

Abstract
Next-generation sequencing (NGS) approaches are highly applicable to clinical studies. We review recent advances in sequencing technologies, as well as their benefits and tradeoffs, to provide an overview of clinical genomics from study design to computational analysis. Sequencing technologies enable genomic, transcriptomic, and epigenomic evaluations. Studies that use a combination of whole genome, exome, mRNA, and bisulfite sequencing are now feasible due to decreasing sequencing costs. Single-molecule sequencing increases read length, with the MinION™ nanopore sequencer, which offers a uniquely portable option at a lower cost. Many of the published comparisons we review here address the challenges associated with different sequencing methods. Overall, NGS techniques, coupled with continually improving analysis algorithms, are useful for clinical studies in many realms, including cancer, chronic illness, and neurobiology. We, and others in the field, anticipate the clinical use of NGS approaches will continue to grow, especially as we shift into an era of precision medicine.
Keywords: Sequencing, genomics, clinical utility
I. INTRODUCTION
Next-generation sequencing (NGS) has become ubiquitous over the past few years, producing a deluge of new data at an unprecedented rate. However, how to incorporate the novel insights from these data into clinical practice is not always obvious. Here, we review the current challenges associated with different genomic, transcriptomic, and epigenomic sequencing approaches and platforms, as well as important considerations when designing sequencing studies to maximize statistical power and clinical utility. We also describe the current applications of these technologies across a range of topics including cancer genomics and precision medicine, with a focus on integrative study design and computational analyses. Collectively, this review provides a guide to experimental and computational methods of using NGS in clinical research. NGS technologies have already improved medical interventions and will continue to transform medicine in the clinic and at a personal level by offering individuals increased opportunities to manage their health throughout a lifetime.
II. CHALLENGES
A. Sequencing Has Promising Clinical Utility
The development of NGS has lowered the cost of sequencing from $100 million in 2001 to $1000 in 2014. The lower cost has made sequencing more accessible to the medical community for diagnostic support (Fig. 1). Sequencing can generate a wide variety of data types, which favors its use over other existing techniques to characterize nucleic acids, including PCR and microarrays. Traditional NGS platforms, such as the Illumina HiSeq sequencer, are widely used for DNA sequencing, RNA sequencing, and bisulfite sequencing. Emerging sequencing techniques from the past few years provide alternatives to the short reads produced by these platforms. Together, the analysis of patients’ genomes, transcriptomes, and DNA methylomes can aid diagnosis and prognostic classifications.
FIG. 1.
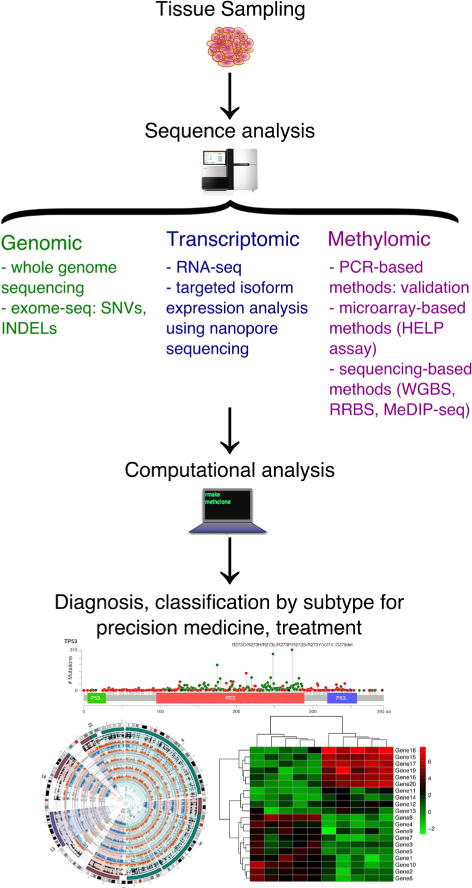
Overview of genomic, transcriptomic, and methylomic sequence analysis workflows for disease characterization and precision medicine. Computational analysis pipelines are further described in Fig. 2.
