myopathy suggests a role for modifier gene(s) Hum Mol Genet. 1999;8:871–7. doi: 10.1093/hmg/8.5.871. [DOI] [PubMed] [Google Scholar]
- 25.Leshinsky-Silver E, Argov Z, Rozenboim L, Cohen S, Tzofi Z, Cohen Y, et al. Dysferlinopathy in the Jews of the Caucasus: A frequent mutation in the dysferlin gene. Neuromuscul Disord. 2007;17:950–4. doi: 10.1016/j.nmd.2007.07.010. [DOI] [PubMed] [Google Scholar]
- 26.Cagliani R, Fortunato F, Giorda R, Rodolico C, Bonaglia MC, Sironi M, et al. Molecular analysis of LGMD-2B and MM patients: Identification of novel DYSF mutations and possible founder effect in the Italian population. Neuromuscul Disord. 2003;13:788–95. doi: 10.1016/s0960-8966(03)00133-0. [DOI] [PubMed] [Google Scholar]
- 27.Argov Z, Sadeh M, Mazor K, Soffer D, Kahana E, Eisenberg I, et al. Muscular dystrophy due to dysferlin deficiency in Libyan Jews. Clinical and genetic features. Brain. 2000;123(Pt 6):1229–37. doi: 10.1093/brain/123.6.1229. [DOI] [PubMed] [Google Scholar]
- 28.Vilchez JJ, Gallano P, Gallardo E, Lasa A, Rojas-García R, Freixas A, et al. Identification of a novel founder mutation in the DYSF gene causing clinical variability in the Spanish population. Arch Neurol. 2005;62:1256–9. doi: 10.1001/archneur.62.8.1256. [DOI] [PubMed] [Google Scholar]
- 29.Pathak P, Sharma MC, Sarkar C, Jha P, Suri V, Mohd H, et al. Limb girdle muscular dystrophy type 2A in India: A study based on semi-quantitative protein analysis, with clinical and histopathological correlation. Neurol India. 2010;58:549–54. doi: 10.4103/0028-3886.68675. [DOI] [PubMed] [Google Scholar]
- 30.Fanin M, Angelini C. Protein and genetic diagnosis of limb girdle muscular dystrophy type 2A: The yield and the pitfalls. Muscle Nerve. 2015;52:163–73. doi: 10.1002/mus.24682. [DOI] [PubMed] [Google Scholar]
- 31.Dinçer P, Leturcq F, Richard I, Piccolo F, Yalnizoglu D, de Toma C, et al. A biochemical, genetic, and clinical survey of autosomal recessive limb girdle muscular dystrophies in Turkey. Ann Neurol. 1997;42:222–9. doi: 10.1002/ana.410420214. [DOI] [PubMed] [Google Scholar]
- 32.Pogoda TV, Krakhmaleva IN, Lipatova NA, Shakhovskaya NI, Shishkin SS, Limborska SA. High incidence of 550delA mutation of CAPN3 in LGMD2 patients from Russia. Hum Mutat. 2000;15:295. doi: 10.1002/(SICI)1098-1004(200003)15:3<295::AID-HUMU15>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 33.Canki-Klain N, Milic A, Kovac B, Trlaja A, Grgicevic D, Zurak N, et al. Prevalence of the 550delA mutation in calpainopathy (LGMD 2A) in Croatia. Am J Med Genet A. 2004;125A:152–6. doi: 10.1002/ajmg.a.20408. [DOI] [PubMed] [Google Scholar]
- 34.Chrobáková T, Hermanová M, Kroupová I, Vondrácek P, Maríková T, Mazanec R, et al. Mutations in Czech LGMD2A patients revealed by analysis of calpain3 mRNA and their phenotypic outcome. Neuromuscul Disord. 2004;14:659–65. doi: 10.1016/j.nmd.2004.05.005. [DOI] [PubMed] [Google Scholar]
- 35.Fanin M, Nascimbeni AC, Fulizio L, Angelini C. The frequency of limb girdle muscular dystrophy 2A in northeastern Italy. Neuromuscul Disord. 2005;15:218–24. doi: 10.1016/j.nmd.2004.11.003. [DOI] [PubMed] [Google Scholar]
