

Abstract
Background
There was no record of Aedes aegypti in Yunnan Province, China, until 2002, but this species is now continuously found in nine cities (or counties). Until now, little was known about the genetic diversity and population structure of this invasive species. Thus, a detailed understanding of the invasion strategies, colonisation and dispersal of this mosquito from a population genetics perspective is urgently needed for controlling and eliminating this disease vector.
Methods
The genetic diversity and population structure of Ae. aegypti communities were analysed by screening nine microsatellite loci from 833 Ae. aegypti mosquitoes sampled from 28 locations in Yunnan Province.
Results
In total, 114 alleles were obtained, and the average polymorphic information content (PIC) value was 0.672. The value of the alleles per locus ranged from 2.90 to 5.18, with an average of 4.04. The value of He ranged from 0.353 to 0.681, and the value of Ho within populations ranged from 0.401 to 0.689. Of the 28 locations, two showed significant departures from the Hardy-Weinberg equilibrium (HWE) with P-values less than 0.05, and a bottleneck effect was detected among locations from Ruili and the border areas with the degree of 60% and 50%, respectively. Combined with the F-statistics (FIT = 0.222; FCT = 0.145), the analysis of molecular variance (AMOVA) revealed that there was substantial molecular variation among individuals, accounting for 77.76% of the sample, with a significant P-value (<0.0001). The results suggest that genetic differences in Ae. aegypti originated primarily among individuals rather than among populations. Furthermore, the STRUCTURE and UPGMA cluster analyses showed that Ae. aegypti from the border areas were genetically isolated compared to those from the cities Ruili and Jinghong, consistent with the results of the Mantel test (R 2 = 0.245, P < 0.0001).
Conclusions
Continuous invasion contributes to the maintenance of Ae. aegypti populations’ genetic diversity and different invasion accidents result in the genetic difference among Ae. aegypti populations of Yunnan Province.
Electronic supplementary material
The online version of this article (doi:10.1186/s13071-017-2213-6) contains supplementary material, which is available to authorized users.
Keywords: Aedes aegypti, Genetic diversity, Population structure, Microsatellite loci, Genetic differences, Yunnan Province
Background
Dengue fever (DF) is an acute systemic viral disease caused by dengue virus and transmitted by Aedes mosquitoes. There are approximately 390 million dengue infections per year, and approximately 40% of the world population is at risk of infection from dengue [1]. Since 2013, local transmission of DF infections has been caused by imported cases of DF that have repeatedly occurred in Yunnan Province, especially in the cities of Ruili and Jinghong [2]. Aedes aegypti is the primary vector of the arboviruses, spreading dengue, yellow fever, chikungunya, Zika fever and other diseases [3–6] that originated in Africa and invaded the Americas and Southeast Asia through the shipping trade in the late nineteenth Century [7–9]. Previous studies have indicated that this species is distributed only in the area south of 22 degrees north latitude in China, which includes southern Taiwan, Hainan Island, and some sections of Guangdong and Guangxi Provinces, including islands [10].
Yunnan Province is located in southwestern China and extends from 21°8′32″ to 29°15′8″N and 97°31′39″ to 106°11′47″E. The climate in most regions of this province is fairly mild in winter and rather cool in summer. Because of its geographical position and natural climate conditions, Yunnan Province has the greatest diversity and abundance of mosquito species in China. However, there was no record of Ae. aegypti in Yunnan Province until 2002 [11].
As Yunnan Province is along the main passageway connecting South Asia and Southeast Asia, the international tourism industry as well as logistics and communications between Yunnan Province and neighbouring countries where Ae. aegypti is widespread [12], such as Myanmar, Cambodia, Vietnam and Thailand, have expanded in recent years. As previously reported, human trade and travel might support Ae. aegypti migration [13], and the invasion risk via human travel has become much more severe. Since the first report of Ae. aegypti in Yunnan Province at Ruili Jiegao Port, the species has been continuously found in nine cities in Yunnan Province [11, 14–18].
Biological invasions are closely associated with human health and genetic factors, such as genetic variation and admixture, and this information has been shown to be extremely useful for managing invasive species [19, 20]. Therefore, knowledge about the genetic diversity and structure among Ae. aegypti populations can help with inferring their invasion processes and with modelling the spread of disease and of insecticide resistance [21], which is useful for maintaining effective vector control strategies and providing insight into the epidemiology of arthropod-borne diseases such as DF [22–24]. Therefore, it is necessary to understand the invasion strategies, colonisation and dispersal of this mosquito species, especially from a population genetics perspective. However, to date, no studies have investigated the genetic diversity and structure of Ae. aegypti populations in Yunnan Province or even in China overall.
Microsatellites are short lengths of DNA sequences in which motifs of 1–6 bases are tandemly repeated, and they occur throughout the eukaryotic genome [25], providing many advantages, such as high polymorphism, co-dominant expression, and broad genome distribution [26]. Since the microsatellite markers for Ae. aegypti were isolated in 2001 [27], they have become a powerful tool that has been widely applied in population genetic studies of Ae. aegypti. Several studies on the genetic structure of Ae. aegypti using microsatellite markers have been conducted in Southeast Asia [28–34].
To explain the invasion, colonisation and dispersal of Ae. aegypti in Yunnan Province, we conducted a population genetics study of 28 locations in this area, using nine microsatellite loci. The results of this study can also provide a theoretical basis for programs for controlling Ae. aegypti in China.
Methods
Mosquito sampling
Based on the results of previous investigations and the distribution of Ae. aegypti in Yunnan Province, 28 locations in Yunnan Province were chosen as research sites (Fig. 1, Table 1). All of the Ae. aegypti adults and larvae used in this study were collected from these regions from 2015 to 2016. The distances between the sampling sites ranged from several hundred meters (> 500 m) to kilometres. The precise locations of these sites were recorded using the global positioning system.
Fig. 1.
