

Abstract
Spatial memory acquisition in Morris water maze was tested in C57BL/6 mice. Animals were injected once daily with different doses of either N6-cyclopentyladenosine (CPA) or 8-cyclopentyl-1,3-dipropylxanthine (CPX). Drugs were administered for 9 days either concurrently with water maze testing (drugs injected 1 h after each trial), or prior to the entire block of trials. In the latter case, 1 day without injections preceded water maze experiments. Chronic administration of CPA resulted in a significant, dose-dependent reduction of target latencies, rapid development of spatial preference, and the absence of animals unable to perform the task. CPX treated animals did not show significant performance changes, and failed to develop spatial preference. Locomotor disturbances were not the cause of the observed effects. Our results indicate that chronic treatment with agents acting at adenosine A1 receptors results in behavioral effects that are significantly different from those observed following their acute administration. Therefore, particular caution is required in development of adenosine-based strategies targeted at neurodegenerative or cognitive disorders in which chronic treatment is advocated.
Keywords: Adenosine, Xanthine, Memory, Learning, Water maze, (Mouse)
1. Introduction
The development of hippocampal long-term potentiation, a stable form of synaptic facilitation involved in acquisition and encoding of memory, is associated with sustained stimulation of postsynaptic NMDA receptors by glutamate and its analogues (Collingridge and Davies, 1991). In vitro experiments have shown that the release of hippocampal glutamate appears to be modulated by extracellular adenosine (Burke and Nadler, 1988). Furthermore, evidence exists for a functional relationship of NMDA and adenosine A1 receptors (Bartrup and Stone, 1990), and particularly dense concentration of both NMDA and adenosine A1 receptors has been demonstrated in the hippocampus (Onodera and Kogure, 1988).
The A1 receptor-rich hippocampus intimately participates in learning paradigms that involve spatial coding (O’Keefe and Nadel, 1978). Predictably, therefore, several authors demonstrated profound in-vitro effect of acutely administered adenosine A1 receptor agonists and antagonists on a wide range of electrophysiological phenomena thought to be related to the process of learning (Daval et al., 1991). Thus, it has been shown that acute administration of adenosine A1 receptor agonists results in a depression of hippocampal excitatory transmission and evoked potentials (Lambert and Teyler, 1991; Okada and Ozawa, 1980; Schubert and Mitzdorf, 1979), and that acute administration of these agents causes impairment in development and consolidation of long-term potentiation (Arai et al., 1990). In-vitro evidence of possible involvement of adenosine in learning has been supported by experiments of Normile and Barraco showing that acute treatment with adenosine A1 receptor agonist L-phenylisopropyl adenosine (L-PIA) attenuates retention in passive avoidance tests (Normile and Barraco, 1991).
While acutely administered agonists appear to attenuate, the antagonists of the adenosine A1 receptor show an augmenting effect upon in-vitro physiological phenomena involved in learning as well as upon the process of learning itself. Several studies indicate that acute exposure to adenosine A1 receptor antagonists assists in development of long-term potentition (Alzheimer et al., 1989; Arai et al., 1990; Tanaka et al., 1990). Moreover, treatment of mice with a specific A1 receptor antagonist KFM 19 results in a significant improvement of learning of tasks in a Y-maze paradigm (Schingnitz et al., 1991).
Chronic administration of either adenosine A1 agonists or antagonists has been reported to cause respectively either a reduction or an increase in the density of adenosine A1 receptors (Parsons and Stiles, 1987; Abbracchio and Cattabeni, 1992; Gonzalez-Calero and Cubero, 1992). Therefore, it is likely that chronic administration of adenosine A1 selective drugs may affect spatial learning and memory in a manner that differs significantly from that observed following acute administration.
We now report that chronic treatment with the selective adenosine A1 receptor agonist, N6-cyclopentyladenosine (CPA), enhances, while the selective antagonist 8-cyclopentyl-1,3-dipropylxanthine (CPX), impairs performance in the water maze (Morris et al., 1982), a paradigm in which intact hippocampal functions are necessary for acquisition of spatial tasks. Our data demonstrate that adenosine A1 receptor plays a significant role in the acquisition and/or retrieval of spatial information. Moreover, in contrast to the results obtained in acute studies, these results also indicate that long-term administration of adenosine A1 receptor agonists rather than antagonists may be a useful therapeutic approach to the treatment of dementing disorders.
2. Materials and methods
2.1. Animals
Male C57BL/6 mice (The Jackson Laboratory, Bar Harbor, Maine) were housed in a constant environment (25 ± 2°C, 12 h light/dark cycle) in groups of 10. Animals had free access to food and water and were acclimatized to the colony for at least 7 days prior to experiments.
2.2. Drugs and administration regimens
Drugs were purchased from Research Biochemicals, Inc. (South Natick, MA), dissolved in a 20:80 (v/v) mixture of Alkamuls EL-620 (Rhone-Poulenc, Cranbury, NJ) and phosphate buffered saline and administered in a volume of 0.15 ml. CPA was given at 5, 10, 20 and 30 μg/kg (n = 10/group), while CPX was administered at 0.2, 0.5 and 1.0 mg/kg (n = 10/group). Controls for each dose (n = 10/group) were injected with the vehicle. All injections were made i.p. using a 25 gauge hypodermic needle.
