

Abstract
Background
Systemic sclerosis (SSc) is characterized by fibrosis of the skin and internal organs. Although the involvement of connective tissue growth factor (CTGF/CCN2) has been well-documented in SSc fibrosis, the therapeutic potential of targeting CTGF in SSc has not been fully investigated. Our aim was to examine the therapeutic potential of CTGF blockade in a preclinical model of SSc using two approaches: smooth muscle cell fibroblast-specific deletion of CTGF (CTGF knockout (KO)) or a human anti-CTGF monoclonal antibody, FG-3019.
Methods
Angiotensin II (Ang II) was administered for 14 days by subcutaneous osmotic pump to CTGF KO or C57BL/6 J mice. FG-3019 was administered intraperitoneally three times per week for 2 weeks. Skin fibrosis was evaluated by histology and hydroxyproline assay. Immunohistochemistry staining was used for alpha smooth muscle actin (αSMA), platelet-derived growth factor receptor β (PDGFRβ), pSmad2, CD45, von Willebrand factor (vWF), and immunofluorescence staining was utilized for procollagen and Fsp1.
Results
Ang II-induced skin fibrosis was mitigated in both CTGF KO and FG-3019-treated mice. The blockade of CTGF reduced the number of cells expressing PDGFRβ, procollagen, αSMA, pSmad2, CD45, and Fsp1 in the dermis. In addition, inhibition of CTGF attenuated vascular injury as measured by the presence of vWF-positive cells.
Conclusions
Our data indicate that inhibition of CTGF signaling presents an attractive therapeutic approach in SSc.
Keywords: Angiotensin II, CTGF/CCN2, FG-3019, Fibrosis, Skin, Systemic sclerosis
Background
Systemic sclerosis (SSc) is a multisystem connective tissue disorder that typically results in fibrosis of the skin and internal organs [1]. SSc is characterized by autoimmunity, inflammation, and widespread microvascular injury, leading to the activation of fibroblasts and excessive accumulation of extracellular matrix (ECM) proteins; however, the causes and the pathogenesis of this disease are not yet fully explained [2]. Accumulation of ECM proteins restricts normal function of the affected tissues and organs leading to high morbidity and mortality. Despite significant improvements in the management of SSc, effective disease modifying therapies are not yet available.
Connective tissue growth factor (CTGF, also known as CCN2), a member of the CCN family of matricellular proteins, is widely known as a hallmark of fibrosis in multiple tissues, including skin, heart, lung, liver, and kidney [3, 4]. A key study in support of the essential role of CTGF in fibrosis was published by Takehara and colleagues who showed that subcutaneous injection of transforming growth factor (TGF)-β and CTGF led to sustained fibrosis, while either factor alone failed to do so [5]. Further support was provided by a recent study that showed that fibroblast-specific ablation of CTGF inhibited development of dermal fibrosis in the bleomycin injection model [6]. The role of CTGF in fibrosis may not be limited to its function as a co-factor of TGF-β, but CTGF may also regulate other aspects of the fibrogenic process, consistent with its multifunctional nature [7, 8]. Accordingly, it has been suggested that CTGF may contribute to myofibroblast recruitment during bleomycin-induced skin fibrosis [6]. Transgenic mice overexpressing CTGF develop skin fibrosis and microvascular abnormalities [9], and loss of CTGF results in a reduction of bleomycin-induced skin fibrosis [6].
Accumulating reports show that CTGF is highly expressed in SSc [10]. Elevated levels of CTGF have been identified in fibrotic skin and serum from patients with SSc, and have been correlated with the severity of skin and lung fibrosis [11, 12]. Elevated CTGF protein expression has been observed in fibroblasts in affected skin in SSc [13]. Furthermore, N-terminal cleavage products of CTGF have been identified in interstitial fluid in the skin of patients with SSc [14].
Given the key role of CTGF in the development of skin fibrosis in SSc, the goal of this study was to evaluate the efficacy of FG-3019 as a potential therapeutic agent for SSc using a murine model of angiotensin II (Ang II)-induced skin fibrosis [15–17]. FG-3019 is a fully human monoclonal antibody specific for CTGF [18]. Clinically, FG-3019 is being evaluated for treatment of idiopathic pulmonary fibrosis, pancreatic cancer, and Duchenne muscular dystrophy [19–21]. In parallel studies, we employed mice with smooth muscle cell fibroblast-specific deletion of CTGF to assess the contribution of CTGF to the process of skin fibrosis in the Ang II model [6]. Together, these studies showed that FG-3019 was comparable to genetic CTGF deletion in attenuating skin fibrosis. The results of this study support the use of FG-3019 as therapy for skin fibrosis.
Methods
Reagents
A fully human IgG1kappa monoclonal antibody recognizing domain 2 of human and rodent CTGF (FG-3019) and whole human IgG control antibody were obtained from Fibrogen (San Francisco, CA, USA).
Mice
Smooth muscle cell fibroblast-specific CTGF knockout (KO) mice were generated as described [6]. In brief, mice with CTGF flanked by loxP sites were crossed with mice containing a Cre recombinase gene located downstream of a collagen α2 (I) promoter enhancer that confers fibroblast-specific gene expression. The expression of Cre is dependent on the presence of tamoxifen. Tamoxifen was diluted in corn oil to 10 mg/ml. Three-week-old mice were given intraperitoneal injections of the tamoxifen suspension (100 μl of 10 mg/ml) over 10 days. Three weeks later, the mice were used for further studies. We used mice homozygous for CTGF gene flanked by loxP sites and heterozygous for Cre as CTGF KO mice. Littermate mice homozygous for loxP-flanked CTGF that were wild for Cre (non Cre) were used as control mice. C57BL/6 J mice for FG-3019 injection were purchased from the Jackson Laboratory (Bar Harbor, ME, USA). All mice experiments were performed in accordance with the National Institutes of Health and institutional guidelines for animal care, and approved by the Committee on the Ethics of Animal Experiments of the Boston University (Protocol AN-15037).