B. Single-Molecule Sequencing
Both the PacBio RS and the MinION™ nanopore sequencer offer longer read lengths than other sequencing technologies, on the order of kilobases or tens of kilobases.1 Pacific Biosciences released the PacBio RS sequencer in 2010, and although accuracy was initially poor at 86%, repeated sequencing of each strand can increase accuracy to 99%. While specific bioinformatics tools have been developed over the past few years to cope with the error rate,2 the high cost of the instrument has limited its adoption.
Oxford Nanopore Technologies began distributing the MinION™ sequencer to researchers through an early-access program in 2014 before releasing the sequencer commercially in 2015. Unlike the PacBio RS, the MinION™ is highly portable, at the size of a large USB stick, and requires a relatively small investment of approximately $1000. These features could help avoid the time and cost of sending samples to reference laboratories by bringing sequencing to clinics themselves, particularly in remote locations. However, nanopore technology is still in the nascent stages of development. Estimates have placed perbase error rates at 10–15%,3 which needs to improve drastically before nanopore sequencers can be considered a viable tool for many diagnostic applications. Efforts to demonstrate the clinical potential of the MinION™ have focused on pathogen identification and characterization, including sequencing of the influenza virus,4 antibiotic resistance genes in Salmonella enterica serovar Typhi,5, and the Ebola virus to identify transmission patterns during the recent outbreak.6 Researchers have also taken advantage of long read lengths to analyze isoform expression of alternatively spliced RNA using cDNA libraries.7 Current coverage depths and error rates best facilitate targeted studies of specific genes or RNA isoforms but not whole-genome or whole-transcriptome analysis. Yet, as the chemistry and analysis continue to evolve, nanopore sequencing shows increasing promise as an accessible and powerful means of evaluating patients and the pathogens that affect them.
C. DNA Sequencing for Clinical Applications
Many consortiums are devoted to standardizing sequencing performance. Accuracy and reproducibility are the two key factors necessary for sequencing technology that are widely used in clinical practice. DNA sequencing enables the detection of germline and somatic mutations. Whole-genome sequencing (WGS) is an approach to determining the complete DNA sequence of a genome with a single assay. A cross-platform WGS performance comparison revealed that 88.1% of SNVs detected were shared by Illumina and Complete Genomics.8 The concordance of insertion and deletion (indel) calling is much lower, with just 26% shared.8 Another study comparing Illumina MiSeq, 454 GS Junior and Ion Torrent PGM from Life Technology for bacteria genome sequencing showed that Illumina has the lowest error rate and no homopolymer-associated indel errors.9
Whole-exome sequencing (WES), which captures only genic regions, provides a cost-efficient alternative to whole genome sequencing. WES shows high accuracy for detecting single-nucleotide variants (SNVs) and short indels; however, when compared to high-coverage WGS, WES has limited power for detecting copy-number variation (CNV).10 A recent assessment of WES and exome array comparative genomic hybridization (CGH) using clinical samples has shown that WES has the potential for clinical CNV detection, but currently, the combination of an array-based approach with WES improves the accuracy of CNV calling, especially for intergenic regions and single-exon changes.11
When using WES, the choice of exome-seq protocol affects results. A comprehensive comparison between Agilent, Roche, and Illumina exome-seq protocols showed varying strengths in the detection of variants across genic and untranslated regions.12 NimbleGen, from Roche, is the only platform that uses high-density overlapping baits and has higher sensitivity in variant detection. A concurrent study also confirmed that the NimbleGen platform has higher coverage of exonic regions, with at least 20× coverage.13 The Agilent and Illumina platforms, however, target a wider range of genomic regions, and with deeper sequencing, these two platforms detect more variants.14 Another advantage to Illumina’s capture method is that it provides coverage for untranslated areas, which might be of interest to researchers who would like to include noncoding variants in their analyses.