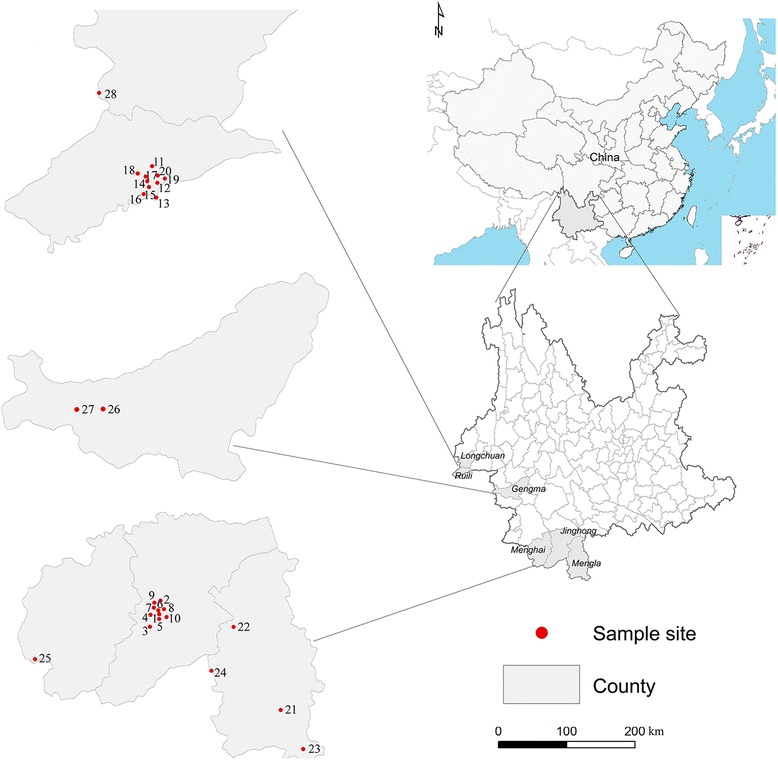
Illustration of the geographical locations of Ae. aegypti collection sites
Table 1.
Sampling information of Ae. aegypti collection in Yunnan Province,China
| Collection region | No. | Location name (code) | No. of samples | Geographical coordinates | Collection date |
|---|---|---|---|---|---|
| Jinghong city | 1 | Linjiangyuan (LJY) | 30 | 22°0′49 “N, 100°48′6”E | 05/12/2015 |
| 2 | Nonken Hospital (NKH) | 30 | 22°1′49″N, 100°47′35″E | 05/15/2015 | |
| 3 | Gasa town (GSZ) | 30 | 21°57′5″N, 100°45′44″E | 05/19/2015 | |
| 4 | Damanyao (DMY) | 30 | 22°0′0″N, 100°46′37″E | 05/24/2015 | |
| 5 | Guanguang Hotel (GGH) | 30 | 22°0′12″N, 100°47′54″E | 06/02/2015 | |
| 6 | Galan Road (GLR) | 30 | 22°1′8″N, 100°47′45″E | 06/05/2015 | |
| 7 | Ganxiusuo (GXS) | 30 | 22°0′4″N, 100°47′24″E | 06/07/2015 | |
| 8 | Jiangbei (JBQ) | 30 | 22°1′51″N, 100°47′15″E | 06/12/2015 | |
| 9 | Yunshu Company (YSC) | 30 | 22°1′30″N, 100°47′15″E | 06/15/2015 | |
| 10 | Pi’er Park (PSS) | 30 | 22°0′58″N, 100°49′3″E | 06/17/2015 | |
| Ruili city | 11 | Munao Road (MNL) | 30 | 24°1′38″N, 97°52′5″E | 08/12/2015 |
| 12 | Jindian Road (JDL) | 30 | 24°1′3″N, 97°52′56″E | 08/15/2015 | |
| 13 | Jiegao Freight Yard (JGH) | 30 | 23°58′40″N, 97°53′24″E | 08/19/2015 | |
| 14 | Candy Company (TCP) | 30 | 24°0′40″N, 97°52′42″E | 08/22/2015 | |
| 15 | Jincheng Steel Plant (JCG) | 30 | 24°0′0″N, 97°52′18″E | 08/24/2015 | |
| 16 | Guomen bridge (GMG) | 30 | 23°59′24″N, 97°52′3″E | 08/27/2015 | |
| 17 | Hongda Freight Yard (HDH) | 30 | 24°0′52″N, 97°52′11″E | 08/28/2015 | |
| 18 | North Bus Terminal (BNS) | 30 | 24°1′20″N, 97°51′35″E | 08/30/2015 | |
| 19 | Automobile Company (CXT) | 30 | 24°00′53″N, 97°53′24″E | 09/02/2015 | |
| 20 | Scrapyard (BFC) | 30 | 24°0′40″N, 97°52′12″E | 09/05/2015 | |
| Border areas | 21 | Nanla Middle School (ML-1) | 30 | 21°27′59″N, 101°34′2″E | 05/10/2016 |
| 22 | Botanical Garden (ML-2) | 30 | 21°56′24″N, 101°15′41″E | 05/13/2016 | |
| 23 | Shangyong (ML-3) | 28 | 21°14′34″N, 101°42′44″E | 05/15/2016 | |
| 24 | Guanlei (ML-4) | 30 | 21°40′47″N, 101°8′11″E | 05/17/2016 | |
| 25 | Daluo (MH-1) | 25 | 21°42′6″N, 100°3′23″E | 07/08/2016 | |
| 26 | Mengding-1 (GM-1) | 30 | 23°33′53″N, 99°5′48″E | 07/12/2016 | |
| 27 | Mengding-2 (GM-2) | 30 | 23°33′00″N, 99°3′33″E | 07/14/2016 | |
| 28 | Longchuan (LC-1) | 30 | 24°11′56″N, 97°43′36″E | 07/20/2016 |
Abbreviation: No number of locations
A human-baited method with handheld aspirators performed according to the Surveillance Methods for Vector Density - Mosquito (GB/T 23797–2009), was used in the collection of adult mosquitoes (i.e. intercepted before biting), and field identification of the samples was performed through analysis of morphological characteristics [35]. To reduce the chance of collecting siblings, we selected only one mosquito per container if the samples were from larvae, and all larvae were reared to adulthood in the laboratory until they could be identified. All adult Ae. aegypti samples were preserved in 100% ethanol at 4 °C for the isolation of genomic DNA.
DNA isolation, PCR amplification and microsatellite genotyping
In the present study, 30 Ae. aegypti samples per population were randomly selected from each site for genetic analyses. For sites with fewer than 30 mosquito samples, all the samples were selected. Genomic DNA was extracted individually from the samples using a TaKaRa Mini-BEST Universal Genomic DNA Extraction Kit (Takara, Dalian, China) and following the standard DNA extraction protocol provided by the manufacturer. The DNA yield was checked with the primers LCO1490 and HCO2198; in total, 833 samples were obtained and were stored at -20 °C until analysis.
Using denaturing polyacrylamide gel electrophoresis, nine microsatellite polymorphic loci were selected from 74 loci that were previously described in related studies [22, 26, 36]. The most suitable annealing temperature for each primer pair was confirmed with preliminary PCR amplifications, and the forward sequence of each primer was fluorescently labelled with FAM, HEX or TAMRA. The primer sequences and information are summarised in Table 2.
Table 2.