Cell culture and immunoblotting
Mouse dermal fibroblasts were obtained from skin from the backs of male CTGF KO and control littermate mice. Fibroblasts between the third and fifth sub passages were used for experiments. Mouse dermal fibroblasts were lysed and subjected to immunoblotting, as described previously [22]. Primary antibodies used were (1:1000): CTGF from Santa Cruz Biotechnology (Dallas, TX, USA) and β-actin from Sigma (St. Louis, MO, USA).
Ang II-induced dermal fibrosis
Alzet osmotic miniature pumps (Model 1002, DURECT, Cupertino, CA, USA) delivering Ang II (EMD Millipore, Billerica, MA, USA) at a rate of 1000 ng/kg/min (pressor dose) or PBS, were implanted subcutaneously on the back of 8-week-old mice, as described previously [17]. After 2 weeks, mice were euthanized and the skin surrounding the pump outlet was collected. FG-3019 (25 mg/kg) or control IgG (25 mg/kg) was administered intraperitoneally three times per week for 2 weeks, after the osmotic pump was installed.
Histologic assessment
Mice skin samples were paraffin-embedded, and sections (5 μm in thickness) were stained with hematoxylin and eosin (HE). Dermal thickness was evaluated by measuring the distance between the epidermal-dermal junction and the dermal-fat junction in HE sections. Skin trichrome staining was performed by Masson’s trichrome stain kit (Polysciences, Warrington, PA, USA). The αSMA positive cells were counted in five random high-power fields using a light microscope. The mean score was used for analysis. The von Willebrand factor (vWF) staining intensity for immunohistochemical assessment was scored semi-quantitatively. The staining intensity (1: negative or weak staining, 2: moderate staining, and 3: strong staining) was evaluated in six randomly selected fields in the subcutaneous area. Then a semi-quantitative score per sample was generated by calculating the average of the six intensity scores per sample.
Hydroxyproline assay
Collagen deposition was determined by measuring total hydroxyproline content in 8-mm skin punch biopsies obtained from PBS and Ang II infusion sites as described previously [16]. Briefly, the skin samples were hydrolyzed with 6 M sodium hydroxide at 120 °C for 16 h. The hydrolysate was then oxidized with oxidation buffer (one part 7% chloramine T and four parts acetate citrate buffer) for 4 minutes at room temperature. Ehrlich’s aldehyde reagent was added to each sample, and the chromophore was developed by incubating the samples at 60 °C for 25 minutes. Absorbance of each sample was read at 560 nm using a spectrophotometer. Results were expressed as relative hydroxyproline content. A standard curve was performed for all hydroxyproline measurements using known quantities of hydroxyproline.
Immunohistochemical assessment
For single antibody staining, formalin-fixed, paraffin-embedded 5-μm skin tissue sections were de-paraffinized and rehydrated through a graded series of ethanol. Antigens were retrieved by incubation with a proteinase K solution (EMD Millipore, Billerica, MA, USA) for 5 minutes. Blocking was done by 2.5% normal horse serum for 1 h. Tissue sections were then incubated with primary antibody to αSMA (1:100, Novus Biologicals, Littleton, CO, USA), PDGFRβ (1:50, Cell Signaling Technology, Danvers, MA, USA), rat anti-mouse CD45 Ab (1:100, BD Pharmingen, San Diego, CA, USA), phospho-Smad2 (Ser465/467) Ab (1:100, Cell Signaling), or vWF (1:1000, DAKO, Santa Clara, CA, USA) at 4 °C for 16 h. Next, sections were incubated with ImmPress horseradish peroxidase (HRP) anti-rabbit IgG (Vector Laboratories, Burlingame, CA, USA) for 30 minutes. The color was developed using 3,3-diaminobenzidine (DAB) substrate (DAKO). Immunohistochemical images were collected using a microscope (BH-2; Olympus, Center Valley, PA, USA).
Immunofluorescence staining
Staining was performed on 5-μm paraffin sections or cryosections. Slides were blocked with a blocking solution (3% BSA, and 0.2% Triton X-100 in PBS) for 2 h. Then, tissue sections were incubated at 4 °C overnight with primary antibodies: procollagen (COL1A1 propeptide, 1:50, Thermo Fisher Scientific) for the cryosections and rabbit anti-mouse FSP1 (1:100, Abcam, Cambridge, MA, USA) for the paraffin sections. Secondary antibodies conjugated with Alexa 594 (Thermo Fisher Scientific) were used. Coverslips were mounted by using Vectashield with 4′,6-diamidino-2-phenylindole (DAPI) (Vector Laboratories, Burlingame, CA, USA). Fluorescence images were recorded with FV10i fluorescence microscope (Olympus, Tokyo, Japan).
Statistical analysis
Values are presented as means ± standard deviation (SD). One-way analysis of variance with Tukey-Kramer test was used to determine significant differences between more than two groups. Analyses were performed with Statcel software (OMS, Tokorozawa, Japan). Significance was defined as P <0.05.