For an even more targeted and affordable method than WES, specific cancer panels are commonly used. These require prior knowledge of recurrent genetic or epigenetic lesions. Recurrent somatic mutations appear in many cancer types and can predict risk levels of the disease.15 In acute myeloid leukemia (AML), 15 biomarkers have been used to further stratify patients who were previously all placed in the intermediate risk group by cytogenetic classification.16 This method helps to develop treatment plans for AML patients tailored to the risk for each group. Indeed, targeted sequencing provides a much deeper view of the known genes and hotspots for mutations. However, with ever-decreasing sequencing cost and increasing detection of possible drug targets, exomeseq covering larger areas of the genome has the potential for wider application in clinical diagnosis and prognostic decisions.
D. RNA Sequencing (RNA-seq): A Promising Candidate for Clinical Applications
This technique enables whole-transcriptome examination, including detection of gene expression, alternative isoforms, fusion genes, and expressed variants.17,18 However, RNA-seq is also very sensitive to systematic bias.19,20 Previously, we and others have defined multiple quality metrics that flag samples with potential gene expression quantification issues, including gene body coverage evenness, GC content, insert size, and base error rate.21 The FDA-led Sequencing Quality Control (SEQC) study for RNA-seq performance evaluation showed that gene body coverage evenness, GC content, and insert size relate to library preparation and that base error rate depends on the sequencer used.19
Multiple software packages exist for gene expression normalization. EDAseq, which corrects for both the intra-group variations and quantification bias caused by GC-content and gene length, offers the best accuracy for differential gene expression analysis.21 PEER and sva show greater power to detect latent variables for the quantification of gene expression among different sites of sequencing data.22 For a statistically powerful RNA-seq study design, consistent experimental strategies are recommended, including sequencer, read length, sequencing depth, and protocol.23 High sequencing depth is critical for the discovery of new genes and accurate gene expression profiling.24 Follow-up studies on differential gene expression analysis have shown that increasing biological replicates improves the accuracy of gene quantifications.25 Therefore, experimental design for RNA-seq analysis is critical for accurate differential gene expression analysis.
E. DNA Methylation Provides a Complementary Approach to Clinical Measures for Patient Classification
In humans, DNA methylation involves the addition of a methyl group to the fifth position of cytosine, which has the specific effect of suppressing gene expression. DNA methylation is one of the hallmarks of cancers and aging.26,27 Many different types of cancers show consistent dysregulation of DNA methylation.28–31 The Cancer Genome Atlas (TCGA) consortium and many other research studies have shown that cancers can be classified based on their degree of DNA methylation.28,32 Subgroups of many cancers exhibit CpG island methylator phenotype (CIMP), including breast cancer,33 brain cancer,28,30,34 blood cancer,29 gastric cancer,35 liver cancer,36 and lung cancer.31 Groups of patients classified based on DNA methylation patterns show distinct clinical outcomes, including overall survival and disease free progression.28,29 The CIMP-positive group can be used to differentiate and stratify patients into groups with distinct clinical outcomes. For example, in glioblastoma patients, a CIMP-positive phenotype is usually associated with distinct copy number changes, appears exclusively in the proneural subtypes, and is associated with IDH1 mutations and improved clinical outcomes.28 In a recent study of ependymoma, which is the third most common pediatric brain tumor, researchers showed that CIMP-positive patients with posterior fossa ependymoma have worse clinical outcomes than CIMP-negative patients.30,34 The genetic background of CIMP-positive patients presents a blended picture and indicates the importance of DNA methylation as an alternative approach for patient risk stratification.30
There are many advantages to using DNA methylation analysis for clinical profiling. First, this analysis does not rely on the genetic alterations of the diseases; thus, it can be applied to diseases with sparse somatic mutations. Second, the material under analysis is DNA, which is advantageous because DNA is less sensitive to heat or enzymatic degradation than RNA, resulting in more accurate profiling.