Two dosing regimens were investigated. In the first, mice were given either CPA, CPX or saline once daily for 9 days followed by one treatment-free (wash-out) day. No injections were administered during the subsequent water maze training (see below). In the second regimen, drugs were administered during the entire length of the experiment, i.e., as a pretreatment prior to water maze trials and then, concurrently with the trials. Pretreatment consisted of 9 daily injections of either CPA, CPX or vehicle. There was no ‘wash-out’ period, and animals were injected 1 h after the first water maze training session on the 10th day of the experiment. Thereafter, injections were given each day, 1 h after the trial. A 24 h interval separated each injection from the following trial.
2.3. Water maze
The circular water maze tank (inner diameter, 1 m) was constructed from black Plexiglas. The tank was filled with warm (30–35°C) tap water to a depth of 15 cm. Immediately before the experiment, dry milk powder was added to the contents of the tank producing an a opaque solution.
The maze was located in a sound-proof room illuminated by fluorescent ceiling light. Two visually distinct target cues were placed on the north (N) and east (E) side of the tank (fig. 1), while the observer stood at its south (S) side. A blank wall faced the west (W) side. A transparent circular target platform (Plexiglas, 8 cm diameter) was covered with white surgical tape and submerged to a depth of 0.5 cm in the center of one quadrant.
Fig. 1.

Diagram of the water maze tank showing quadrant subdivisions and their geographical coordinates, position of the invisible target (T) during the initial set of trials, and the starting position opposite to the target. During trials to a reversed, invisible target, it was placed in the SE quadrant, with the starting position in the NW quadrant.
2.4. Training
Each set of trials consisted of four phases: invisible target acquisition, probe trial, reversed target acquisition, and visible target acquisition. The invisible target acquisition phase tested the global learning in the water maze paradigm. The spatial component of the learned task was isolated and determined during the probe trials during which perseverence in execution of a spatially oriented search in the absence of the original target was studied. This was further tested during reversal trials which provide an additional measure of both search perseverence and the ability of the animals to learn a new target position in a task simplified by the preceding target acquisition phase. The visible trials tested both visual and motor abilities, and elements of motivation to perform in the water maze paradigm.
All home cages were moved to the testing room 30–45 min before the trial. Mice were removed randomly from their cages and gently lowered into the water facing the wall of the quadrant opposite to the target quadrant. They were allowed 120 s to reach and mount the platform. Fifteen seconds after mounting, each animal was removed and placed in a holding cage warmed by means of an infrared heating lamp. Mice failing to locate and mount the target within 120 s were placed on the platform for 15 s prior to their transfer to the holding cage. After all animals from a cage had been tested, they were placed again in their home cage. The order in which the groups were run was randomly varied for each trial. After all subjects were tested, animals were returned to the animal facility.
Based on previous experiments of Sei et al. (1992), the performance of animals in reaching the target (target latency) was defined as ‘good’ (≤40 s), ‘average’ (41–90 s) and ‘poor’ (> 90 s). Animals were trained once daily beginning at 3 p.m. The initial target acquisition phase was continued until the control group reached a stable performance plateau (typically 7–9 days). The acquisition phase was followed by a single probe trial during which the target was removed from the maze and quadrant preference (time spent in each quadrant during 120 s testing period) was recorded. Trials to a submerged target placed in the quadrant diagonally opposite (SE) to the original one (NW) were conducted during the following 5 days (‘target reversal phase’ of the experiment) During this phase, the starting position along the wall of the tank was selected at random. Five days training to a visible target placed again in the NW quadrant followed target reversal trials. The visible target was made of black Plexiglas and its platform protruded 1 cm above the water surface. Starting position was again in the SE quadrant.
2.5. Tracking and data collection
Mice were tracked by the observer using the Water-maz data acquisition program (Infallible Software, Research Triangle Park, NC) which recorded the time to target (TT), number of quadrant crossings (X), the overall duration of immobility, as well as a breakdown of these indices by quadrant. The overall search strategy employed by the animal was rated by the observer for thigmotaxis (‘wall-hugging’), center-crossing, or target-direction.
2.6. Locomotor performance and activity testing
The effect of chronic administration of CPA and CPX on locomotor activity was studied in three separate groups of animals (n = 10/group). Animals were tested during the entire course of drug administration and each test preceded injection of the drug. A 24 h interval separated each injection from the subsequent test.
To investigate the effect of drugs or saline injections upon motor coordination animals (n = 10/group) were placed on a rotorod revolving at 10 rpm. For each animal the number of falls during a 2 min rotorod session was recorded. Open-field locomotor performance was tested for 20 min in 2 min epochs using an automated system (Omnitech Electronics, Columbus, OH).
2.7. Statistics
Parametric data were initially analyzed using single factor repeated measures analysis of variance. Comparisons between individual groups were defined a priori as follows: (a) trial 1 vs. subsequent trials within drug treatment groups (acquisition and reversal trials); (b) target quadrant vs. other quadrants within drug treatment groups (‘probe trials’), and (c) saline vs. drug treatment groups within an individual trial (all trials). Planned comparisons were conducted using contrast analyses employing univariate F tests (Systat 5.0, Evanston, IL). Post-test corrections, such as Bonferroni/Dunn, were not performed for any individual group comparisons. Nonparametric data were analyzed with Fisher’s exact test (Genstat 3.0, Infallible Software). Ρ < 0.05 was the criterion of statistical significance.
3. Results
3.1. The effect of motor coordination and locomotor performance in the open field
Repeated administration of either CPA or CPX at any of the studied doses produced no disturbances of either motor coordination or locomotor performance at any time (table 1).
TABLE 1.
Locomotor effect (horizontal activity ± S.E.M.) of chronic administration of CPA, CPX, and vehicle. CPA (20 μg/kg), CPX (1.0 mg/kg) and vehicle injected for 9 days prior to testing followed by concurrent testing and administration.