Results
Fibroblast-specific deletion of CTGF alleviates Ang II-induced skin fibrosis
To evaluate the therapeutic effects of CTGF blockade in the in vivo model of SSc, we used a mouse model of Ang II-induced skin fibrosis [17]. Ang II-induced skin fibrosis is accompanied by diverse pathogenic mechanisms, including collagen accumulation, CTGF upregulation, myofibroblast accumulation, endothelial cell injury, inflammation, and fibrosis [15–17]. In an initial experiment, we examined the contribution of CTGF to Ang II-induced skin fibrosis using mice with smooth muscle cell fibroblast-specific deletion of CTGF (CTGF KO mice). We observed >80% reduction in CTGF protein levels in skin fibroblasts cultured from CTGF KO mice when compared to control mice (Fig. 1a).
Fig. 1.
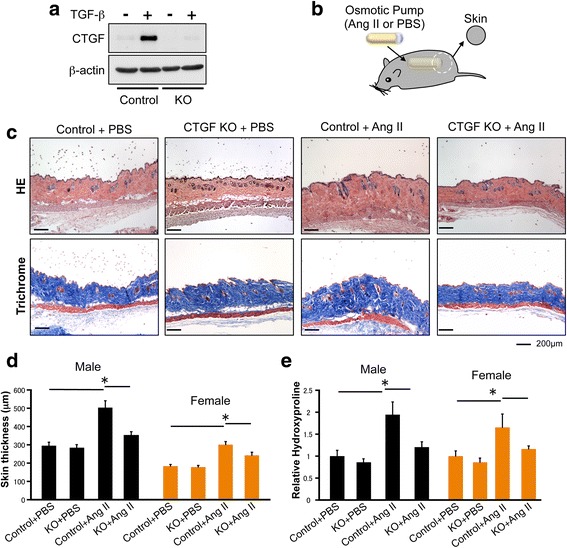
Fibroblast-specific connective tissue growth factor (CTGF) knockout inhibits angiotensin II (Ang II)-induced skin fibrosis. a Mice skin fibroblasts from CTGF knockout (CTGF KO) and control mice were treated for 24 h with transforming growth factor beta 1 (TGF-β1) (2.5 ng/ml). Cell lysates were subjected to immunoblotting. b Skin fibrosis in mice was induced by Ang II administered via subcutaneously installed osmotic pumps. After 2 weeks, the skin surrounding the pump outlet was collected. c Mice skin sections were stained with HE and trichrome. Scale bar 200 μm. d Dermal thickness is summarized. e Collagen contents were measured by hydroxyproline assay. Values are normalized relative to the PBS control group. Each graph represents mean ± SD; n = 3 per group; *P < 0.05
Because gender differences have been shown to affect pathogenic features, including the severity of fibrosis in SSc [23], we used both male and female mice in our experiments. CTGF KO and control male and female mice were then subjected to Ang II-induced skin fibrosis. Ang II or PBS was administered by subcutaneous osmotic pump, and the skin surrounding the pump outlet was collected on day 14 (Fig. 1b). In control mice, Ang II increased dermal thickness and the hydroxyproline content of lesional skin (Fig. 1c–e). Both male and female mice responded similarly to Ang II treatment. In contrast, male and female CTGF KO mice were resistant to the Ang II-induced changes (Fig. 1c–e). We next compared the number of αSMA-positive cells in the upper dermis of CTGF KO and control mice. The number of αSMA-positive cells in the upper dermis of CTGF KO mice challenged with Ang II was significantly decreased compared with control mice (Fig. 2a). Likewise, the number of PDGFRβ and procollagen positive cells were decreased in those mice (Fig. 2b and c). Collectively, these results indicated that CTGF is required for Ang II-induced skin fibrosis.
Fig. 2.
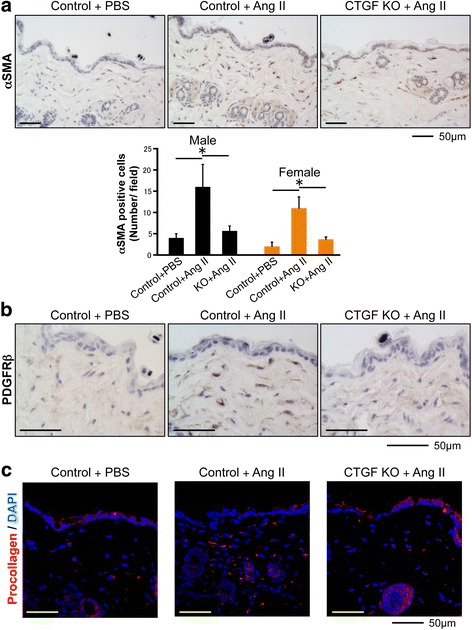
Fibroblast-specific connective tissue growth factor knockout (CTGF KO) reduces alpha smooth muscle actin (αSMA)-positive cells number. a Skin sections were stained with anti-αSMA antibody. Representative images are shown. The αSMA-positive cells were counted in five random high-power fields using a light microscope. The mean score was used for analysis. Each graph represents mean ± SD. b Skin sections were stained with anti-platelet-derived growth factor receptor β (PDGFRβ) antibody. Representative images are shown. c Immunofluorescent staining was performed on cryosections using procollagen antibody. Representative images are shown. Scale bar 50 μm; n = 3 per group; *P < 0.05. DAPI 4′,6-diamidino-2-phenylindole
FG-3019 attenuates Ang II-induced skin fibrosis
We next investigated the effects of FG-3019 on Ang II-induced skin fibrosis. Ang II or PBS was administered by subcutaneous osmotic pump and FG-3019 (25 mg/kg) or control IgG (25 mg/kg) was administered intraperitoneally three times per week for 2 weeks. The skin surrounding the pump outlet was collected on day 14 (Fig. 3a). Treatment with FG-3019 significantly reduced dermal thickness and collagen content in skin from the backs of Ang II-challenged mice in both male and female animals (Fig. 3b and c). FG-3019 significantly decreased the number of αSMA-positive cells in the upper dermis of mice challenged with Ang II (Fig. 4a). FG-3019 also reduced PDGFRβ and procollagen expression in the upper dermis of mice challenged with Ang II (Fig. 4b and c). We only used male mice in subsequent experiments because we did not notice any apparent differences in responses to Ang II or the blockade of CTGF in male and female mice.
