

Abstract
Neuroblastoma amplified sequence (NBAS) gene mutation or infantile liver failure syndrome type 2 (ILFS type 2) is an extremely rare disease characterized by episodic liver failure precipitated by intercurrent febrile illness, and liver function recovering completely. Here, we report a 4-year-old girl with recurrent hepatitis. A diagnosis of ILFS type 2 was made based on NBAS mutation gene found by whole-exome sequencing. Our case provides a new insight toward considering NBAS mutation as a part of the differential diagnoses of any infant presenting with recurrent liver failure or hepatitis. We recommend sequencing NBAS in cases of recurrent hepatitis in infancy of unknown cause, especially in individuals with fever-associated hepatic dysfunction.
Keywords: Hepatitis, infantile liver failure syndrome type 2, liver crisis
INTRODUCTION
Neuroblastoma amplified sequence (NBAS) gene mutation or infantile liver failure syndrome type 2 (ILFS type 2) is an extremely rare disease characterized by episodic liver failure precipitated by intercurrent febrile illness, and liver function recovered completely with conservative management.[1] ILFS type 2 is an autosomal recessive disorder diagnosed by mutations in the NBAS mutation gene found by whole-exome sequencing.[2] We report the case of a 4-year-old girl presenting with recurrent hepatitis found to have NBAS mutation. A review of the literature is also provided.
CASE REPORT
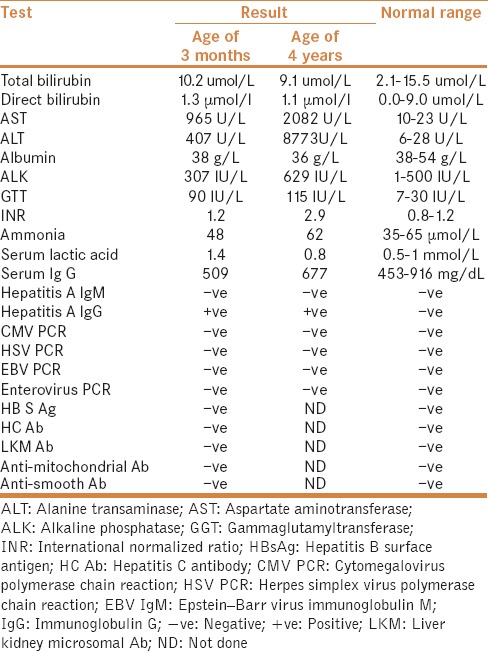
A 4-year-old girl was referred in January 2012 for recurrent episodes of hepatitis. At the age of 3 months, she presented with a history of fever, decreased activity and elevated liver enzymes. Infectious causes and drug-induced hepatitis were ruled out. Liver function tests were normalized on subsequent days. She had multiple hospital admissions with febrile illnesses associated with liver dysfunction. At the age of 12 months, she presented with salmonella bacteremia. At the age of 13 months, she presented with gastroenteritis. At the age of 15 months, she presented with follicular tonsillitis. At the age of 3 years, she presented with pneumonia. Past medical history revealed that she was born of a full term, spontaneous vaginal delivery, and an uncomplicated neonatal course. The parents were first degree cousins. Two sibling boys died at ages of 12 months due to hepatomegaly and jaundice of unknown cause. The patient had normal developmental milestones. She was not taking medications. On admission to the hospital, physical examination revealed poor growth and hepatomegaly. She was not jaundiced and not dysmorphic. There was no splenomegaly or ascites. The rest of the systemic examination was otherwise unremarkable. The laboratory findings are shown in Table 1. Ultrasonography revealed mild hepatomegaly without splenomegaly. Immunological workup including serum immunoglobulin, complement C3 and C4 functions, T cells subset, and B cell function were normal. Workup for autoimmune hepatitis was normal [Table 1]. Extensive metabolic workup including serum amino acids, urine organic acids, carnitine, acylcarnitine, galactose-1-phosphate uridyle transferase, carbohydrate deficient transferrin, very long chain fatty acid, ceruloplasmin, copper, and alpha 1 antitrypsin were all normal. As there was no definitive infectious or metabolic cause of hepatitis, percutaneous liver biopsy was undertaken, which showed nonspecific acute hepatocellular injury. There was mild acute hepatitis with small foci of lobular necrosis and chronic inflammation. No portal fibrosis or bile duct damage was identified. Immunohistochemistry for cytomegalovirus and herpes simplex virus was negative. Iron and periodic acid-Schiff (PAS) were not identified. During the last admission, she presented at the age of 4 years with pneumonia, hepatitis, and coagulopathy. She was diagnosed as liver failure and managed with conservative treatment. In each liver failure episode, the patient required fluid resuscitation and correction of electrolyte imbalances, hypoglycemia, coagulopathy, and intravenous antibiotics. As the patient had episodic hepatitis precipitated by undercurrent febrile illness, and liver function recovered completely with conservative management in the interval, whole-exome sequencing was performed and showed mutations in the NBAS gene. Genotype demonstrated c.2819A>C (p. His940Pro). The type of mutation was missense deletion and homozygous mutation. A diagnosis of infantile ILFS type 2 was made and there was no recurrence of symptoms during the 6-month follow-up period.
Table 1.