Analysis of variance showed no significant drug effect in any group. Horizontal activity: [F(l,16)= 0.001, Ρ > 0.05)].
| Day 1 | Day 2 | Day 3 | Day 4 | Day 5 | Day 6 | Day 7 | Day 8 | |
|---|---|---|---|---|---|---|---|---|
| Vehicle | 431/72 | 319/32 | 315/38 | 354/66 | 379/46 | 345/53 | 371/30 | 410/35 |
| CPA | 371/38 | 389/45 | 354/65 | 309/57 | 400/45 | 450/22 | 369/46 | 312/48 |
| CPX | 420/37 | 429/47 | 348/32 | 382/34 | 322/47 | 328/39 | 271/47 | 343/38 |
3.2. Target latencies
Beginning with the second trial, CPA produced a dose-dependent decrease in target latency (time to target) which persisted throughout the initial acquisition period (fig. 2A). CPA at a dose of 20 μg/kg was used in all subsequent experiments since this was the lowest dose to consistently reduce target latency.
Fig. 2.

Training to an invisible target placed in the NW quadrant. (A) Dose-response curves for CPA. Open square: controls; open circles: CPA, 5 μg/kg; black circles: CPA 10 μg/kg; open triangles: CPA, 20 μg/kg; black diamonds: CPA, 30 μg/kg. Analysis of variance revealed significant main effects of both drug treatment [F(3,35) = 9.788 Ρ < 0.0001] and training trials [F(7,245) = 13.595, Ρ < 0.0001]. No interation was observed between drug treatment and training trials (P > 0.5). Bars: S.E.M. (B) Dose-response curves for CPX. Open squares: controls; black circles: CPX, 0.2 mg/kg; open triangles: CPX, 0.5 mg/kg; open circles: CPX, 1 mg/kg. Analysis of variance revealed significant main effects of both drug treatment [F(3,46) = 3.859, Ρ < 0.02] and training trials [F(7,322) = 7.965, Ρ < 0.0001] No interation was observed between drug treatment and training trials (P > 0.5). Bars: S.E.M.
CPX given at 0.2 mg/kg did not affect target latencies (fig. 2B), while in 0.5 and 1.0 mg/kg groups the effect was minimal and appeared late into trial sequence. Doses higher than 1 mg/kg CPX produced locomotor disturbances (data not shown). Therefore, CPX at 1 mg/kg was used in all following experiments.
3.2. Performance and spatial preference
CPA at doses ≤ 10 μg/kg did not produce significant performance changes (data not shown). However, when the drug was given at 20 μg/kg (fig. 3) or 30 μg/kg (data not shown), a significant reduction in the number of animals performing poorly (TT > 90 s) was observed during the second trial (fig. 3, left panel). Concurrently, the number of animals performing well (TT < 40 s) was increased (P < 0.05). At the end of the acquisition phase (trials 7, 8), target latencies > 90 s were still observed in 15% of the control, while poor performers were entirely eliminated in the CPA group. Furthermore, the number of good performers (i.e., TT < 40 s) in the CPA group exceeded controls by 33% (fig. 3, right panel). As noted above, target latencies of CPX-treated animals were reduced by the last two trials of the acquisition phase (fig. 2B). However, in contrast to the CPA-treated mice, no significant performance differences between controls and CPX animals were seen during either the first two or the last two trials (fig. 3, left and right panels).
Fig. 3.
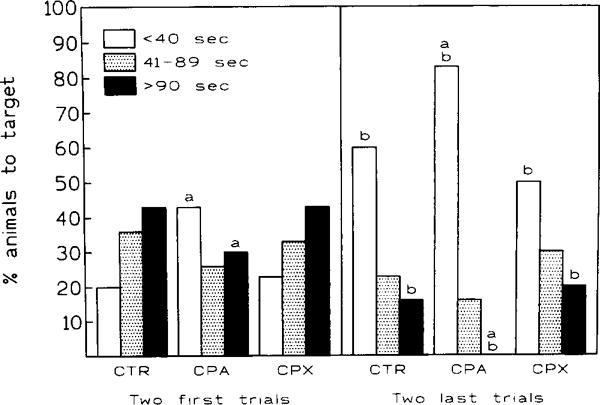
The effect of chronic co-treatment (8 days) and training (7 days) on the ability of animals to reach an invisible target (performance) in NW quadrant within specified time limits. Comparison of the first two and the last two trials. CPA administered at 20 μg/kg, CPX at 1 mg/kg daily. Statistical significance:a Ρ < 0.05 within the trial group,b Ρ < 0.05 between both trial groups. Fisher’s exact test.
At the conclusion of the initial acquisition phase, both controls and animals treated with CPA spent a significantly greater amount of time in the target quadrant (NW) than in any other quadrant (fig. 4A). However, CPA-treated animals spent more time in the target quadrant than did either the controls or CPX-treated mice (fig. 4A). In contrast, CPX-treated animals showed no preference for any of the maze quadrants (fig. 4A).
Fig. 4.
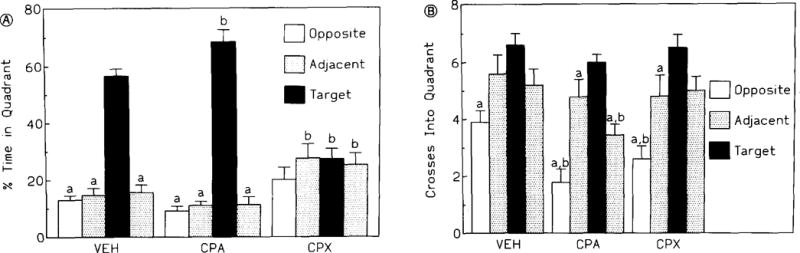
Quadrant preference following a set of nine trials to an invisible target placed in the NW quadrant (A), and frequency of crosses into each quadrant of the maze (B) Drugs (CPA: 20 μg/kg, CPX: 1 mg/kg) and vehicle were administered concurrently with training. Statistical significance for both figures:a Ρ < 0.05 within each treatment group; b Ρ < 0.05 among groups. Fisher’s exact test.