Fig. 3.
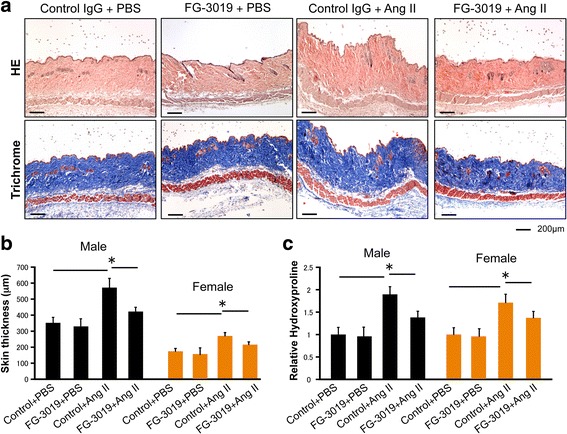
FG-3019 ameliorates angiotensin II (Ang II)-induced skin fibrosis. a Mice skin sections were stained with HE and trichrome. Scale bar 200 μm. b Dermal thickness is summarized. c Collagen contents were measured by hydroxyproline assay. Values are normalized relative to the PBS control group. Each graph represents mean ± SD; n = 4 per group; *P < 0.05
Fig. 4.
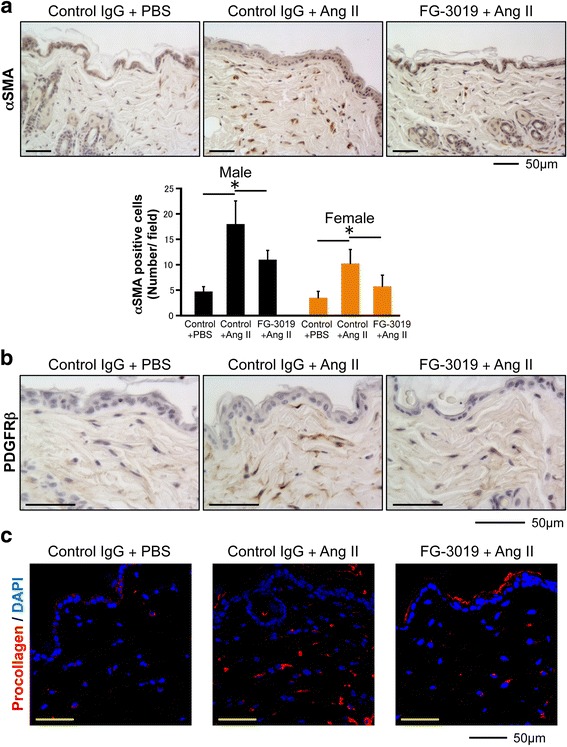
FG-3019 reduces alpha smooth muscle actin (αSMA)-positive cell numbers. a Skin sections were stained with anti-αSMA antibody. Representative images are shown. The αSMA-positive cells were counted in five random high-power fields using a light microscope. The mean score was used for analysis. Each graph represents mean ± SD. b Skin sections were stained with anti-platelet-derived growth factor receptor β (PDGFRβ) antibody. Representative images are shown. c Immunofluorescent staining was performed on cryosections using procollagen antibody. Representative images are shown. Scale bar 50 μm; n = 4 per group; *P < 0.05. DAPI 4′,6-diamidino-2-phenylindole
Inhibition of CTGF ameliorates activation of TGF-β signaling in Ang II-induced skin fibrosis
We have previously shown activation of the TGF-β signaling pathway in the skin of mice challenged with Ang II [17]. As reported, Ang II induced a significant increase in pSmad2-positive cells distributed throughout the dermis. However, the number of pSmad2-positive cells was markedly reduced in CTGF KO mice (Fig. 5a). Interestingly, treatment with FG-3019 was significantly more effective than CTGF KO in reducing the number of pSmad2-positive cells comparable to the levels observed in control mice (Fig. 5b).
Fig. 5.