- 36.Milic A, Canki-Klain N. Calpainopathy (LGMD2A) in Croatia: Molecular and haplotype analysis. Croat Med J. 2005;46:657–63. [PubMed] [Google Scholar]
- 37.Balci B, Aurino S, Haliloglu G, Talim B, Erdem S, Akcören Z, et al. Calpain-3 mutations in Turkey. Eur J Pediatr. 2006;165:293–8. doi: 10.1007/s00431-005-0046-3. [DOI] [PubMed] [Google Scholar]
- 38.Todorova A, Georgieva B, Tournev I, Todorov T, Bogdanova N, Mitev V, et al. A large deletion and novel point mutations in the calpain 3 gene (CAPN3) in Bulgarian LGMD2A patients. Neurogenetics. 2007;8:225–9. doi: 10.1007/s10048-007-0083-3. [DOI] [PubMed] [Google Scholar]
- 39.Hanisch F, Müller CR, Grimm D, Xue L, Traufeller K, Merkenschlager A, et al. Frequency of calpain-3 c.550delA mutation in limb girdle muscular dystrophy type 2 and isolated hyperCKemia in German patients. Clin Neuropathol. 2007;26:157–63. doi: 10.5414/npp26157. [DOI] [PubMed] [Google Scholar]
- 40.Dadali EL, Shagina OA, Ryzhkova OP, Rudenskaia GE, Fedotov VP, Poliakov AV. Clinical-genetic characteristics of limb girdle-muscular dystrophy type 2A. Zh Nevrol Psikhiatr Im S S Korsakova. 2010;110:79–83. [PubMed] [Google Scholar]
- 41.Nadaj-Pakleza AA, Dorobek M, Nestorowicz K, Ryniewicz B, Szmidt-Salkowska E, Kaminska AM. Muscle pathology in 31 patients with calpain 3 gene mutations. Neurol Neurochir Pol. 2013;47:214–22. doi: 10.5114/ninp.2013.35490. [DOI] [PubMed] [Google Scholar]
- 42.Stehlíková K, Skálová D, Zídková J, Mrázová L, Vondrácek P, Mazanec R, et al. Autosomal recessive limb-girdle muscular dystrophies in the Czech Republic. BMC Neurol. 2014;14:154. doi: 10.1186/s12883-014-0154-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fardeau M, Hillaire D, Mignard C, Feingold N, Feingold J, Mignard D, et al. Juvenile limb-girdle muscular dystrophy. Clinical, histopathological and genetic data from a small community living in the Reunion Island. Brain. 1996;119(Pt 1):295–308. doi: 10.1093/brain/119.1.295. [DOI] [PubMed] [Google Scholar]
- 44.Young K, Foroud T, Williams P, Jackson CE, Beckmann JS, Cohen D, et al. Confirmation of linkage of limb-girdle muscular dystrophy type 2 to chromosome 15. Genomics. 1992;13:1370–1. doi: 10.1016/0888-7543(92)90074-3. [DOI] [PubMed] [Google Scholar]
- 45.Richard I, Broux O, Allamand V, Fougerousse F, Chiannilkulchai N, Bourg N, et al. Mutations in the proteolytic enzyme calpain 3 cause limb-girdle muscular dystrophy type 2A. Cell. 1995;81:27–40. doi: 10.1016/0092-8674(95)90368-2. [DOI] [PubMed] [Google Scholar]
- 46.Urtasun M, Sáenz A, Roudaut C, Poza JJ, Urtizberea JA, Cobo AM, et al. Limb-girdle muscular dystrophy in Guipúzcoa (Basque Country, Spain) Brain. 1998;121(Pt 9):1735–47. doi: 10.1093/brain/121.9.1735. [DOI] [PubMed] [Google Scholar]
- 47.de Paula F, Vainzof M, Passos-Bueno MR, de Cássia M, Pavanello R, Matioli SR, et al. Clinical variability in calpainopathy: What makes the difference? Eur J Hum Genet. 2002;10:825–32. doi: 10.1038/sj.ejhg.5200888. [DOI] [PubMed] [Google Scholar]