All three groups showed similar patterns of quadrant crossings, with the greatest number of crosses into the target quadrant and the least into the opposite (starting quadrant, fig. 4B). No significant effects of drug treatment were observed, although the CPA-treated animals tended to show the most pronounced preference compared to either vehicle- or CPX-treated mice (fig. 4B).
3.3. Training to a reversed target
During the first day of target reversal training (target placed in a new, SE quadrant), target latencies increased in all groups (fig. 5A). Subsequently, with repeated training to the new target position, all groups showed a progressive reduction in target latencies. By the second reversal trial, CPA-treated mice displayed significantly shorter latencies, reaching the new target significantly more rapidly than controls (fig. 5A). The difference between CPA- and vehicle-treated animals disappeared by the third reversal trial but significant differences persisted between CPA and CPX groups (fig. 5A).
Fig. 5.

Target latencies (A) and performance (B) during the target reversal phase, i.e., training to the invisible, target placed in SE quadrant with a random start position. The target reversal phase (starting with trial 1) followed immediately after the probe phase. ‘Time to original target’ (fig. 5A, abscissa) indicates target latencies attained at the last trial of the preceding target acquisition phase during which the invisible target was placed in the opposite (NW) quadrant. In fig. 5A, there are significant main effects of drug treatment [F(2,27) = 7.437, Ρ < 0.003] but no trials effect [F(4,1O8)= 1.960, P>0.1], No interation was observed between drug treatment and training trials (P > 0.5). Bars: S.E.M. Statistical significance in fig. B: a Ρ < 0.05 within the trial group; b Ρ < 0.05 between both trial groups. Fisher’s exact test. In both A and B, drugs (CPA: 20 μg/kg, CPX: 1 mg/kg) and vehicle administered for 9 days prior to and then concurrently with training.
The performance of CPA-treated mice was greatly improved already during the second trial (fig. 5B, left panel). At the end of the training period, no animals required more than 90 s to reach the target, and at least 90% of the CPA mice attained the criterion (i.e., target latencies ≤ 40 s). Compared to the first two trials, the performance of CPX-treated mice was also significantly improved during the last two trials. Although there was no significant increase in the proportion of good performers (TT ≤ 40 s), the number of moderately performing mice (41 < TT < 90 s) increased significantly together with equally significant reduction in the proportion of poor performing animals (TT ≥ 90 s). However, CPX animals continued to perform much worse than either the control or CPA-treated mice (fig. 5B).
3.4. Training to a visible target
When the invisible target was replaced with a visible target in a new (NW) quadrant, target latency during the first trial again increased in all groups (fig. 6A). Both controls and CPA-treated mice rapidly acquired the new task with mean target latencies ≤ 40 s (fig. 6A). In contrast, CPX-treated animals showed no improvement despite repeated trials (fig. 6A). Their target latencies varied around the mean observed at the end of training to an invisible target (~ 60 s, figs. 2B and 4A). Higher target latencies in the CPX-treated group may have been related to the failure to recognize the target evidenced by frequent brushes against the platform prior to its mounting. The performance pattern during visible target experiments was very similar to that seen during training to a reversed target (figs. 5B and 6B).
Fig. 6.
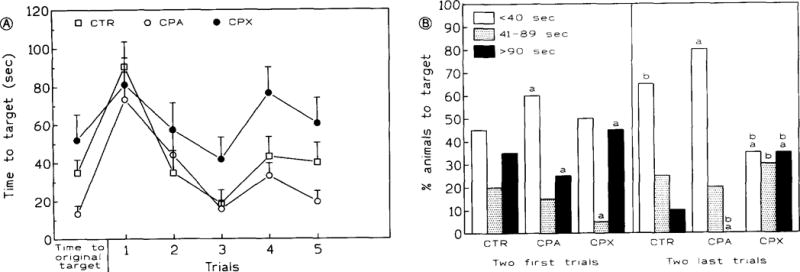
Target latencies (A) and performance (B) to a visible target placed again in the NW quadrant. This phase (starting with trial 1) was the last in a series consisting of the target acquisition phase (nine trials to an invisible target in NW quadrant) and target reversal phase (five trials to an invisible target in SE quadrant with a random starting position. ‘Time to original target’ (fig. 6A, abscissa) indicates target latencies attained at the last trial of the reversal phase in which the invisible target was placed in the opposite (SE) quadrant. There are significant main effects of drug treatment [F(2,26)= 5.921, P< 0.008] and trials effect [F(4,104)= 10.156, P< 0.0001] in 6A. No interation was observed between drug treatment and training trials (P > 0.5). Bars: S.E.M. Statistical significance in fig. B: a P < 0.05 within the trial group; b P < 0.05 between both trial groups. Fisher’s exact test. In both A and B, drugs (CPA: 20 μg/kg, CPX: 1 mg/kg) and vehicle administered for 9 days prior to and then concurrently with training. Bars: S.E.M.