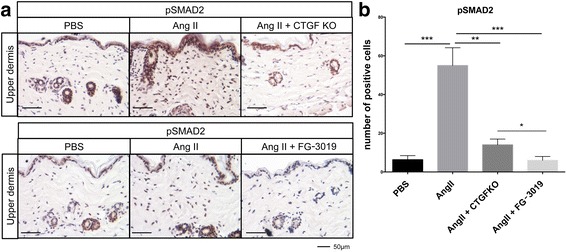
Blockade of connective tissue growth factor (CTGF) reduces the number of pSmad-positive cells. a Representative results of immunostaining for pSmad2. Scale bar 50 μm. b The pSmad2-positive cells were counted in five random high-power fields using a light microscope. The mean score was used for analysis. Each graph represents mean ± SD; *P < 0.05, **P < 0.01, ***P < 0.001. Ang II angiotensin II, KO knockout
Inhibition of CTGF reduces inflammation in the skin of Ang II treated mice
Ang II-induced skin fibrosis is accompanied by the increased presence of inflammatory cells in the dermis [17]. We next evaluated the effect of CTGF blockade on the recruitment of inflammatory cells. As shown in Fig. 6a, a significant increase in CD45-positive cells was observed in the skin of Ang II-challenged mice. The number of inflammatory cells was significantly reduced in the Ang II-challenged CTGF KO mice. Treatment with FG-3019 was somewhat more efficient in reducing the number of CD45-positive cells; however, the difference between the two treatments was not statistically significant (Fig. 6b).
Fig. 6.
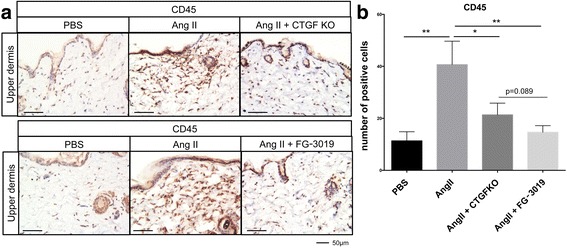
Blockade of connective tissue growth factor (CTGF) reduces the number of CD45-positive cells. a Representative results of immunostaining for CD45. Scale bar 50 μm. The CD45-positive cells were counted in five random high-power fields using a light microscope. The mean score was used for analysis. Each graph represents mean ± SD; *P < 0.05, **P < 0.01. Ang II angiotensin II, KO knockout
We have previously shown that the Ang II model of skin fibrosis is characterized by the infiltration of fibrocytes (CD45+/FSP1+ double-positive cells) [17]. Consistent with these findings, immunofluorescent staining showed numerous FSP1-positive cells in the dermis of Ang II-infused mice, while inhibition of CTGF significantly reduced their number (Fig. 7a and b). Both treatments similarly reduced fibrocyte infiltration.
Fig. 7.
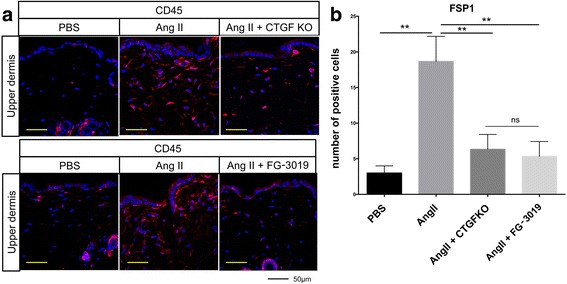
Blockade of connective tissue growth factor (CTGF) reduces the number of Fsp1 positive cells. a Representative results of immunostaining for Fsp1. Scale bar 50 μm. b The Fsp1-positive cells were counted in five random high-power fields using a fluorescent microscope. The mean score was used for analysis. Each graph represents mean ± SD; *P < 0.05, **P < 0.01. Ang II angiotensin II, KO knockout
Inhibition of CTGF reduces vascular damage in Ang II-induced skin fibrosis
Vascular damage is one of the key events in the pathogenesis of SSc. Mice challenged with Ang II develop prominent vascular injury [16]. We next examined whether inhibition of CTGF reduced vascular injury in this model. To evaluate the vascular injury, we used immunohistochemical staining of vWF, a widely used injury marker known to be elevated in SSc [24]. Consistent with the previous study [16], increased vWF staining was present in the dermal vessels of Ang II mice compared with the PBS control (Fig. 8a and b). The staining intensities were reduced in both CTGF KO and FG-3019-treated mice (Fig. 8a and b). Scoring of the staining intensity in the subcutaneous vessels and the representative images of each score (score 1–3) are shown in Fig. 8c. Although the intensity of vWF staining was significantly reduced in both CTGF KO and FG-3019-treated mice (Fig. 8d and e), the effect was more pronounced in CTGF KO mice.
Fig. 8.
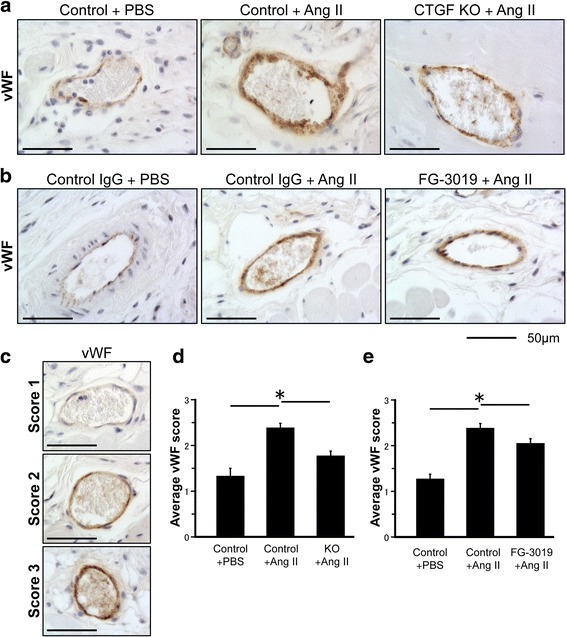
Inhibition of connective tissue growth factor (CTGF) ameliorates vascular damage in angiotensin II (Ang II)-induced fibrotic skin. a, b Representative results of immunostaining for von Willebrand factor (vWF). Scale bar 50 μm. c Representative staining intensity (1: negative or weak staining, 2: moderate staining, and 3: strong staining). d, e Semi-quantitative vWF vascular staining scores. Each graph represents mean score ± SD; n = 3 per group; *P < 0.05. KO knockout
Discussion
Currently, there are only limited treatment options for patients with SSc. Recent studies have provided evidence of an important role of CTGF in the development of organ fibrosis, including the skin [25]. Blockade of CTGF using the human anti-CTGF antibody, FG-3019, effectively prevented the development of skin fibrosis in response to Ang II challenge. The anti-fibrotic effect of FG-3019 was comparable to the genetic deletion of CTGF in collagen-producing cells. In addition, both treatments were very effective in reducing inflammation and lessening the damaging effects of Ang II on skin microvasculature.