- 48.Cobo AM, Sáenz A, Poza JJ, Urtasun M, Indakoetxea B, Urtizberea JA, et al. A common haplotype associated with the Basque 2362AG –> TCATCT mutation in the muscular calpain-3 gene. Hum Biol. 2004;76:731–41. doi: 10.1353/hub.2005.0002. [DOI] [PubMed] [Google Scholar]
- 49.Chae J, Minami N, Jin Y, Nakagawa M, Murayama K, Igarashi F, et al. Calpain 3 gene mutations: Genetic and clinico-pathologic findings in limb-girdle muscular dystrophy. Neuromuscul Disord. 2001;11:547–55. doi: 10.1016/s0960-8966(01)00197-3. [DOI] [PubMed] [Google Scholar]
- 50.Kawai H, Akaike M, Kunishige M, Inui T, Adachi K, Kimura C, et al. Clinical, pathological, and genetic features of limb-girdle muscular dystrophy type 2A with new calpain 3 gene mutations in seven patients from three Japanese families. Muscle Nerve. 1998;21:1493–501. doi: 10.1002/(sici)1097-4598(199811)21:11<1493::aid-mus19>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- 51.Minami N, Nishino I, Kobayashi O, Ikezoe K, Goto Y, Nonaka I. Mutations of calpain 3 gene in patients with sporadic limb-girdle muscular dystrophy in Japan. J Neurol Sci. 1999;171:31–7. doi: 10.1016/s0022-510x(99)00245-2. [DOI] [PubMed] [Google Scholar]
- 52.Fanin M, Benedicenti F, Fritegotto C, Nascimbeni AC, Peterle E, Stanzial F, et al. An intronic mutation causes severe LGMD2A in a large inbred family belonging to a genetic isolate in the Alps. Clin Genet. 2012;82:601–2. doi: 10.1111/j.1399-0004.2012.01873.x. [DOI] [PubMed] [Google Scholar]
- 53.Ankala A, Kohn JN, Dastur R, Gaitonde P, Khadilkar SV, Hegde MR. Ancestral founder mutations in calpain-3 in the Indian Agarwal Community: Historical, clinical, and molecular perspective. Muscle Nerve. 2013;47:931–7. doi: 10.1002/mus.23763. [DOI] [PubMed] [Google Scholar]
- 54.Khadilkar SV, Chaudhari CR, Dastur RS, Gaitonde PS, Yadav JG. Limb-girdle muscular dystrophy in the Agarwals: Utility of founder mutations in CAPN3 gene. Ann Indian Acad Neurol. 2016;19:108–11. doi: 10.4103/0972-2327.175435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sharma MC, Mannan R, Singh NG, Gulati S, Kalra V, Sarkar C. Sarcoglycanopathies: A clinicopathological study of 13 cases [corrected] Neurol India. 2004;52:446–9. Erratum in: Neurol India 2005;53:98. [PubMed] [Google Scholar]
- 56.Meena AK, Sreenivas D, Sundaram C, Rajasekhar R, Sita JS, Borgohain R, et al. Sarcoglycanopathies: A clinico-pathological study. Neurol India. 2007;55:117–21. doi: 10.4103/0028-3886.32781. [DOI] [PubMed] [Google Scholar]
- 57.Khadilkar SV, Singh RK, Hegde M, Urtizberea A, Love DR, Chong B. Spectrum of mutations in sarcoglycan genes in the Mumbai region of Western India: High prevalence of 525del T. Neurol India. 2009;57:406–10. doi: 10.4103/0028-3886.55603. [DOI] [PubMed] [Google Scholar]
- 58.Chakrabarty B, Sharma MC, Gulati S, Kabra M, Pandey RM, Sarkar C. Skin biopsy: A new tool to diagnose sarcoglycanopathy. J Child Neurol. 2014;29:NP5–8. doi: 10.1177/0883073813488662. [DOI] [PubMed] [Google Scholar]