Several types of methods have emerged to quantitatively measure DNA methylation, grouped here into three categories: (1) PCR-based methods, (2) microarray-based methods, and (3) sequencing-based methods. The PCR-based methods are usually used as a validation approach for high-throughput quantification. Among microarray-based methods, the HpaII tiny fragment Enrichment by Ligation-mediated PCR Assay (HELP Assay) is a common regional DNA methylation quantification approach for research and clinical sample profiling.37 It is based on the restriction enzyme Hpall’s ability to exclusively recognize and cleave methylated CpG DNA sites. Another common microarray-based DNA methylation quantification approach with single-base resolution is the Illumina Infinium BeadChip Kit. The BeadChip array platform uses two different bead types to measure DNA methylation levels at single cytosine. The Infinium HumanMethylation450 BeadChip Kit (450K array) is one of the Infinium Kits that covers the most methylation sites for human samples (485,000 sites). This kit covers 99% of RefSeq genes, which, on average, have 17 CpG sites per gene. The 450K array has been widely used in DNA methylation quantification over the past few years, with more than 10,000 entries in the Gene Expression Omnibus (GEO) database, providing a valuable international resource for comparison among different cohorts of patient samples.38
Sequencing-based methods provide either single-base resolution or regional quantifications of DNA methylation levels.39–41 Single-base resolution methods mainly use bisulfite conversion sequencing, where bisulfite converts cytosines without methylation into uracil but leaves cytosines with methylation intact as cytosines. In the final sequencing readout, unmethylated CpG sites appear as thymine instead of cytosine.39,40 CpG methylation levels for individual sites are calculated based on the percentage of reads with cytosine among the total number of reads mapped. Bisulfite-based methods include whole genome bisulfite sequencing (WGBS),39 reduced representation bisulfite sequencing (RRBS),40 and targeted methylation sequencing (TMS).42 WGBS requires high sequencing depth, as at least four reads must cover each base in the whole genome to achieve accurate quantification. WGBS enables the inclusion of regions with both high and low CG density. RRBS and TMS each cover a subset of regions in the genome, providing cheaper alternatives to WGBS, and accurately quantify of approximately 15% of higher density CpG sites, including CpG islands and promoter regions.40,42. These targeted approaches make it possible to profile more patients with regions that are of particular interest in transcriptome regulation.
Regional quantification approaches for methylation analysis mainly use affinity-based DNA methylation sequencing, such as methylated DNA immunoprecipitation sequencing (MeDIP-seq).41 This approach uses antibodies that recognize genomic locations with methylated CpGs. Comparison of the 450K array and WGS approaches showed sufficient correlation (Spearman correlation coefficient = 0.68).43 Another study showed the 450K array generated highly reproducible data between seven technical replicates of clinical samples.37 However, a large-scale effort comparing different platforms remains to be done.
F. Computational Analysis of Multi-Omics Data
One of the biggest challenges in going from bench to bedside in sequencing studies is the accurate and reproducible analysis of the resulting terabytes of data. Sequencing data analysis is a multistep process that researchers often need to adapt for specific experiments or scientific questions. For any sequencing data, computational analysis generally begins with aligning reads to a reference genome (Fig. 2). Commonly used programs include BWA for DNA reads, STAR for RNA-seq data, and Bismark, BSMAP, or BSmapper for bisulfite sequencing data.44–48 The choice between different aligners available for a specific type of data depends on such factors as sequencing platform, read length, and desired SNP tolerance, with various programs optimized for different read characteristics.49 After mapping to the genome, analysis depends on the scientific question, with specific programs designed broadly to call variants, identify differential expression, or quantify the extent of methylation. For example, the Genome Analysis Toolkit (GATK) offers variant calling algorithms for both DNA- and RNA-seq data, the results of which are often followed with annotation using SnpEff or Oncotator for cancer studies.50–52 Another method useful for the analysis of variants in cancer data, specifically intra-tumor heterogeneity, is PyClone.53 For RNA-seq data, the pipeline r-make, mediated by GNU Make, provides an easy, one-step method to align data with STAR, perform quality assessments, and generate gene counts, which can then be used for differential expression analysis with tools such as edgeR.19,54 Software for downstream analysis of methylation data includes methylKit, eDMR, and methclone.55–57 When multiple data types are available, their integration can identify a network of interacting and interdependent processes contributing to disease states using tools such as iCluster and Cytoscape.58,59 Indeed, clustering patient samples using models that computationally combine different data types has revealed new subtypes not seen when evaluating a single data type.60,61 Despite challenges in cost, cross-platform comparisons, technical standards, and analysis methods, advances in massively parallel sequencing techniques present new opportunities to improve clinical research, which we explore in the next section.