The current study supports the findings of Liu et al. [6] who demonstrated the essential role of CTGF in bleomycin-induced fibrosis. In both studies, deletion of CTGF from fibroblasts and smooth muscle cells/pericytes prevented increased collagen deposition and myofibroblast accumulation in the skin. In addition, we also showed that blockade of CTGF significantly reduced the number of PDGFRβ-positive cells in the fibrotic lesion. Although the origin of these PDGFRβ collagen-producing cells in this model is not known, these cells may represent the expansion of local fibroblasts. Alternatively, they could originate from pericytes or mesenchymal stem cells. The role of CTGF in recruitment and expansion of these cells is not clear at present. Side by side comparison of the anti-fibrotic effects of FG-3019 and genetic deletion of CTGF showed that antibody directed against CTGF was comparable to CTGF deletion in blocking the CTGF profibrotic function in this study. Our results are also consistent with previous findings that showed therapeutic effects of FG-3019 in bleomycin-induced lung fibrosis [26].
Although profibrotic functions of CTGF are most widely recognized, CTGF has also been shown to induce inflammatory response in various cell types in vitro [7]. The pro-inflammatory role of CTGF was further confirmed in in vivo models of pancreatic and renal inflammation [27, 28]. In our model of Ang II-induced fibrosis, which is also characterized by increased inflammation, blockade of CTGF almost completely abrogated infiltration of inflammatory cells, including fibrocytes. Interestingly, treatment with FG-3019 appeared to be more potent than genetic CTGF deletion in reducing infiltration of CD45-positive cells and pSmad2 cells. However, both treatments were similarly effective in reducing the number of fibrocytes. These findings are consistent with a recent report that showed an inhibitory effect of FG-3019 on acute and delayed immune responses in a model of radiation-induced pulmonary fibrosis [29], where the effect appears to be indirect via modulation of myofibroblast activation and chemokine secretion (K. Lipson, personal communication). Thus, in this study, it is conceivable that blockade of CTGF inhibits recruitment of inflammatory cells from the circulation either by preventing secretion of the pro-inflammatory chemokines/cytokines by the activated resident cells or by reducing vascular injury and vessel permeability.
Ang II has a detrimental effect on the vasculature by affecting both endothelial and vascular smooth muscle cells (vSMCs) [30]. CTGF plays an important role in the activation of vSMCs by promoting their growth, migration, and production of collagen. Furthermore, Ang II is known to induce CTGF in vSMCs [8]. It has also been reported that CTGF is overexpressed in microvascular endothelial cells in SSc and that conditioned medium from microvascular endothelial cells stimulates the proliferation and migration of fibroblasts in SSc [31]. Although the association between CTGF and cardiovascular diseases is well-documented [8, 32], less is known about the potential contribution of CTGF to vascular disease in SSc. In this study, we performed a limited assessment of the effects of CTGF blockade on vascular damage by measuring the vascular injury marker, vWF. We showed that inhibition of CTGF partially reduced vWF expression. The effect was more pronounced in the CTGF KO than in FG-3019-treated mice. Together, these data suggest that blockade of CTGF may have a beneficial effect on vascular injury. The limitation of this study is that it only assessed the preventive effect of FG-3019, and further studies are needed to evaluate whether FG-3019 is effective in reversing the established fibrosis. A phase II clinical trial evaluating FG-3019 as a treatment for idiopathic pulmonary fibrosis has recently been completed. Considering the anti-fibrotic effects of FG-3019 in the skin, our study strongly supports the testing of FG-3019 as a therapeutic agent for SSc dermal fibrosis.
Conclusions
We showed that FG-3019 is effective in reducing Ang II-induced inflammation and skin fibrosis in mice. FG-3019 also partially reduced the Ang II-induced vascular damage. This study supports the testing of FG-3019 as therapy for skin fibrosis in patients with SSc.
Acknowledgements
Not applicable.
Funding
This work was supported in part by the NIH NIAMS grant R01 AR44883 (MT).
Availability of data and materials
The datasets supporting the conclusions of this article are included in this published article.
Authors’ contributions
KM and TM performed the research and wrote the manuscript. LS contributed to the experimental design and performed some experiments. AL contributed analytic tools. KL contributed reagents. MT was the principal investigator and was involved in conception and design of the study, data analysis, and manuscript writing. All authors read and approved the final manuscript.
Competing interests
K.E. Lipson is an employee of FibroGen, Inc. No other conflicts of interest exist for any of the authors other than K.E. Lipson.