- 59.Tanveer N, Sharma MC, Sarkar C, Gulati S, Kalra V, Singh S, et al. Diagnostic utility of skin biopsy in dystrophinopathies. Clin Neurol Neurosurg. 2009;111:496–502. doi: 10.1016/j.clineuro.2009.01.011. [DOI] [PubMed] [Google Scholar]
- 60.Marbini A, Gemignani F, Bellanova MF, Guidetti D, Ferrari A. Immunohistochemical localization of utrophin and other cytoskeletal proteins in skin smooth muscle in neuromuscular diseases. J Neurol Sci. 1996;143:156–60. doi: 10.1016/s0022-510x(96)00210-9. [DOI] [PubMed] [Google Scholar]
- 61.Sewry CA, Philpot J, Sorokin LM, Wilson LA, Naom I, Goodwin F, et al. Diagnosis of merosin (laminin-2) deficient congenital muscular dystrophy by skin biopsy. Lancet. 1996;347:582–4. doi: 10.1016/s0140-6736(96)91274-x. [DOI] [PubMed] [Google Scholar]
- 62.Marbini A, Bellanova MF, Ferrari A, Lodesani M, Gemignani F. Immunohistochemical study of merosin-negative congenital muscular dystrophy: Laminin alpha 2 deficiency in skin biopsy. Acta Neuropathol. 1997;94:103–8. doi: 10.1007/s004010050680. [DOI] [PubMed] [Google Scholar]
- 63.Manilal S, Sewry CA, Man N, Muntoni F, Morris GE. Diagnosis of x-linked emery-dreifuss muscular dystrophy by protein analysis of leucocytes and skin with monoclonal antibodies. Neuromuscul Disord. 1997;7:63–6. doi: 10.1016/s0960-8966(96)00405-1. [DOI] [PubMed] [Google Scholar]
- 64.Jimenez-Mallebrera C, aioli MA, Kim J, Brown SC, Feng L, Lampe AK, et al. A comparative analysis of collagen VI production in muscle, skin and fibroblasts from 14 Ullrich congenital muscular dystrophy patients with dominant and recessive COL6A mutations. Neuromuscul Disord. 2006;16:571–82. doi: 10.1016/j.nmd.2006.07.015. [DOI] [PubMed] [Google Scholar]
- 65.Ben Hamida M, Fardeau M, Attia N. Severe childhood muscular dystrophy affecting both sexes and frequent in Tunisia. Muscle Nerve. 1983;6:469–80. doi: 10.1002/mus.880060702. [DOI] [PubMed] [Google Scholar]
- 66.Lim LE, Duclos F, Broux O, Bourg N, Sunada Y, Allamand V, et al. Beta-sarcoglycan: Characterization and role in limb-girdle muscular dystrophy linked to 4q12. Nat Genet. 1995;11:257–65. doi: 10.1038/ng1195-257. [DOI] [PubMed] [Google Scholar]
- 67.Duclos F, Broux O, Bourg N, Straub V, Feldman GL, Sunada Y, et al. Beta-sarcoglycan: Genomic analysis and identification of a novel missense mutation in the LGMD2E Amish isolate. Neuromuscul Disord. 1998;8:30–8. doi: 10.1016/s0960-8966(97)00135-1. [DOI] [PubMed] [Google Scholar]
- 68.Fanin M, Hoffman EP, Angelini C, Pegoraro E. Private beta- and gamma-sarcoglycan gene mutations: Evidence of a founder effect in Northern Italy. Hum Mutat. 2000;16:13–7. doi: 10.1002/1098-1004(200007)16:1<13::AID-HUMU3>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- 69.Noguchi S, McNally EM, Ben Othmane K, Hagiwara Y, Mizuno Y, Yoshida M, et al. Mutations in the dystrophin-associated protein gamma-sarcoglycan in chromosome 13 muscular dystrophy. Science. 1995;270:819–22. doi: 10.1126/science.270.5237.819. [DOI] [PubMed] [Google Scholar]