FIG. 2.
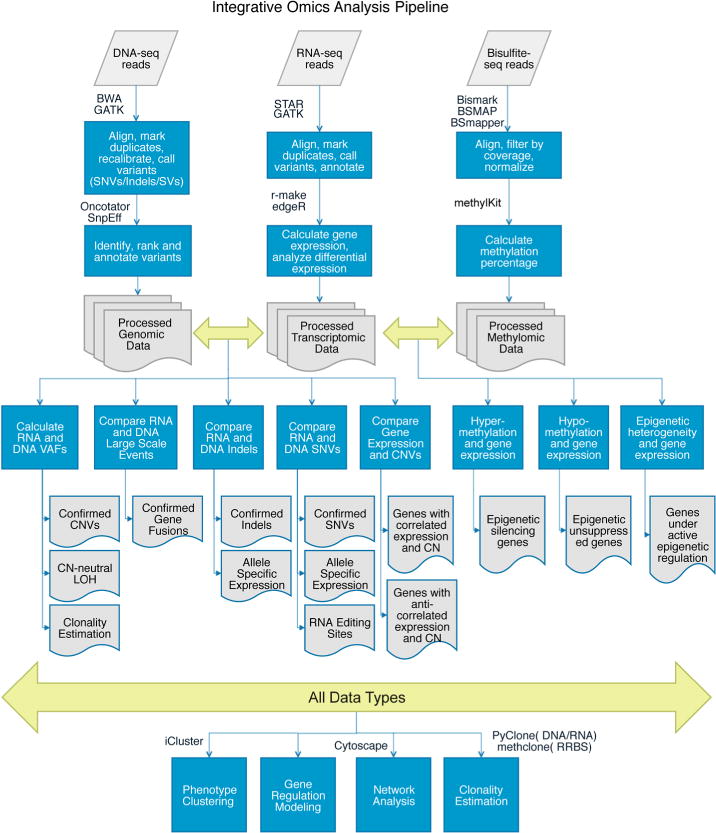
Computational analysis pipelines for integrative genomic, transcriptomic, and methylomic data. SNV, singlenucleotide variant; SV, structural variant; VAF, variant allele frequency; CN, copy number; CNV, copy-number variant; LOH, loss of heterozygosity.