3.5. Movement patterns
Movement patterns were very similar to those previously described by Sei et al. (1992). During the first two trials thigmatactic movement was dominant. In both controls and CPA-treated mice, center crossing became prevalent during trial 3. During subsequent trials the pattern evolved into either center-crossing/target-oriented motion or a swift translocation from the point of immersion to the target. In the CPX mice, changes of the search pattern occurred slowly, and purposeful center-crossing was not apparent before the 4–5th trial. Target-oriented movement was infrequent and over 50% CPX-treated animals required several brushes against the platform edge before mounting it. At the end of the training cycle, CPX-treated mice showed a significant (P < 0.05) increase of crossing into the target quadrant (NW). However, the time spent in the target quadrant did not increase beyond chance levels (figs. 4A,B).
4. Discussion
The well documented impact of adenosine on the function of individual neurons is reflected by its ability to elicit equally significant changes in a variety of behavioral responses. Acute stimulation of A1 receptors produces anticonvulsant effects in the kindling seizure model (Barraco et al., 1984), results in sedation (Crawley et al., 1981), depression of spontaneous and/or evoked locomotor activity (Barraco et al., 1983; Barraco et al., 1987; Martin et al., 1989), and in decrements in schedule-controlled operant behaviors (Commissaris et al., 1990). Xanthines antagonize these effects, and caffeine and theophylline were shown to increase locomotor activity (Coffin et al., 1984; Nikodijevic et al., 1991; Kaplan et al., 1992), to act as proconvulsants (Dragunow, 1991; Dragunow and Goddard, 1984), and to enhance mood and memory (Loke et al., 1988; Normile and Barraco 1991). Furthermore, Schingnitz et al. (1991) demonstrated that acutely administered selective adenosine A1 receptor antagonist KFM 19 improved learning in a Y-maze.
Administration of a highly selective adenosine receptor A1 agonist CPA either as chronic pretreatment or treatment simultaneous with training resulted enhanced acquisition in the water maze reflected by a dose-dependent reduction of target latencies during the initial acquisition trials (3–6), and a complete elimination of animals who failed to acquire the task (target latency > 90 s) by the last two trials. In contrast to control mice, CPA treatment resulted in a faster transition from thigmotactic to center-crossing and target-oriented search strategies. These effects did not appear to be the result of CPA-induced increases in locomotor activity, since target latencies of trained CPA-treated mice (trials 7, 8) and search patterns did not differ from those of the trained controls.
Both CPA- and vehicle-treated animals displayed clear evidence of spatial learning. After eight trials, both groups showed a significant spatial preference although spatial preference in CPA-treated mice developed much earlier than in controls. Already by the third trial, the CPA group spent most of the test time in the target quadrant. Likewise, animals in both groups crossed more frequently into the target rather than any other quadrant of the maze. These data indicate that the rapid improvement in target latency observed among CPA-treated animals is a consequence of spatial learning and/or memory.
In contrast to CPA-treated mice, target acquisition in CPX group was significantly slower than in controls. Notably, although CPX-treated mice slowly shifted from thigmotactic to a center-crossing search pattern, they rarely developed the target-directed search strategy characteristic of trained control or CPA-treated animals. Upon reaching a stable target latency (trials 7, 8), CPX-treated animals equally divided their search time among all quadrants rather than focussing on the target quadrant as seen in both control and CPA groups. However, similarly to control and CPA groups, CPX mice made more crosses into the target quadrant than any other. Although these data may appear confusing, it must be remembered that mice in the water maze do not swim at a constant rate. Rather, the movement is episodic and consists of periods of immobility and/or restricted area search alternating with wider searches of the maze. Moreover, even within such pattern, the swimming speed is not constant throughout the maze area but varies within individual quadrants and also within the entire time spent by the animal in the maze. Unfortunately, our monitoring system is not equipped to quantitate these variables. Thus, these data are not inconsistent and, in fact, detect a relatively subtle effect of chronic CPX treatment which is not readily apparent from examination of either the target latency or locomotor activity alone. In conclusion, it is possible that chronic CPX treatment does not simply disrupt acquisition or retrieval of spatial memory, but instead, interferes with its expression.
Target latency of CPX-treated mice increased significantly during the subsequent target reversal trial. Moreover, the difference in latency between control and CPX animals was not statistically significant (P < 0.1). This finding is baffling when seen only in the context of equal time spent by CPX-treated animals in each quadrant during the preceding probe trial. It must be remembered, however, that at the end of the target acquisition segment, CPX-treated mice reached the target as easily as the controls. Furthermore, despite a clear lack of temporal quadrant preference during the subsequent probe trial, CPX-treated mice crossed more frequently into the target quadrant. Therefore, significant increase in target latency seen during the target reversal segment may indicate a possibility that chronically administered CPX causes subtle modifications in the manner spatial memory is acquired, retrieved and expressed. Possibility of such modifications is also supported by the fact that, although at the end of target reversal trials the number of well performing animals did not change, the CPX treatment resulted in a small but statistically significant (15%, P < 0.05) reduction of poorly performing animals, and in an increased number of mice with an average performance. These observations indicate that more detailed studies of the effect of chronic administration of A1 antagonists on memory may be necessary.
The mechanism behind cognitive modifications introduced by treatment with CPA or CPX given at doses well below those required to elicit gross behavioral changes (Normile and Barraco, 1991; Baumgold et al., 1992) remains to be demonstrated. Receptor-associated phenomena and their ultimate behavioral expression may explain some of the effects seen in the regimen of training and coadministration of either drug, especially that prolonged exposure to CPA has been shown to uncouple striatal adenosine A1 receptors from their effector systems (Parsons and Stiles, 1987; Abracchio and Cattabeni, 1992).