Consent for publication
Not applicable.
Ethics approval and consent to participate
All mice experiments were performed in accordance with the National Institutes of Health and institutional guidelines for animal care, and were approved by the Committee on the Ethics of Animal Experiments of the Boston University.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Abbreviations
- Ang II
Angiotensin II
- BSA
Bovine serum albumin
- CTGF
Connective tissue growth factor
- DAPI
4′,6-Diamidino-2-phenylindole
- ECM
Extracellular matrix
- HE
Hematoxylin and eosin
- KO
Knockout
- PBS
Phosphate-buffered saline
- PDGFRβ
Platelet-derived growth factor receptor β
- SSc
Systemic sclerosis
- vSMCs
vascular smooth muscle cells
- vWF
von Willebrand factor
- αSMA
Alpha smooth muscle actin
References
- 1.Allanore Y, Simms R, Distler O, Trojanowska M, Pope J, Denton CP, et al. Systemic sclerosis. Nat Rev Dis Primers. 2015;1:15002. doi: 10.1038/nrdp.2015.2. [DOI] [PubMed] [Google Scholar]
- 2.Lafyatis R. Transforming growth factor β–at the centre of systemic sclerosis. Nat Rev Rheumatol. 2014;10(12):706–19. doi: 10.1038/nrrheum.2014.137. [DOI] [PubMed] [Google Scholar]
- 3.Leask A, Parapuram SK, Shi-Wen X, Abraham DJ. Connective tissue growth factor (CTGF, CCN2) gene regulation: a potent clinical bio-marker of fibroproliferative disease? J Cell Commun Signal. 2009;3(2):89–94. doi: 10.1007/s12079-009-0037-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hall-Glenn F, Lyons KM. Roles for CCN2 in normal physiological processes. Cell Mol Life Sci. 2011;68(19):3209–17. doi: 10.1007/s00018-011-0782-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mori T, Kawara S, Shinozaki M, Hayashi N, Kakinuma T, Igarashi A, et al. Role and interaction of connective tissue growth factor with transforming growth factor-β in persistent fibrosis: a mouse fibrosis model. J Cell Physiol. 1999;181(1):153–9. doi: 10.1002/(SICI)1097-4652(199910)181:1<153::AID-JCP16>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 6.Liu S, Shi-wen X, Abraham DJ, Leask A. CCN2 is required for bleomycin-induced skin fibrosis in mice. Arthritis Rheum. 2011;63(1):239–46. doi: 10.1002/art.30074. [DOI] [PubMed] [Google Scholar]
- 7.Kular L, Pakradouni J, Kitabgi P, Laurent M, Martinerie C. The CCN family: a new class of inflammation modulators? Biochimie. 2011;93(3):377–88. doi: 10.1016/j.biochi.2010.11.010. [DOI] [PubMed] [Google Scholar]
- 8.Ponticos M. Connective tissue growth factor (CCN2) in blood vessels. Vascul Pharmacol. 2013;58(3):189–93. doi: 10.1016/j.vph.2013.01.004. [DOI] [PubMed] [Google Scholar]
- 9.Sonnylal S, Shi-Wen X, Leoni P, Naff K, Van Pelt CS, Nakamura H, et al. Selective expression of connective tissue growth factor in fibroblasts in vivo promotes systemic tissue fibrosis. Arthritis Rheum. 2010;62(5):1523–32. doi: 10.1002/art.27382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rice LM, Ziemek J, Stratton EA, McLaughlin SR, Padilla CM, Mathes AL, et al. A longitudinal biomarker for the extent of skin disease in patients with diffuse cutaneous systemic sclerosis. Arthritis Rheumatol. 2015;67(11):3004–15. doi: 10.1002/art.39287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Igarashi A, Nashiro K, Kikuchi K, Sato S, Ihn H, Fujimoto M, et al. Connective tissue growth factor gene expression in tissue sections from localized scleroderma, keloid, and other fibrotic skin disorders. J Invest Dermatol. 1996;106(4):729–33. doi: 10.1111/1523-1747.ep12345771. [DOI] [PubMed] [Google Scholar]
- 12.Sato S, Nagaoka T, Hasegawa M, Tamatani T, Nakanishi T, Takigawa M, et al. Serum levels of connective tissue growth factor are elevated in patients with systemic sclerosis: association with extent of skin sclerosis and severity of pulmonary fibrosis. J Rheumatol. 2000;27(1):149–54. [PubMed] [Google Scholar]
- 13.Holmes A, Abraham DJ, Chen Y, Denton C, Shi-wen X, Black CM, et al. Constitutive connective tissue growth factor expression in scleroderma fibroblasts is dependent on Sp1. J Biol Chem. 2003;278(43):41728–33. doi: 10.1074/jbc.M305019200. [DOI] [PubMed] [Google Scholar]
- 14.Dziadzio M, Usinger W, Leask A, Abraham D, Black CM, Denton C, et al. N-terminal connective tissue growth factor is a marker of the fibrotic phenotype in scleroderma. QJM. 2005;98(7):485–92. doi: 10.1093/qjmed/hci078. [DOI] [PubMed] [Google Scholar]
- 15.Makino K, Makino T, Stawski L, Mantero JC, Lafyatis R, Simms R, et al. Blockade of PDGF receptors by crenolanib has therapeutic effect in patient fibroblasts and in preclinical models of systemic sclerosis. J Invest Dermatol. 2017. doi: 10.1016/j.jid.2017.03.032 [DOI] [PMC free article] [PubMed]