Primer information for nine microsatellite loci screened in Ae. aegypti from Yunnan Province, China
| Locus | Repeat motif | Primer sequence (5′-3′) | Annealing temperature (°C) | Allele size (bp) | Fluorescence labeling | Reference |
|---|---|---|---|---|---|---|
| SQM1 | CT10(TT)CT | F: AATCGTGACGCGTCTTTTG; R: TAACTGCATCGAGGGAAACC |
54 | 233–239 | 5′-HEX | [27] |
| SQM2 | GA15 | F: CAAACAACGAACTGCTCACG; R: TCGCAATTTCAACAGGTAGG |
53 | 157–183 | 5′-FAM | [27] |
| SQM3 | CAT7 | F: ATTGGCGTGAGAACATTTTG; R: GAGGAGTGAGCAGATAGGAGTG |
58 | 156–186 | 5′-FAM | [27] |
| SQM4 | TAGA8 | F: GCCAAAAACCAACAAACAGG; R: AATCGACCCGACCAATAACA |
53 | 286–290 | 5′-TAMRA | [27] |
| SQM5 | ATA36 | F: GGAGCATTCATAGAGAATTGTCA; R: GAGATGAACCAGTCATAGGGC |
56 | 110–116 | 5′-FAM | [27] |
| SQM6 | (TTTA)7(T)14 | F: CGACAGATGGTTACGGACGG; R: GTC CCG CTC CAA AAA TGC CC |
58 | 228 | 5′-FAM | [23] |
| SQM7 | AG4 | F: AAAACCTGCGCAACAATCAT; R: AAGGACTCCGTATAATCGCAAC |
56 | 147–169 | 5′-FAM | [36] |
| SQM8 | AG5 | F: TGATCTTGAGAAGGCATCCA; R: CGTTATCCTTTCATCACTTGTTTG |
55 | 170–180 | 5′-FAM | [36] |
| SQM9 | AC1 | F: TCCGGTGGGTTAAGGATAGA; R: ACTTCACGCTCCAGCAATCT |
55 | 193–209 | 5′-FAM | [36] |
All PCR amplifications were conducted in a total volume of 20 μl using an S1000 Thermal Cycler (BIO-RAD, CA, USA). Each reaction contained 10 μl of 2× PCR Master Mix (Thermo Fisher Scientific, MA, USA), each primer at 0.8 μM, and 10 pg −1 μg of template DNA, with ddH2O, added to reach a volume of 20 μl. The PCR reaction conditions were as follows: 95 °C for 8 min, followed by 30 cycles at 94 °C for 45 s, a different annealing temperature for each locus (Table 2) for 30 s and 72 °C for 45 s. A final extension was conducted at 72 °C for 10 min. All the PCR products were diluted 1:10, mixed with LIZ 500-labeled size standard and formamide at a 1:9 ratio, and then run on an ABI3730XL (Applied Biosystems, Foster City, USA) capillary sequencer. Allele sizes for each locus were read with Gene Marker software (version 2.2.0).
Microsatellite analysis
Genetic diversity within each population
To determine the genetic diversity within each sampling location, standard genetic parameters were calculated using several software programs. FSTAT (version 2.9.3.2) was used to assess allelic richness (r) in each population [37]. The inbreeding coefficient (FIS), observed heterozygosity (Ho) and expected heterozygosity (He) were calculated with Arlequin software (version 3.5.2.2) [38]. The linkage disequilibrium, departures from the Hardy-Weinberg equilibrium (HWE) and the P-value for each location were assessed using Fisher’s exact test in Gene pop (version 4.2) [39]. Null allele frequency for each microsatellite locus was estimated using Micro-checker software (version 2.2.3) [40]. The stepwise mutation model (SMM) were used to assess deviation from expected heterozygote excess related to allelic diversity across all nine loci and based on the loss of allelic diversity exceeding heterozygosity during a bottleneck effect in BOTTLENECK software (version 1.2.02) [41]. To determine whether the individuals in each location used in this study were sufficient for the research, the mean number of alleles per locus (NA) for each population was calculated with a rarefaction method by using the software Allelic Diversity Analyzer (version 1.0) [42]. P-values less than 0.05 were considered significant in each analysis. Polymorphic information content (PIC) across all nine loci was assessed using PIC-Calc 0.6 [43].
Population genetics structure and differences among populations
With regard to the analysis of the population genetics structure, the pair fixation indices (FST) between populations and genetic distance [FST/(1- FST)] and the analysis of molecular variance (AMOVA) were performed with Arlequin (version 3.5.2.2). The software STRUCTURE (version 2.3.4) was used to assess the genetic structure differences among populations based on a Bayesian clustering approach with a Markov chain Monte Carlo algorithm (100,000 iterations and 1,000,000 repetitions). The K value was set from 1 to 31 (the true number of populations plus 3) with 20 runs for each K value [44, 45], and the ΔK methods were executed to determine the most likely K value [46]. A graphical display of the genetic structure was produced using DISTRUCT (version 1.1) [47]. To test the relationship between genetic distance [FST/(1- FST)] and geographic distance [ln (km)], a Mantel test with 10,000 permutations was performed using the ZT software package, and a graphical display of the results was produced with Excel 2016 (Microsoft Corporation, WA, USA) [48]. Based on the unweighted pair group method, UPGMA cluster analysis was conducted with the software NTSYS pc (version 2.2), and the polygenetic tree was modified with the software Tree View [49]. Principal component analysis (PCA) and non-metric multidimensional scaling (NMDS) analyses were conducted via the Vegan and Permute packages in R software [50].
Results
Genetic diversity
In total, 114 alleles were obtained for nine microsatellite loci. The highest number of alleles (20) was found at locus SQM6, and the lowest number (7) was found at locus SQM1. PIC was high, with values ranging from 0.392 to 0.886 (mean value 0.672). Micro-checker results suggest that null alleles were found at the loci SQM3, SQM4 and SQM9 and that the null allele frequency varied from 0 to 0.2956 among the 9 loci in all locations (Table 3). No consistency was found between any pair of loci across all locations, though 166 of 1008 (16.47%) pairwise tests for linkage disequilibrium remained significant after Bonferroni correction (Additional file 1: Figure S1).
Table 3.