Acute agonist stimulation of adenosine A1 receptors at micromolar doses severely impairs both acquisition and retention of memory (Normile and Barraco, 1991). As noted above, chronic administration of adenosine receptor A1 agonist results in disturbances of normal function of these receptors. Therefore, it is not surprising that a series of daily injections of CPA results in facilitation of learning possibly caused by downregulation of A1 receptor sites. Conversely, therefore, chronic exposure to an adenosine receptor A1 antagonist may upregulate them (Ramkumar et al., 1988) with the consequent impairment of memory functions. Interpretation of the results obtained when drugs were administered prior to training is more difficult. Following the last injection, the effects persist for at least 19 days (duration of the entire trial sequence). Pilot studies indicate that this limit may be extended even further. Although adenosine A1 receptor density shifts caused by chronic administration of selective agonists and antagonists have been described (Abbracchio and Cattabeni, 1992; Ramkumar et al., 1988), virtually nothing is known about the subsequent duration of these phenomena. In chronic treatment with caffeine, receptor levels still have not normalized 2 weeks after cessation of drug administration (Boulenger and Marangos, 1989). If the same obtains with CPA and CPX, the assumption that late cognitive effects of these drugs are based solely on receptor-related phenomena may still be correct. However, one may also argue that only the initial effects are the result of a receptor-mediated facilitation of memory acquisition and retrieval. Once the receptors revert to their normal levels, any subsequent improvement in performance will be related to memory consolidation in a manner that is independent of changes in adenosine A1 receptors. The results of training to reversed and to visible targets mitigate against such contention. In these trials, animals were exposed to a novel environment that either demanded modification of an already learned response (reversed target), or association of old cues with a modified task (visible target). Trials to a reversed target began 9–10 days, and to a visible target 14–15 days after the last injection. Nonetheless, in both cases a dramatic improvement of performance was seen in the CPA-treated group after only one trial, while no changes were present among CPX-treated animals. Since chronic exposure to agents acting at the adenosine A1 receptor site will affect neuronal metabolism (Rudolphi, 1991), peripheral and cerebral blood flow (Van Wylen et al., 1991; Gorman et al., 1991) and cardiac (Collis, 1991) and pulmonary action (Hyman et al., 1991), it is possible that such functions may synergistically augment or impair cognitive responses. Hence, it can not be excluded that prolonged cognitive effects of these drugs may result, at least in part, from persistent biological changes in systems that support neuronal activity.
Adenosinergic therapies have been proposed for neurodegenerative disorders of both acute and chronic etiology e.g., stroke (Von Lubitz et al., 1988; Daval et al., 1989), Alzheimer’s (Schingnitz et al., 1991), Parkinson’s (Schiffman and Vanderhaegen, in press) and Huntington’s (Von Lubitz and Marangos, 1992)) diseases, and epilepsy (Dragunow, 1991). Treatment of cardiovascular (Sollevi, 1986), (Barone et al., 1989) and sleep disorders (Wauquier et al., 1987), and of cystic fibrosis (Eidelman et al., 1992) has been suggested as well. For the most part, suggestions of therapeutic applicability of adenosinergic agents (Daval et al., 1991) have been based on the results of studies in which acute treatment regimens were investigated. However, in most of these disorders chronic administration is clearly the regimen of choice.
Paradoxically, very few studies on the chronic effect of treatment with agents selective at the adenosine A1 receptor have appeared in literature. The data presented in this paper demonstrate that chronic treatment of mice with a potent and selective adenosine A1 receptor agonist or antagonist results in effects that are directly opposite to those observed after acute administration. Although additional studies are necessary, particular caution is required when treatment of both central (Von Lubitz and Marangos, 1992; Daval et al., 1991; Schingnitz et al., 1991) and peripheral (Barone et al., 1989; Sollevi, 1986) disorders based on the stimulation of adenosine A1 receptors is advocated. Implementation of such therapies based solely on results of acute studies may result in disturbances of brain function.
References
- Abracchio M, Cattabeni F. Prolonged agonist exposure induces unbalance of A1 and A2 receptor mediated functions in rat brain slices. Abstr Int J Purine Pyrimid Res. 1992;1:39. [Google Scholar]
- Alzheimer C, Sutor N, Bruggencate G. Transient and selective blockade of adenosine A1 receptors by 8-cyclopentyl-1,3-dipropylxanthine (DPCPX) causes sustained epileptiform activity in hippocampal CA3 neurons of guinea pigs. Neurosci Lett. 1989;99:107. doi: 10.1016/0304-3940(89)90273-5. [DOI] [PubMed] [Google Scholar]