- 16.Stawski L, Haines P, Fine A, Rudnicka L, Trojanowska M. MMP-12 deficiency attenuates angiotensin II-induced vascular injury, M2 macrophage accumulation, and skin and heart fibrosis. PLoS One. 2014;9(10):e109763. doi: 10.1371/journal.pone.0109763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stawski L, Han R, Bujor AM, Trojanowska M. Angiotensin II induces skin fibrosis: a novel mouse model of dermal fibrosis. Arthritis Res Ther. 2012;14(4):R194. doi: 10.1186/ar4028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brenner MC, Krzyzanski W, Chou JZ, Signore PE, Fung CK, Guzman D, et al. FG-3019, a human monoclonal antibody recognizing connective tissue growth factor, is subject to target-mediated drug disposition. Pharm Res. 2016;33(8):1833–49. doi: 10.1007/s11095-016-1918-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Raghu G, Scholand MB, de Andrade J, Lancaster L, Mageto Y, Goldin J, et al. FG-3019 anti-connective tissue growth factor monoclonal antibody: results of an open-label clinical trial in idiopathic pulmonary fibrosis. Eur Respir J. 2016;47(5):1481–91. doi: 10.1183/13993003.01030-2015. [DOI] [PubMed] [Google Scholar]
- 20.Neesse A, Frese KK, Bapiro TE, Nakagawa T, Sternlicht MD, Seeley TW, et al. CTGF antagonism with mAb FG-3019 enhances chemotherapy response without increasing drug delivery in murine ductal pancreas cancer. Proc Natl Acad Sci U S A. 2013;110(30):12325–30. doi: 10.1073/pnas.1300415110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Morales MG, Gutierrez J, Cabello-Verrugio C, Cabrera D, Lipson KE, Goldschmeding R, et al. Reducing CTGF/CCN2 slows down mdx muscle dystrophy and improves cell therapy. Hum Mol Genet. 2013;22(24):4938–51. doi: 10.1093/hmg/ddt352. [DOI] [PubMed] [Google Scholar]
- 22.Hattori T, Stawski L, Nakerakanti SS, Trojanowska M. Fli1 is a negative regulator of estrogen receptor α in dermal fibroblasts. J Invest Dermatol. 2011;131(7):1469–76. doi: 10.1038/jid.2011.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Peoples C, Medsger TA, Lucas M, Rosario BL, Feghali-Bostwick CA. Gender differences in systemic sclerosis: relationship to clinical features, serologic status and outcomes. J scleroderma relat disord. 2016;1(2):204–12. [DOI] [PMC free article] [PubMed]
- 24.Springer TA. von Willebrand factor, Jedi knight of the bloodstream. Blood. 2014;124(9):1412–25. doi: 10.1182/blood-2014-05-378638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kubota S, Takigawa M. Cellular and molecular actions of CCN2/CTGF and its role under physiological and pathological conditions. Clin Sci (Lond) 2015;128(3):181–96. doi: 10.1042/CS20140264. [DOI] [PubMed] [Google Scholar]
- 26.Wang Q, Usinger W, Nichols B, Gray J, Xu L, Seeley TW, et al. Cooperative interaction of CTGF and TGF-β in animal models of fibrotic disease. Fibrogenesis Tissue Repair. 2011;4(1):4. doi: 10.1186/1755-1536-4-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Charrier A, Chen R, Kemper S, Brigstock DR. Regulation of pancreatic inflammation by connective tissue growth factor (CTGF/CCN2) Immunology. 2014;141(4):564–76. doi: 10.1111/imm.12215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sanchez-Lopez E, Rayego S, Rodrigues-Diez R, Rodriguez JS, Rodrigues-Diez R, Rodriguez-Vita J, et al. CTGF promotes inflammatory cell infiltration of the renal interstitium by activating NF-kappaB. J Am Soc Nephrol. 2009;20(7):1513–26. doi: 10.1681/ASN.2008090999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bickelhaupt S, Erbel C, Timke C, Wirkner U, Dadrich M, Flechsig P, et al. Effects of CTGF blockade on attenuation and reversal of radiation-induced pulmonary fibrosis. J Natl Cancer Inst. 2017;109(8):djw339. [DOI] [PubMed]
- 30.Montezano AC, Nguyen Dinh Cat A, Rios FJ, Touyz RM. Angiotensin II and vascular injury. Curr Hypertens Rep. 2014;16(6):431. doi: 10.1007/s11906-014-0431-2. [DOI] [PubMed] [Google Scholar]
- 31.Serrati S, Chilla A, Laurenzana A, Margheri F, Giannoni E, Magnelli L, et al. Systemic sclerosis endothelial cells recruit and activate dermal fibroblasts by induction of a connective tissue growth factor (CCN2)/transforming growth factor β-dependent mesenchymal-to-mesenchymal transition. Arthritis Rheum. 2013;65(1):258–69. doi: 10.1002/art.37705. [DOI] [PubMed] [Google Scholar]
- 32.Alapati D, Rong M, Chen S, Hehre D, Rodriguez MM, Lipson KE, et al. Connective tissue growth factor antibody therapy attenuates hyperoxia-induced lung injury in neonatal rats. Am J Respir Cell Mol Biol. 2011;45(6):1169–77. doi: 10.1165/rcmb.2011-0023OC. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets supporting the conclusions of this article are included in this published article.