Null allele frequency, number of alleles and PIC information for genetic markers of Ae. aegypti from Yunnan Province, China
| Locus | Number | Null allele frequency | Number of allele | PIC |
|---|---|---|---|---|
| SQM1 | 833 | – | 7 | 0.392 |
| SQM 2 | 833 | – | 16 | 0.804 |
| SQM 3 | 833 | 0.1703 | 13 | 0.629 |
| SQM 4 | 833 | 0.2956 | 12 | 0.633 |
| SQM 5 | 833 | – | 9 | 0.679 |
| SQM 6 | 833 | – | 20 | 0.886 |
| SQM 7 | 833 | – | 10 | 0.657 |
| SQM 8 | 833 | – | 11 | 0.614 |
| SQM9 | 833 | 0.1024 | 16 | 0.755 |
| Mean | 12.67 | 0.672 |
The genetic diversity and difference analysis of Ae. aegypti within 28 locations showed that the mean number of alleles per locus of each location ranged from 2.90 to 5.18, with an average of 4.04, and the allelic richness ranged from 3.34 to 5.72, with an average of 4.53. The average NA value for Ruili City (5.15) was greater than in Jinghong City (4.88) and the border areas (4.63). The largest number of alleles (5.18) was observed at site HDH from Ruili City, while the lowest numbers were observed at M3 L (2.90) and M4 L (3.15) from the border areas. The He values for each location ranged from 0.353 to 0.681, and the Ho values for each location ranged from 0.401 to 0.689. The two locations M2 L and M3 L from the border areas had the highest and the lowest observed individual heterozygosity, respectively (M2 L, 0.689; M3 L, 0.401) (Additional file 2: Table S1).
The Hardy-Weinberg equilibrium (HWE) test revealed that only 2 of the 28 locations (7.14%) had significant departures from HWE, and the values of FIS ranged from -0.147 to 0.157 (Additional file 2: Table S1). Among the three sampling areas, two locations from Ruili City had the highest mean allelic richness (JGH, 5.67; HDH, 5.72), and the lowest richness was observed in the border areas (M4 L, 3.67; M3 L, 3.34) (Additional file 3: Figure S2). The interaction analysis of the mean number of alleles per locus, Ho values and allelic richness of all locations also showed that the locations from Ruili City had higher genetic diversity than those from Jinghong City and the border areas. The SMM model analyses (Additional file 4: Table S2) also revealed that nearly 50% and 60% of sampling locations from the border areas and Ruili City showed significant population expansion, while all P-values (P > 0.05) remained non-significant among the populations of Jinghong City after Bonferroni correction.
Genetic structure and differences
The Mantel test for the 28 locations showed a significant positive correlation (49.5%, P < 0.0001) between genetic distances and geographical distances. Using the isolation-by-distance model, the genetic distribution of all 28 locations was illustrated and is shown in Fig. 2a (R 2 = 0.245, P < 0.01). The genetic distances are positively correlated with geographical distances within the 10 locations in the cities Jinghong and Ruili (R 2 = 0.178 and 0.479, respectively; P < 0.01; Fig. 2b, c), but no such significant correlation was observed within the 8 locations of the border areas (R 2 = 0.072, P = 0.167; Fig. 2d).
Fig. 2.
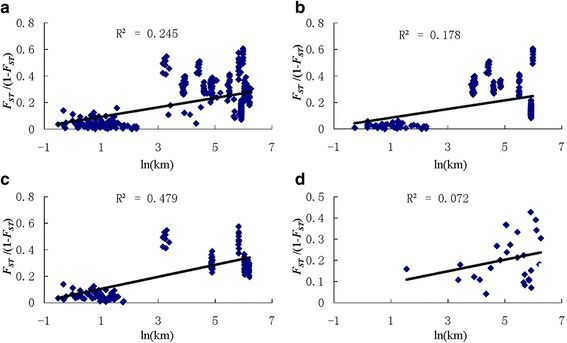
Analysis on the relationship between genetic distance [FST/(1-FST)] and geographical distance [ln(km)] for (a) all locations (R 2 = 0.245, P < 0.001); (b) locations from Jinghong city only (R 2 = 0.178, P < 0.001); (c) locations from Ruili city only (R 2 = 0.479, P < 0.001); and (d) locations from border area only (R 2 = 0.072, P = 0.167)
The FIT and FCT values for Ae. aegypti were 0.222 and 0.145, respectively (both P < 0.01). This result indicates that genetic differences may exist mainly among individuals and groups. The AMOVA results (Additional file 5: Table S3) also indicated that the major variation in Ae. aegypti was mainly found among individuals and groups, accounting for 77.76% and 14.51% of the variation, respectively, while the variations among populations within groups and individuals within populations accounted for only 6.03% and 1.73%, respectively. Fisher’s exact tests showed that there was significant genetic variation in these four levels.
The pairwise FST values of Ae. aegypti ranged from -0.004 to 0.377, as displayed in Additional file 6: Table S4. Among these comparisons, 361 out of 378 (95.5%) showed significant genetic differences. The FST values were lower than 0.05 among 10 locations from Jinghong City, between 0.05 and 0.10 among 10 locations from Ruili City and above 0.10 among 8 locations from the border areas. All P-values were significant (P < 0.05) after Bonferroni corrections were applied.
Based on Evanno et al.’s ΔK methods [44], all locations in this study were assigned to two genetically differentiated groups (K = 2, Fig. 3a, b) that overall showed a considerable mixture and that Ae. aegypti samples from the border areas were more genetically isolated compared to the two other sampling areas. Bayesian clustering analysis on all these collections also identified that the Ae. aegypti from the border areas were genetically isolated from Ruili and Jinghong cities (Fig. 4). Additionally, the UPGMA cluster analysis (Fig. 5) displayed similar results showing that 8 locations from the border areas clustered first and then grouped with 20 locations from Jinghong City and Ruili City, which were also separated into two different sub-clusters with strong support. PCA and NMDS analyses were illustrated in Fig. 6a, b. The results revealed that the genetic structures among the locations from the border areas were significantly different from the structures in Ruili and Jinghong.
Fig. 3.
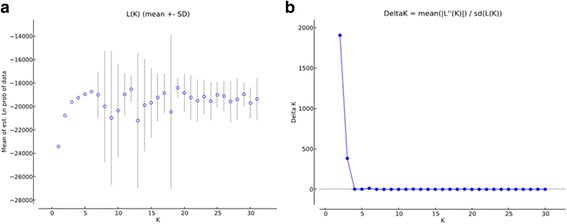
Scatter plots of Log probability of the data (a) and △K (b) for all Ae. aegypti populations analysed. Delta K plots are based on the rate of change in the log probability of the data between successive K values
Fig. 4.
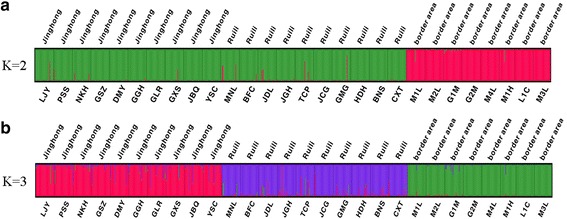
Genetic structure within 28 locations of Ae. aegypti. STRUCTURE bar plots are indicating relatedness of Ae. aegypti populations based on nine microsatellite loci. Each vertical bar represents an individual. The height of each bar represents the probability of assignment to each of Koptimal clusters (different colours) determined using Evanno et al.’s ΔK methods. a K = 2. b K = 3
Fig. 5.