- Arai A, Kessler M, Lynch G. The effects of adenosine on the development of long-term potentiation. Neurosci Lett. 1990;119:41. doi: 10.1016/0304-3940(90)90750-4. [DOI] [PubMed] [Google Scholar]
- Barone S, Churchill PC, Jacobson KA. Adenosine receptor prodrugs: towards kidney-selective dialkylxanthines. J Expt Pharmacol Ther. 1989;1:79. [PMC free article] [PubMed] [Google Scholar]
- Barraco RA, V, Coffin L, Altman HJ, Phillis JW. Central effects of adenosine analogs on locomotor activity in mice and antagonism of caffeine. Brain Res. 1983;272:392. doi: 10.1016/0006-8993(83)90591-7. [DOI] [PubMed] [Google Scholar]
- Barraco RA, Aggarawai AK, Phillis JW, Moran MA, Wu PH. Dissociation of locomotor and hypertensive effects of adenosine analogs in rat. Neurosci Lett. 1984;48:139. doi: 10.1016/0304-3940(84)90009-0. [DOI] [PubMed] [Google Scholar]
- Bartrup JT, Stone TW. Activation of NMDA receptor coupled channels suppresses the inhibitory action of adenosine on hippocampal slices. Brain Res. 1990;530:330. doi: 10.1016/0006-8993(90)91305-z. [DOI] [PubMed] [Google Scholar]
- Baumgold J, Nikodijevic O, Jacobson KA. Penetration of adenosine antagonists into mouse brain as determined by ex vivo binding. Biochem Pharmacol. 1992;43:889. doi: 10.1016/0006-2952(92)90257-j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulenger JP, Marangos PJ. Ceffeine withdrawal affects central adenosine receptors but not benzodiazepine receptors. J Neural Transm. 1989;78:9. doi: 10.1007/BF01247109. [DOI] [PubMed] [Google Scholar]
- Burke SP, Nadler JV. Regulation of glutamate and aspartate release from slices of the hippocampal CA1 area: effects of adenosine and baclofen. Neurochem. 1988;51:154. doi: 10.1111/j.1471-4159.1988.tb01123.x. [DOI] [PubMed] [Google Scholar]
- Coffin VL, Taylor JA, Phillis JW, Altman HJ, Barraco RA. Behavioral interaction of adenosine and methylxanthines on central purinergic systems. Neurosci Lett. 1984;47:91. doi: 10.1016/0304-3940(84)90412-9. [DOI] [PubMed] [Google Scholar]
- Collingridge GL, Davies SN. NMDA receptors and long term potentiation in the hippocampu, in the hippocampu. In: Watkins JC, Collingridge GL, editors. The NMDA Receptor. IRL Press; Oxford: 1991. p. 123. [Google Scholar]
- Collis MG. Influence of adenosine on cardiac activity. In: Phillis JW, editor. Adenosine and Adenine Nucleotides as Regulators of Cellular Function. CRC Press; Boca Raton: 1991. p. 249. [Google Scholar]
- Commissaris RL, McCloskey TC, Damian TC, Brown BD, Barraco RA, Altman HJ. Antagonism of the anti-conflict effects of phenobarbital, but not diazepam by the A1 adenosine agonist L-PIA. Psychopharmacologia. 1990;102:283. doi: 10.1007/BF02244091. [DOI] [PubMed] [Google Scholar]
- Crawley JN, Patel J, Marangos PJ. Behavioral characterization of long-lasting adenosine analogs: sedative properties and interaction with diazepam. Neurosci Lett. 1983;36:169. doi: 10.1016/0024-3205(81)90636-6. [DOI] [PubMed] [Google Scholar]
- Daval JL, Von Lubitz DKJE, Deckert J, Redmond DJ, Marangos PJ. Protective effect of cyclohexyl adenosine on adenosine A1 receptors, guanine nucleotide and forskollin binding sites following transient brain ischemia: a quantitative autoradiographic study. Brain Res. 1989;491:212. doi: 10.1016/0006-8993(89)90058-9. [DOI] [PubMed] [Google Scholar]
- Daval JL, Nehling A, Nicolas F. Physiological and pharmacological properties of adenosine: therapeutic implications. Life Sci. 1991;49:1435. doi: 10.1016/0024-3205(91)90043-b. [DOI] [PubMed] [Google Scholar]
- Dragunow M. Adenosine and epileptic seizures. In: Phillis JW, editor. Adenosine and Adenine Nucleotides as Regulators of Cellular Function. CRC Press; Boca Raton: 1991. p. 367. [Google Scholar]
- Dragunow M, Goddard GV. Adenosine modulation of amygdala kindling. Exp Neurol. 1984;84:654. doi: 10.1016/0014-4886(84)90212-7. [DOI] [PubMed] [Google Scholar]
- Eidelman O, Guay-Broder C, Van Galen PJM, Jacobson KA, Fox C, Turner RJ, Cabantchik ZI, Pollard HB. A1 adenosine receptor antagonists activate chloride efflux from cystic fibrosis cells. Proc Natl Acad Sci. 1992;89:5562. doi: 10.1073/pnas.89.12.5562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Calzero MR, Cubero A. Chronic treatment with R-N6 phenylisopropyl adenosine causes modification of the A1 receptor adenylate cyclase system in rat brain synaptic plasma membranes. Abstr J Purine Pyrimid Res. 1992;1:70. [Google Scholar]
- Gorman MW, Kelley SS, Kaiser L, Sparks HV., Jr . Purinergic control of skeletal muscle blood flow. In: Phillis JW, editor. Adenosine and Adenine Nucleotides as Regulators of Cellular Function. CRC Press; Boca Raton: 1991. p. 181. [Google Scholar]
- Hyman A, Cai B, Feng C, Hao Q, Lippton H. Effects of adenosine and adenine nucleotides on the pulmonary circulation. In: Phillis JW, editor. Adenosine and Adenine Nucleotides as Regulators of Cellular Function. CRC Press; Boca Raton: 1991. p. 221. [Google Scholar]