- 70.Piccolo F, Jeanpierre M, Leturcq F, Dodé C, Azibi K, Toutain A, et al. A founder mutation in the gamma-sarcoglycan gene of gypsies possibly predating their migration out of India. Hum Mol Genet. 1996;5:2019–22. doi: 10.1093/hmg/5.12.2019. [DOI] [PubMed] [Google Scholar]
- 71.Nigro V, de Sá Moreira E, Piluso G, Vainzof M, Belsito A, Politano L, et al. Autosomal recessive limb-girdle muscular dystrophy, LGMD2F, is caused by a mutation in the delta-sarcoglycan gene. Nat Genet. 1996;14:195–8. doi: 10.1038/ng1096-195. [DOI] [PubMed] [Google Scholar]
- 72.Angelini C, Fanin M, Freda MP, Duggan DJ, Siciliano G, Hoffman EP. The clinical spectrum of sarcoglycanopathies. Neurology. 1999;52:176–9. doi: 10.1212/wnl.52.1.176. [DOI] [PubMed] [Google Scholar]
- 73.Hackman P, Juvonen V, Sarparanta J, Penttinen M, Aärimaa T, Uusitalo M, et al. Enrichment of the R77C alpha-sarcoglycan gene mutation in Finnish LGMD2D patients. Muscle Nerve. 2005;31:199–204. doi: 10.1002/mus.20267. [DOI] [PubMed] [Google Scholar]
- 74.Tétreault M, Srour M, Allyson J, Thiffault I, Loisel L, Robitaille Y, et al. Founder mutation for a-sarcoglycan-LGMD2D in a Magdalen Islands Acadian cluster. Can J Neurol Sci. 2011;38:747–52. doi: 10.1017/s0317167100054135. [DOI] [PubMed] [Google Scholar]
- 75.Liang WC, Chou PC, Hung CC, Su YN, Kan TM, Chen WZ, et al. Probable high prevalence of limb-girdle muscular dystrophy type 2D in Taiwan. J Neurol Sci. 2016;362:304–8. doi: 10.1016/j.jns.2016.02.002. [DOI] [PubMed] [Google Scholar]
- 76.Reddy HM, Hamed SA, Lek M, Mitsuhashi S, Estrella E, et al. Homozygous nonsense mutation in SGCA is a common cause of limb-girdle muscular dystrophy in Assiut, Egypt. Muscle Nerve. 2016;54:690–5. doi: 10.1002/mus.25094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ben Othmane K, Speer MC, Stauffer J, Blel S, Middleton L, Ben Hamida C, et al. Evidence for linkage disequilibrium in chromosome 13-linked Duchenne-like muscular dystrophy (LGMD2C) Am J Hum Genet. 1995;57:732–4. [PMC free article] [PubMed] [Google Scholar]
- 78.Nalini A, Polavarapu K, Sunitha B, Kulkarni S, Gayathri N, Srinivas Bharath MM, et al. A prospective study on the immunophenotypic characterization of limb girdle muscular dystrophies 2 in India. Neurol India. 2015;63:548–60. doi: 10.4103/0028-3886.162048. [DOI] [PubMed] [Google Scholar]
- 79.Stensland E, Lindal S, Jonsrud C, Torbergsen T, Bindoff LA, Rasmussen M, et al. Prevalence, mutation spectrum and phenotypic variability in Norwegian patients with limb girdle muscular dystrophy 2I. Neuromuscul Disord. 2011;21:41–6. doi: 10.1016/j.nmd.2010.08.008. [DOI] [PubMed] [Google Scholar]
- 80.Nalini A, Gayathri N, Dawn R. Distal myopathy with rimmed vacuoles: Report on clinical characteristics in 23 cases. Neurol India. 2010;58:235–41. doi: 10.4103/0028-3886.63804. [DOI] [PubMed] [Google Scholar]