Laboratory results
DISCUSSION
NBAS mutations or ILFS type 2 is an extremely rare disease characterized by episodic liver failure precipitated by intercurrent febrile illness, and liver function recovering completely with conservative management.[1] ILFS type 2 is caused by a mutation in the NBAS gene on the chromosome 2p24.[2] ILFS type 1 is caused by mutation in the Leucyl-tRNA synthetase (LARS) gene on the chromosome 5q32.[3]
The differential diagnoses of any infant presenting with recurrent liver failure or hepatitis includes autoimmune hepatitis, Wolcott–Rallison syndrome, perforin deficiency, fatty acid oxidation defects, mitochondrial hepatopathy, dihydrolipoamide dehydrogenase deficiency, and other inborn errors of metabolic liver disease. Our patient's feature is similar to Wolcott–Rallison syndrome because of recurrent liver failure following viral infections, however, it is excluded because there is no insulin-dependent diabetes mellitus, bone dysplasia, and developmental delay, which are the features of Wolcott–Rallison syndrome.
NBAS mutation is a rare condition, with only 16 cases reported in the English literature.[2] Haack et al. and Staufner et al.[1,2] have examined 16 patients with NBAS mutation. They showed that phenotypic spectrum of NBAS deficiency ranges between isolated recurrent acute liver failure and multisystemic disease, which includes short stature, skeletal dysplasia, optic atrophy, and immunological abnormalities. Liver crises of NBAS mutation are usually triggered by febrile illness and complete recovery is typical between the attacks. Hepatic phenotype episodes usually start with massive increase in alanine transaminase (ALT) and aspartate transaminase (AST), followed by severe coagulopathy and mild-to-moderate jaundice.[1,2] However, hypoglycemia, hyperammonemia, and hepatic encephalopathy are transiently observed in some patients. Extrahepatic phenotype of NBAS mutation presents with the following: Short stature, osteopenia, dysmorphism, autoimmune disease, optic atrophy, brain atrophy, and delayed development.[4,5,6] The data of Haack et al. and Staufner et al.[1,2] taken together with the observation in our patient suggest that most patients had onset in the first 2 years of life, with episodic liver failure in these patients precipitated by intercurrent febrile illness, and liver function recovering completely with conservative management in the interval. However, our patient mostly presented with recurrent episodes of hepatitis (liver crisis without liver failure) during intercurrent febrile illness. The data of Haack et al.[1] taken together with the observation in our patient suggests that crises were manifest by vomiting, lethargy, increased liver enzymes, jaundice, and coagulopathy. Haack et al.[1] examined 11 patients with NBAS mutation; 4 patients had comorbid features, such as cardiomyopathy, autoimmune gastrointestinal disease, and epilepsy, however, none of them died. Our patient and the data of Staufner et al.[2] would suggest that NBAS mutation has a favorable prognosis. However, liver transplant was performed in one patient aged 3 years; no further crisis occurred after liver transplant. Thermal susceptibility of the syntaxin 18 complex is the basis of fever dependency of liver failure episodes and could be the possible mechanism of NBAS mutation. NBAS deficiency is the first disease related to a primary defect of retrograde transport.[2] In each liver failure episode, our patient received early administration of antipyretics, intravenous antibiotics, and intravenous fluids with high glucose and lipids to prevent progression to fulminant liver failure.[2]
CONCLUSION
Although ILFS type 2 or NBAS mutation is a rare disease, we suggest considering NBAS mutation as a part of the differential diagnoses of any infant presenting with recurrent liver failure or hepatitis. We recommend sequencing NBAS in any patient with unexplained recurrent hepatitis, especially in individuals with fever-associated hepatic dysfunction. Our case provides a new insight for understanding NBAS mutation presenting as recurrent hepatitis and further studies are required to elucidate the pathogenesis and hepatic mechanism for NBAS mutation.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
REFERENCES
- 1.Haack TB, Staufner C, Köpke MG, Straub BK, Kölker S, Thiel C, et al. Biallelic mutations in NBAS cause recurrent acute liver failure with onset in infancy. Am J Hum Genet. 2015;97:163–9. doi: 10.1016/j.ajhg.2015.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Staufner C, Haack TB, Köpke MG, Straub BK, Kölker S, Thiel C, et al. Recurrent acute liver failure due to NBAS deficiency: Phenotypic spectrum, disease mechanisms, and therapeutic concepts. J Inherit Metab Dis. 2016;39:3–16. doi: 10.1007/s10545-015-9896-7. [DOI] [PubMed] [Google Scholar]
- 3.Casey JP, McGettigan P, Lynam-Lennon N, McDermott M, Regan R, Conroy J, et al. Identification of a mutation in LARS as a novel cause of infantile hepatopathy. Mol Genet Metab. 2012;106:351–8. doi: 10.1016/j.ymgme.2012.04.017. [DOI] [PubMed] [Google Scholar]
- 4.Segarra NG, Ballhausen D, Crawford H, Perreau M, Campos-Xavier B, van Spaendonck-Zwarts K, et al. NBAS mutations cause a multisystem disorder involving bone, connective tissue, liver, immune system, and retina. Am J Med Genet. 2015;167:2902–12. doi: 10.1002/ajmg.a.37338. [DOI] [PubMed] [Google Scholar]
- 5.Capo-Chichi JM, Mehawej C, Delague V, Caillaud C, Khneisser I, Hamdan FF, et al. Neuroblastoma Amplified Sequence (NBAS) mutation in recurrent acute liver failure: Confirmatory report in a sibship with very early onset, osteoporosis and developmental delay. Eur J Med Genet. 2015;58:637–41. doi: 10.1016/j.ejmg.2015.11.005. [DOI] [PubMed] [Google Scholar]
- 6.Maksimova N, Hara K, Nikolaeva I, Chun-Feng T, Usui T, Takagi M, et al. Neuroblastoma amplified sequence gene is associated with a novel short stature syndrome characterised by optic nerve atrophy and Pelger-Huet anomaly. J Med Genet. 2010;47:538–48. doi: 10.1136/jmg.2009.074815. [DOI] [PMC free article] [PubMed] [Google Scholar]