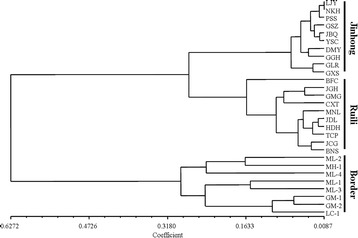
UPGMA cluster analysis of 28 sampling locations based on the genetic distance of all populations
Fig. 6.
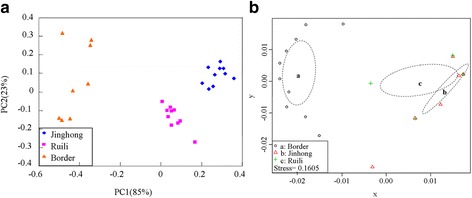
Correlation analysis among Ruili, Jinghong and the border sites. Principal coordinates analyses (a PCoA analysis) and non-metric multidimensional scaling (b NMDS analysis), based on co-dominant alleles at nine microsatellite loci, displaying genetic similarities among individuals of Ae. aegypti sampled from Ruili, Jinghong and the border sites
Discussion
Most species can be transported to a new geographical area via human activities but either fail to establish or maintain their population with minimal impact [51]. By definition, the progress of one exotic species to the level of a biological invasion involves a significant increase within its new territory [52]. Aedes aegypti, an exotic mosquito species in China was not discovered in Yunnan Province until it was first reported in Ruili in 2002. This species is now established in nine cities in Yunnan Province [11, 14–18, 53], and its distribution range and abundance have increased significantly. Thus, Ae. aegypti has become an invasive mosquito species in Yunnan Province.
Our results indicate that the Ae. aegypti populations in the border areas most likely came recently from neighbouring countries, and that continuous invasion still exists, which may explain the negative values of FIS and why the values of Ho are higher than those of He [54–56]. Meanwhile, natural selection might explain the existence of a bottleneck effect in half of these locations.
Jinghong and Ruili, famous for their tourist and jewellery trading industries, are two major cities in Yunnan Province and are also closely connected with neighbouring countries where Ae. aegypti is widespread, such as Myanmar, Thailand and Vietnam [12]. Based on the invasion history, it is most likely that the invasion of Ruili started from neighbouring countries; nevertheless, although Ae. aegypti was first reported in Jinghong in 2013; it remains unclear whether the Ae. aegypti population also came from neighbouring countries or simply dispersed from Ruili or border areas because of the continuous tourist and business activities between these areas.
As the values of FIS are all positive for the sampling locations of Jinghong and Ruili, and because the FIS values of Jinghong locations are lower than those of Ruili locations, the difference may come from different invasion times of Ae. aegypti in these two cities, since the Ae. aegypti population was reported nearly 11 years earlier in Ruili than in Jinghong. This hypothesis can also be reached from the bottleneck effect analysis results, with a bottleneck effect observed in nearly 60% of the Ae. aegypti populations in Ruili and showed significant heterozygosity deficit for population expansion but in none of those in Jinghong.
Bayesian analysis revealed high genetic structure among the populations studied, especially between the Ae. aegypti population of the border area and the populations of Ruili and Jinghong cities. This segregation can also be observed in the UPGMA dendrogram, NMDS and PCA analysis. This may be because of the Ae. aegypti population of Ruili, Jinghong and the border area came from different invasion accidents, and their dispersal ability was limited by the short flight range of this mosquito.
Our analysis also suggests that Ae. aegypti may have been introduced to Jinghong from Ruili or that the original locations of the Ae. aegypti populations in these two cities are close together. This idea was supported by UPGMA clustering analysis, which indicates that the genetic distance between Ae. aegypti populations in the cities of Jinghong and Ruili is closer than that of the border area population. Furthermore, multiple invasions can help the allelic richness of invasive populations to recover, but the richness is usually lower than the original population [57, 58]. In this study, the allelic richness of the Ae. aegypti population in Jinghong is higher than that in border areas but lower than that in Ruili.
Passive dispersal is the most common way for this mosquito to spread in southwestern China, given that Ae. aegypti has a very close association with humans and shelters in indoor habitats [59] and has a very short flight range, usually limited to several hundred meters [60–62]. Results of isolation by distance analysis reveal that the genetic distances are positively correlated with geographical distances within the sampling locations of Jinghong and Ruili (r = 0.422 and 0.692, respectively; P < 0.01), while no such significant correlation was observed within the locations of the border areas (r = 0.269, P = 0.167). This may reflect the translocation of mosquito eggs and aquatic stages in containers as a result of human travel throughout the cities [63]. As for the border areas without natural barriers [64], Ae. aegypti may be introduced to these areas from bordering countries through active dispersal or population expansion, but the colonisation and dispersal time has not been long enough to cause genetic differentiation.
The genetic diversity and population structure of Ae. aegypti found in this study revealed a significant population expansion within Yunnan Province, which resulted in the similarity of Ae. aegypti population between Ruili and Jinghong cities. Based on UPGMA, NMDS and PCA analysis, our study also suggests that the Ae. aegypti population of Ruili, Jinghong and the border area came from different invasion accidents that we believe to be associated with passive dispersal, aided by human activities and transportation, representing the invasion risk of Ae. aegypti is still severe, and there is an ever-greater risk of arthropod-borne diseases such as DF.
Conclusion
In conclusion, high genetic diversity and genetic structure were demonstrated among two Ae. aegypti populations from Yunnan Province. Continuous invasion, aided by human activities and transportation, appears to contribute to the maintenance of genetic diversity in Ae. aegypti in this region. The genetic difference among these two Ae. aegypti populations may have arisen from different invasion incidents with subsequent dispersal limited by the short flight range of this mosquito. Overall, the results of this study can provide a theoretical basis for programs for controlling Ae. aegypti in China.
Additional files
Figure S1. Analysis of linkage disequilibrium in 28 locations (TIFF 7443 kb)
Table S1. Population statistics for Ae. aegypti investigated using nine microsatellite loci. (DOCX 17 kb)
Figure S2. The mean number of distinct alleles per locus for 28 sample locations. (TIFF 1958 kb)
Table S2. Aedes aegypti heterozygosity tests. (DOCX 15 kb)
Table S3. Analysis of molecular variance of populations from Jinghong, Ruili and the border areas. (DOCX 15 kb)
Table S4. Pairwise population differentiation estimates (FST) and geographical distance [ln(km)] between all locations of Ae. aegypti. (DOCX 22 kb)
Acknowledgments
Not applicable.