- 81.Das B, Goyal MK, Bhatkar SR, Vinny PW, Modi M, Lal V, et al. Hereditary inclusion body myopathy: A myopathy with unique topography of weakness, yet frequently misdiagnosed: Case series and review of literature. Ann Indian Acad Neurol. 2016;19:119–22. doi: 10.4103/0972-2327.167709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Khadilkar SV, Nallamilli BR, Bhutada A, Hegde M, Gandhi K, Faldu HD, et al. A report on GNE myopathy: Individuals of Rajasthan ancestry share the Roma gene. J Neurol Sci. 2017;375:239–40. doi: 10.1016/j.jns.2017.02.005. [DOI] [PubMed] [Google Scholar]
- 83.Eisenberg I, Avidan N, Potikha T, Hochner H, Chen M, Olender T, et al. The UDP-N-acetylglucosamine 2-epimerase/N-acetylmannosamine kinase gene is mutated in recessive hereditary inclusion body myopathy. Nat Genet. 2001;29:83–7. doi: 10.1038/ng718. [DOI] [PubMed] [Google Scholar]
- 84.Nishino I, Noguchi S, Murayama K, Driss A, Sugie K, Oya Y, et al. Distal myopathy with rimmed vacuoles is allelic to hereditary inclusion body myopathy. Neurology. 2002;59:1689–93. doi: 10.1212/01.wnl.0000041631.28557.c6. [DOI] [PubMed] [Google Scholar]
- 85.Tomimitsu H, Ishikawa K, Shimizu J, Ohkoshi N, Kanazawa I, Mizusawa H. Distal myopathy with rimmed vacuoles: Novel mutations in the GNE gene. Neurology. 2002;59:451–4. doi: 10.1212/wnl.59.3.451. [DOI] [PubMed] [Google Scholar]
- 86.Chamova T, Guergueltcheva V, Gospodinova M, Krause S, Cirak S, Kaprelyan A, et al. GNE myopathy in Roma patients homozygous for the p.I618T founder mutation. Neuromuscul Disord. 2015;25:713–8. doi: 10.1016/j.nmd.2015.07.004. [DOI] [PubMed] [Google Scholar]
- 87.Lu X, Pu C, Huang X, Liu J, Mao Y. Distal myopathy with rimmed vacuoles: Clinical and muscle morphological characteristics and spectrum of GNE gene mutations in 53 Chinese patients. Neurol Res. 2011;33:1025–31. doi: 10.1179/1743132811Y.0000000070. [DOI] [PubMed] [Google Scholar]
- 88.Liewluck T, Pho-Iam T, Limwongse C, Thongnoppakhun W, Boonyapisit K, Raksadawan N, et al. Mutation analysis of the GNE gene in distal myopathy with rimmed vacuoles (DMRV) patients in Thailand. Muscle Nerve. 2006;34:775–8. doi: 10.1002/mus.20583. [DOI] [PubMed] [Google Scholar]
- 89.Nalini A, Gayathri N, Nishino I, Hayashi YK. GNE myopathy in India. Neurol India. 2013;61:371–4. doi: 10.4103/0028-3886.117609. [DOI] [PubMed] [Google Scholar]
- 90.Chaouch A, Brennan KM, Hudson J, Longman C, McConville J, Morrison PJ, et al. Two recurrent mutations are associated with GNE myopathy in the North of Britain. J Neurol Neurosurg Psychiatry. 2014;85:1359–65. doi: 10.1136/jnnp-2013-306314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Jadhav KB, Karpe KK, Maramattom BV. An Indian family with an emery-dreifuss myopathy and familial dilated cardiomyopathy due to a novel LMNA mutation. Ann Indian Acad Neurol. 2012;15:344–6. doi: 10.4103/0972-2327.104355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Jain P, Rajeshwari SM, Singh J, Kumar T, Agarwal SP, Das P. Myofibrillar myopathy presenting as neonatal intestinal pseudo-obstruction: An extremely rare entity. Fetal Pediatr Pathol. 2016;35:124–8. doi: 10.3109/15513815.2015.1131783. [DOI] [PubMed] [Google Scholar]