- Kaplan GB, Greenblatt DJ, Kent MA, Cotreau MM, Arcelin G, Shader R. Caffeine-induced behavioral stimulation is dose-dependent and associated with A1 adenosine receptor occupancy. Neuropsychopharmacology. 1992;1:145. [PubMed] [Google Scholar]
- Lambert NA, Teyler TJ. Adenosine depresses excitatory but not fast inhibitory synaptic transmission in area CA1 of the rat hippocampus. Neurosci Lett. 1991;122:50. doi: 10.1016/0304-3940(91)90190-5. [DOI] [PubMed] [Google Scholar]
- Loke WH, Hinrichs JV, Ghoneim MM. Caffeine and diazepam: separate and combined effects on mood, memory and psychomotor performance. Psychopharmacology. 1985;87:344. doi: 10.1007/BF00432719. [DOI] [PubMed] [Google Scholar]
- Martin JV, Berman KF, Skolnick P, Mendelson WB. Behavioral and electroencephalographic effects of adenosine A1 agonist L-PIA. Pharmacol Biochem Behav. 1989;34:507. doi: 10.1016/0091-3057(89)90549-2. [DOI] [PubMed] [Google Scholar]
- Morris RGM, Garrud P, Rawlins JNP, O’Keefe JO. Place navigation impaired in rats with hippocampal lesions. Nature. 1982;297:681. doi: 10.1038/297681a0. [DOI] [PubMed] [Google Scholar]
- Nikodijević O, Sarges R, Daly JW, Jacobson KA. Behavioral effects of A1 and A2-selective adenosine agonists and antagonists: evidence for synergism and antagonism. J Exp Pharmacol Ther. 1991;1:286. [PMC free article] [PubMed] [Google Scholar]
- Normile HJ, Barraco RA. N6-Cyclopentyladenosine impairs passive avoidance retention by selective action at A1 receptors. Brain Res Bull. 1991;27:101. doi: 10.1016/0361-9230(91)90288-u. [DOI] [PubMed] [Google Scholar]
- Okada Y, Ozawa S. Inhibitory action of adenosine on synaptic transmission in the hippocampus of the guinea pig in vitro. Eur J Pharmacol. 1980;68:483. doi: 10.1016/0014-2999(80)90424-0. [DOI] [PubMed] [Google Scholar]
- O’Keefe J, Nadel L. The Hippocampus as a Cognitive Map. Oxford University Press; Oxford: 1978. [Google Scholar]
- Onodera H, Kogure K. Differential localization of adenosine A1 receptors in the rat hippocampus: quantitative autoradiographic study. Brain Res. 1980;458:212. doi: 10.1016/0006-8993(88)90463-5. [DOI] [PubMed] [Google Scholar]
- Parsons WJ, Stiles GL. Heterologous desensitization of the inhibitory A1 adenosine receptor adenylate cyclase system in rat adipocytes. J Biol Chem. 1987;262:841. [PubMed] [Google Scholar]
- Ramkumar V, Bumgarner JR, Jacobson KA, Stiles GL. Multiple component of the A1 adenosine adenylate cyclase system are regulated in rat cerebral cortex by chronic caffeine ingestion. J Clin Invest. 1988;82:242. doi: 10.1172/JCI113577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudolphi KA. Manipulation of purinergic tone as a mechanism for controlling ischemic brain damage. In: Phillis JW, editor. Adenosine and Adenine Nucleotides as Regulators of Cellular Function. CRC Press; Boca Raton: 1991. p. 423. [Google Scholar]
- Schiffman SN, Vanderhaegen JJ. Adenosine A2 receptors regulate the gene expression of striatopallidal and striatonigral neurons. J Neurosci. 1993 doi: 10.1523/JNEUROSCI.13-03-01080.1993. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schingnitz G, Kufner-Muhl U, Ensinger H, Lehr E, Kuhn FJ. Selective A1-antagonists for treatment of cognitive deficits. Nucleosides Nucleotides. 1991;5:1067. [Google Scholar]
- Sollevi A. Cardiovascular effects of adenosine in man: possible clinical implications. Prog Neurobiol. 1986;26:319. doi: 10.1016/0301-0082(86)90005-5. [DOI] [PubMed] [Google Scholar]
- Schubert P, Mitzdorf U. Analysis and quantitative evaluation of the depressive effect of adenosine on evoked potentials in hippocampal slices. Brain Res. 1979;172:186. doi: 10.1016/0006-8993(79)90910-7. [DOI] [PubMed] [Google Scholar]
- Sei Y, Arora PK, Skolnick P, Paul IA. Spatial learning in a murine model of AIDS. FASEB J. 1992;6:3008. doi: 10.1096/fasebj.6.11.1644264. [DOI] [PubMed] [Google Scholar]
- Tanaka Y, Sakurai M, Goto M, Hayashi S. Effect of xanthine derivatives on hippocampal long term potentiation. Brain Res. 1990:522. doi: 10.1016/0006-8993(90)91577-4. [DOI] [PubMed] [Google Scholar]
- Van Wylen DGL, V, Sciotti M, Winn HR. Adenosine and the regulation of cerebral blood flow. In: Phillis JW, editor. Adenosine and Adenine Nucleotides as Regulators of Cellular Function. CRC Press; Boca Raton: 1991. p. 191. [Google Scholar]
- Von Lubitz DKJE, Marangos PJ. Self defense of the brain: adenosinergic strategies in neurodegeneration. In: Marangos JP, Lai H, editors. Emerging Strategies in Neuroprotection. Birkhauser; Boston: 1992. 151 pp. [Google Scholar]
- Von Lubitz DKJE, Dambrosia JM, Kempski O, Redmond DJ. Cyclohexyl adenosine protects against neuronal death following ischemia in the CA1 region of gerbil hippocampus. Stroke. 1988;19:1133. doi: 10.1161/01.str.19.9.1133. [DOI] [PubMed] [Google Scholar]
- Wauquier A, Van Belle H, Van den Broeck WAE, Janssen PAJ. Sleep improvement in dogs after oral administration of mioflazine, a nucleoside transport inhibitor. Psychopharm. 1987;91:434. doi: 10.1007/BF00216007. [DOI] [PubMed] [Google Scholar]