Funding
This work was funded by grants from the National Natural Science Foundation of China (No. U1402261), the Infective Diseases Prevention and Cure Project of China (No. 2012ZX10004219) and the National Key Technology Support Program (No. 2014BAI13B01).
Availability of data and materials
Data are available on request to the corresponding author.
Authors’ contributions
QMS, CXL and TYZ jointly designed and coordinated the study, with contributions from XXG and YDD. QMS drafted the article with contributions from LX and JG. QMS, HDZ and GW collected samples from Yunnan Province. QMS, DX, QML, AJS, and JG carried out the laboratory work and performed the statistical analysis. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Abbreviations
- AMOVA
Analysis of molecular variation
- DF
Dengue fever
- R
Richness
- FCT
Genetic differences among groups
- FIS
Inbreeding coefficient
- FIT
Global heterozygote deficit across of all populations
- FST
Genetic differences among populations
- He
Expected heterozygosity
- Ho
Observed heterozygosity
- HWE
Hardy-Weinberg equilibrium
- NA
Number of alleles per locus
- NMDS
Non-metric multidimensional scaling
- PCA
Principal components analysis
- PIC
Polymorphic information content
- SMM
Stepwise mutation model
- TPM
Two-phase model
- UPGMA
Unweighted pair-group method with arithmetic averaging
Footnotes
Electronic supplementary material
The online version of this article (doi:10.1186/s13071-017-2213-6) contains supplementary material, which is available to authorized users.
Contributor Information
Qing-Ming Shi, Email: 8438643@qq.com.
Heng-Duan Zhang, Email: 453149452@qq.com.
Gang Wang, Email: wanggang_3314@163.com.
Xiao-Xia Guo, Email: guoxx99@163.com.
Dan Xing, Email: xingdan93@163.com.
Yan-De Dong, Email: dongyd99@sina.com.
Li Xiao, Email: 18692990@qq.com.
Jian Gao, Email: camgao_006@163.com.
Qin-Mei Liu, Email: qmliu_edu@163.com.
Ai-Juan Sun, Email: 1064415830@qq.com.
Chun-Xiao Li, Email: vectorlcx@126.com.
Tong-Yan Zhao, Email: aedes@126.com, Email: tongyanzhao@126.com.
References
- 1.Bhatt S, Gething PW, Brady OJ, Messina JP, Farlow AW, Moyes CL, et al. The global distribution and burden of dengue. Nature. 2013;496:504–507. doi: 10.1038/nature12060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yang DM, Jiang JY, Guo XF, Wu C, Zhou HN. Epidemiological analysis and investigation of dengue fever in Yunnan Province from 2009 to 2014. J Pathog Biol. 2015;10:738–742. [Google Scholar]
- 3.Zheng XL, Luo L. Research progress on flavivirus susceptibility in Aedes aegypti. Acta Parasitol Med Entomol Sinica. 2010;17:47–54.
- 4.Wu JY, Lun ZR, James AA, Chen XG. Dengue fever in mainland China. Am J Trop Med Hyg. 2010;83:664–71. [DOI] [PMC free article] [PubMed]
- 5.Severson DW, Behura SK. Genome investigations of vector competence in Aedes aegypti to inform novel arbovirus sisease control approaches. Insects. 2016;7:58. doi: 10.3390/insects7040058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tabachnick WJ. Evolutionary genetics and arthropod-borne disease: the yellow fever mosquito. Am Entomol. 1991;37:14–26. doi: 10.1093/ae/37.1.14. [DOI] [Google Scholar]
- 7.Smith CE. The history of dengue in tropical Asia and its probable relationship to the mosquito Aedes aegypti. J Trop Med Hyg. 1956;59:243–251. [PubMed] [Google Scholar]
- 8.Tabachnick WJ, Powell JR. A world-wide survey of genetic variation in the yellow fever mosquito, Aedes aegypti. Genet Res. 1979;34:215–229. doi: 10.1017/S0016672300019467. [DOI] [PubMed] [Google Scholar]
- 9.Gloria-Soria A, Ayala D, Bheecarry A, Calderon-Arguedas O, Chadee DD, Chiappero M, et al. Global genetic diversity of Aedes aegypti. Mol Ecol. 2016; doi:10.1111/mec.13866. [DOI] [PMC free article] [PubMed]
- 10.Lu BL. Transmission vectors of dengue fever and control in China. Guiyang: Guizhou People’s Press; 1990. [Google Scholar]
- 11.Dong XS, Cai FC, Zhou HN, Wang X, Dong LM, Wu C, et al. Investigation of mosquitoes at frontier ports in Yunnan Province. Chin J Vector Bio Control. 2004;15:142–145. [Google Scholar]
- 12.Urdaneta-Marquez L, Failloux AB. Population genetic structure of Aedes aegypti, the principal vector of dengue viruses. Infect Genet Evol. 2011;11:253–261. doi: 10.1016/j.meegid.2010.11.020. [DOI] [PubMed] [Google Scholar]
- 13.Powell JR, Tabachnick WJ. History of domestication and spread of Aedes aegypti - a review. Mem Inst Oswaldo Cruz. 2013;108(Suppl 1):11–7. [DOI] [PMC free article] [PubMed]
- 14.Wang PY, Wu C, Zhang CL. Survey of the transmission vectors of dengue fever in some areas in Yunnan Province. China Trop Med. 2006;6:1933–1934. [Google Scholar]
- 15.Chen YW, Wang RG, Li S, Yang LX, Li P, Wang PY, et al. The survey and analysis of Aedes aegypti in Ruili. Chin J Vector Biol Control. 2007;18:61. [Google Scholar]
- 16.Li HC, Yang GR, Shi AJ, Shi J, Yang P, Li Y, et al. Investigation of distribution of dengue vectors in Lincang border area. China Trop Med. 2015;15:186–188. [Google Scholar]
- 17.Yang DM, Jiang JY, Zheng YT, Zhou HN. Distribution survey on Aedes aegypti in the border areas of Yunnan province, China. Chin J Vector Bio Control. 2015;26:406–8.
- 18.Dong SH, Pan SH, Ma L, Lei J, Zhou HN. Survery of Aedes aegypti distribution in Mangshi, Yunnan. Chin J Vector Bio Control. 2011;22:592–594. [Google Scholar]
- 19.Chown SL, Hodgins KA, Griffin PC, Oakeshott JG, Byrne M, Hoffmann AA. Biological invasions, climate change and genomics. Evol Appl. 2015;8:23–46. doi: 10.1111/eva.12234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kosasa TS, Mcnamee PI, Morton C, Huang TTF. Founding events in species invasions: genetic variation, adaptive evolution, and the role of multiple introductions. Mol Ecol. 2008;17:431–449. doi: 10.1111/j.1365-294X.2007.03538.x. [DOI] [PubMed] [Google Scholar]
- 21.Rasheed SB, Boots M, Frantz AC, Butlin RK. Population structure of the mosquito Aedes aegypti (Stegomyia aegypti) in Pakistan. Med Vet Entomol. 2013;27:430–440. doi: 10.1111/mve.12001. [DOI] [PubMed] [Google Scholar]
- 22.Paupy C, Orsoni A, Mousson L, Huber K. Comparisons of amplified fragment length polymorphism (AFLP), microsatellite, and isoenzyme markers: population genetics of Aedes aegypti (Diptera: Culicidae) from Phnom Penh (Cambodia) J Med Entomol. 2004;41:664–671. doi: 10.1603/0022-2585-41.4.664. [DOI] [PubMed] [Google Scholar]
- 23.Mousson L, Vazeille M, Chawprom S, Prajakwong S, Rodhain F, Failloux AB. Genetic structure of Aedes aegypti populations in Chiang Mai (Thailand) and relation with dengue transmission. Tropical Med Int Health. 2002;7:865–872. doi: 10.1046/j.1365-3156.2002.00939.x. [DOI] [PubMed] [Google Scholar]
- 24.Ravel S, Monteny N, Velasco OD, Escalante VJ, Cuny G. A preliminary study of the population genetics of Aedes aegypti (Diptera: Culicidae) from Mexico using microsatellite and AFLP markers. Acta Trop. 2001;78:241–250. doi: 10.1016/S0001-706X(01)00083-3. [DOI] [PubMed] [Google Scholar]
- 25.Schlotterer C. Evolutionary dynamics of microsatellite DNA. Chromosoma. 2000;109:365–371. doi: 10.1007/s004120000089. [DOI] [PubMed] [Google Scholar]
- 26.Chambers EW, Meece JK, McGowan JA, Lovin DD, Hemme RR, Chadee DD, et al. Microsatellite isolation and linkage group identification in the yellow fever mosquito Aedes aegypti. J Hered. 2007;98:202–210. doi: 10.1093/jhered/esm015. [DOI] [PubMed] [Google Scholar]
- 27.Huber K, Mousson L, Rodhain F, Failloux AB. Isolation and variability of polymorphic microsatellite loci in Aedes aegypti, the vector of dengue viruses. Mol Ecol Notes. 2005;1:219–222. doi: 10.1046/j.1471-8278.2001.00077.x. [DOI] [Google Scholar]
- 28.Huber K, Loan LL, Hoang TH, Ravel S, Rodhain F, Failloux AB. Genetic differentiation of the dengue vector, Aedes aegypti (ho chi Minh City, Vietnam) using microsatellite markers. Mol Ecol. 2002;11:1629–1635. doi: 10.1046/j.1365-294X.2002.01555.x. [DOI] [PubMed] [Google Scholar]
- 29.Huber K, Loan LL, Hoang TH, Tien TK, Rodhain F, Failloux AB. Temporal genetic variation in Aedes aegypti populations in ho chi Minh City (Vietnam) Heredity (Edinb) 2002;89:7–14. doi: 10.1038/sj.hdy.6800086. [DOI] [PubMed] [Google Scholar]
- 30.Huber K, Loan LL, Chantha N, Failloux AB. Human transportation influences Aedes aegypti gene flow in Southeast Asia. Acta Trop. 2004;90:23–29. doi: 10.1016/j.actatropica.2003.09.012. [DOI] [PubMed] [Google Scholar]
- 31.Paupy C, Chantha N, Huber K, Lecoz N, Reynes JM, Rodhain F, et al. Influence of breeding sites features on genetic differentiation of Aedes aegypti populations analyzed on a local scale in Phnom Penh municipality of Cambodia. AmJTrop Med Hyg. 2004;71:73–81. [PubMed] [Google Scholar]
- 32.Hlaing T, Tun-Lin W, Somboon P, Socheat D, Setha T, Min S, et al. Spatial genetic structure of Aedes aegypti mosquitoes in mainland Southeast Asia. Evol Appl. 2010;3:319–339. doi: 10.1111/j.1752-4571.2009.00113.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.da Costa-Ribeiro MC, Lourenco-de-Oliveira R, Failloux AB. Higher genetic variation estimated by microsatellites compared to isoenzyme markers in Aedes aegypti from Rio de Janeiro. Mem Inst Oswaldo Cruz. 2006;101:917–921. doi: 10.1590/S0074-02762006000800015. [DOI] [PubMed] [Google Scholar]
- 34.Endersby NM, Hoffmann AA, White VL, Lowenstein S, Ritchie S, Johnson PH, et al. Genetic structure of Aedes aegypti in Australia and Vietnam revealed by microsatellite and exon primed intron crossing markers suggests feasibility of local control options. J Med Entomol. 2009;46:1074–1083. doi: 10.1603/033.046.0514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lu BL. Integrated mosquito control. Beijing: Beijing Science Press; 1999. [Google Scholar]
- 36.Slotman MA, Kelly NB, Harrington LC, Kitthawee S, Jones JW, Scott TW, et al. Polymorphic microsatellite markers for studies of Aedes aegypti (Diptera: Culicidae), the vector of dengue and yellow fever. Mol Ecol Notes. 2006;7:168–171. doi: 10.1111/j.1471-8286.2006.01533.x. [DOI] [Google Scholar]
- 37.Mkize LS, Mukaratirwa S, Zishiri OT. Population genetic structure of the freshwater snail, Bulinus globosus (Gastropoda: Planorbidae) from selected habitats of KwaZulu-Natal, South Africa. Acta Trop. 2016;161:91–99. doi: 10.1016/j.actatropica.2016.06.001. [DOI] [PubMed] [Google Scholar]
- 38.Excoffier L, Laval G, Schneider S. Arlequin (version 3.0): an integrated software package for population genetics data analysis. Evol Bioinformatics Online. 2005;1:47–50. [PMC free article] [PubMed] [Google Scholar]
- 39.Rousset F. Genepop ‘007: a complete re-implementation of the genepop software for windows and Linux. Mol Ecol Res. 2008;8:103–106. doi: 10.1111/j.1471-8286.2007.01931.x. [DOI] [PubMed] [Google Scholar]
- 40.Van Oosterhout C, Hutchinson WF, Wills DPM, Shipley P. Micro - checker: software for identifying and correcting genotyping errors in microsatellite data. Mol Ecol Res. 2004;4:535–8.
- 41.Men QL, Chen MH, Zhang YL, Feng JN. Genetic structure and diversity of a newly invasive species, the codling moth, Cydia pomonella (L.) (Lepidoptera: Tortricidae) in China. Biol Invasions. 2013;15:447–458. doi: 10.1007/s10530-012-0299-5. [DOI] [Google Scholar]
- 42.Szpiech ZA, Jakobsson M, Rosenberg NA. ADZE: a rarefaction approach for counting alleles private to combinations of populations. Bioinformatics. 2008;24:2498–2504. doi: 10.1093/bioinformatics/btn478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shao K, Xiong M, Xu N, Zhu B, Shi F. Characterization of microsatellite loci in Sinilabeo rendahli and cross-amplification in four other Chinese cyprinid species. Conserv Genet Resour. 2012;5:9–13. doi: 10.1007/s12686-012-9717-3. [DOI] [Google Scholar]
- 44.Evanno G, Regnaut S, Goudet J. Detecting the number of clusters of individuals using the software structure: a simulation study. Mol Ecol. 2005;14:2611–2620. doi: 10.1111/j.1365-294X.2005.02553.x. [DOI] [PubMed] [Google Scholar]
- 45.Pritchard JK, Stephens M, Donnelly P. Inference of population structure using multilocus genotype data. Genetics. 2000;155:945–959. doi: 10.1093/genetics/155.2.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yin M, Hongyan Li. Geographical genetic structure of Schistosoma japonicum revealed by analysis of mitochondrial DNA and microsatellite markers. Parasites & Vectors, 2015;8(1):150. [DOI] [PMC free article] [PubMed]
- 47.Rosenberg NA. Distruct: a program for the graphical display of population structure. Mol Ecol Notes. 2004;4:137–138. doi: 10.1046/j.1471-8286.2003.00566.x. [DOI] [Google Scholar]
- 48.Bonnet E, Peer YVD. Zt: a Sofware tool for simple and partial Mantel tests. J Stat Softw. 2002;07:2002. doi: 10.18637/jss.v007.i10. [DOI] [Google Scholar]
- 49.Khan HA, Arif IA, Bahkali AH, Al Farhan AH, Al Homaidan AA. Bayesian, maximum parsimony and UPGMA models for inferring the phylogenies of antelopes using mitochondrial markers. Evol Bioinformatics Online. 2008;4:263–270. doi: 10.4137/ebo.s934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gentleman R, Ihaka R. R: a language and environment for statistical computing. Computing. 2011;14:12–21. [Google Scholar]
- 51.Egizi A, Kiser J, Abadam C, Fonseca DM. The hitchhiker's guide to becoming invasive: exotic mosquitoes spread across a US state by human transport not autonomous flight. Mol Ecol. 2016;25:3033–3047. doi: 10.1111/mec.13653. [DOI] [PubMed] [Google Scholar]
- 52.Richardson DM, Pyšek P, Carlton JT. A compendium of essential concepts and terminology in invasion ecology. In: Richardson DM, editor. Fifty years of invasion ecology: the legacy of Charles Elton. New Jersey: Hoboken Wiley and Sons; 2010. pp. 409–420. [Google Scholar]
- 53.Li Y, Zhu J, Li H. Distribution of the dengue fever vector in Xishuangbanna prefecture of Yunnan. China Trop Med. 2016;16:237–265. [Google Scholar]
- 54.Frankham R, Ballou JD, Briscoe DA. Introduction to conservation genetics. Cambridge: Cambridge University Press; 2010. [Google Scholar]
- 55.Wang L, Zuo SM, Zhang YF, Chen ZX, Huang SW, Pan XB. SSR analysis of population genetic structure of rice sheath blight causing agent Rhizoctonia solani AG1-IA collected from eight provinces (autonomous region) in southern China. Sci Agric Sin. 2015;48:2538–2548. [Google Scholar]
- 56.Xu ZQ, Ge JC, Li YH, Li XH, Bai RF, Zhu QS, et al. The population genetic structure analysis on four stocks of Eriocheir sinensis from different water systems using microsatel lite markers. J Nanjing University (Natural Sciences) 2011;47:82–90. [Google Scholar]
- 57.Tobias U, Roosa L. Founder events predict changes in genetic diversity during human-mediated range expansions. Glob Chang Biol. 2011;17:3478–3485. doi: 10.1111/j.1365-2486.2011.02509.x. [DOI] [Google Scholar]
- 58.Dlugosch KM, Parker IM. Founding events in species invasions: genetic variation, adaptive evolution, and the role of multiple introductions. Mol Ecol. 2008;17:431–449. doi: 10.1111/j.1365-294X.2007.03538.x. [DOI] [PubMed] [Google Scholar]
- 59.Reiter P. Oviposition, dispersal, and survival in Aedes aegypti: implications for the efficacy of control strategies. Vector Borne Zoonotic Dis. 2007;7:261–273. doi: 10.1089/vbz.2006.0630. [DOI] [PubMed] [Google Scholar]
- 60.McDonald PT. Population characteristics of domestic Aedes aegypti (Diptera: Culicidae) in villages on the Kenya coast I. Adult survivorship and population size. J Med Entomol. 1977;14:42–8. [DOI] [PubMed]
- 61.Trpis M, Hausermann W. Dispersal and other population parameters of Aedes aegypti in an African village and their possible significance in epidemiology of vector-borne diseases. AmJTrop Med Hyg. 1986;356:1263–1279. doi: 10.4269/ajtmh.1986.35.1263. [DOI] [PubMed] [Google Scholar]
- 62.Reiter P, Amador MA, Anderson RA, Clark GG. Short report: dispersal of Aedes aegypti in an urban area after blood feeding as demonstrated by rubidium-marked eggs. Am J Trop Med Hyg. 1995;52:177–9. [DOI] [PubMed]
- 63.Endersby NM, Hoffmann AA, White VL, Lowenstein S, Ritchie S, Johnson PH, et al. Genetic structure of Aedes aegypti in Australia and Vietnam revealed by microsatellite and exon primed intron crossing markers suggests feasibility of local control options. J Med Entomol. 2009;46(5):1074–1083. doi: 10.1603/033.046.0514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Li HC, Yang GR. Survey of the first outbreak of dengre fever in Lincang City, Yunnan Province. China Trop Med. 2009;9:2013–2014. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Analysis of linkage disequilibrium in 28 locations (TIFF 7443 kb)
Table S1. Population statistics for Ae. aegypti investigated using nine microsatellite loci. (DOCX 17 kb)
Figure S2. The mean number of distinct alleles per locus for 28 sample locations. (TIFF 1958 kb)
Table S2. Aedes aegypti heterozygosity tests. (DOCX 15 kb)
Table S3. Analysis of molecular variance of populations from Jinghong, Ruili and the border areas. (DOCX 15 kb)
Table S4. Pairwise population differentiation estimates (FST) and geographical distance [ln(km)] between all locations of Ae. aegypti. (DOCX 22 kb)
Data Availability Statement
Data are available on request to the corresponding author.