III. OPPORTUNITIES
A. Leveraging Electronic Health Records Data
Many aspects of patient care increasingly incorporate genomics and informatics, especially with the transition to electronic health records (EHR). Despite the relatively recent shift to EHR, large-scale studies using machine learning and data mining methods are already leveraging the data, as EHR offers unprecedented access to large sample sizes and diverse patient cohorts. These studies include mining for adverse drug effects,62 and developing a classifier for disease phenotype severity.63 The implications of a transition to EHR for clinical genomics, including genetic testing, have been reviewed previously.64
B. Genomics and Chronic Illnesses
Genomic approaches are important for preventing and managing chronic illnesses, such as diabetes and inflammatory bowel disease. The Human Microbiome Project and other metagenomic studies have revealed the role of gut microbiota in health. Fecal microbiota transplants for treating Clostridium difficile infections, ulcerative colitis, Crohn’s disease, and other digestive illnesses represent the translation of this finding into clinical practice.65,66
C. Personalized Healthcare and Direct-to-Consumer Genomics
Statistical models can incorporate genomic features and family history, coupled with factors such as age, weight, and ethnicity, for disease risk prediction in healthy individuals. These models have been especially useful for early intervention in individuals at high risk for diabetes and cardiovascular disease. Clinical genomics platforms such as Foundation Medicine, Ingenuity, and Personalis facilitate the implementation of genetic testing in clinical platforms.67 As of August 2015, the NIH’s genetic testing registry catalogued 28,542 tests spanning 4,726 genes for the purpose of diagnosing any of 9,927 conditions. This registry not only includes classical Mendelian diseases, such as Huntington’s chorea, but also predicts predisposition to complex diseases, such as Alzheimer’s, and drug response, such as sensitivity to the anticoagulant warfarin. With direct-to-consumer tools like 23andme and ancestry.com, which make this type of information accessible to interested individuals, people are more empowered than ever to advocate for their own health. Research continues into disease risk prediction through computational methods that use patients’ genetic information, coupled with EHR in some cases.68 Federal policies are changing to reflect the shift to clinical genomics, as evidenced by the 2015 repeal of the FDA’s shutdown of 23andme’s genetic testing arm, and by the 2013 landmark supreme court case that barred the previously common practice of patenting genes.69 Other legal and ethical issues surrounding clinical genomics include those relating to genetic testing in children and adolescents, previously reviewed by Botkin et al.70
D. Genomics and Cancer
Despite challenges, genomics has produced a paradigm shift in medicine, especially in the treatment of cancer. Where historically cancer was categorized by the tissue type it affects, it is now increasingly being defined by genetic alterations. The vast breadth of knowledge we have gained from large national and international cancer sequencing efforts, mainly The Cancer Genome Atlas and the International Cancer Genome Consortium, has immeasurably increased our understanding of the genetic mechanisms, molecular subtypes, and heterogeneity of cancers.71,72 These data are easily accessible to the scientific community. Tools like the cBio Portal, for example, allow anyone to query the mutation load of any given gene in all assayed cancer types (Figure 1). Thus, cancer genomics is continuously being translated to clinical settings.73 One such case is recurrent mantle cell lymphoma, for which researchers used an integrative genomics and transcriptomics approach coupled with extensive functional studies to attribute the cause of relapse after ibrutinib treatment to a relapse-specific SNV in the drug target, BTK.74 The therapeutic decision-making pipeline can now incorporate this discovery by testing for this BTK mutation. Similar efforts in a wide variety of cancers have categorized subtypes of cancers based on genetic information, and these classifications are actively used in diagnoses, prognoses, and therapeutics.
A classic success story surrounding the use of genomics in cancer therapy relates to the BRAF inhibitor vemurafenib in metastatic melanoma. Genomic screening of metastatic melanoma patients identified BRAF V600 mutations in half of all patients that increased the sensitivity of cancer cells to BRAF inhibitors.75 One of the common challenges of targeted therapies, however, has been the development of resistance, which occurred in cases of melanoma treated with BRAF inhibitors. Combinations of drugs as opposed to monotherapies lower the risk of resistance and relapse. For example, dabrafenib in combination with trametinib prolongs progressionfree survival and increases response rates in BRAF V600 melanoma compared to monotherapy.76
Combination therapies often perform more successfully, as developing resistance is less likely. Computational methods for predicting effective drug combinations alleviate the enormous cost of exhaustive experimental testing in every cancer model. Instead, these machine learning methods can use data from cell line assays as training sets and predict successful combinations for genetically defined subtypes that researchers can then test in patient-derived xenograft models.77 Some of the experimental data sets currently available for use in computational models are the NCI 60 cancer cell line and drug screening data,78 NIH’s Library of Integrated Cellular Signatures (LINCS), and the Broad Institute’s connectivity map.79 By modeling drug-gene interactions coupled with the genomic alterations of a patient’s tumor, doctors are now able to predict the efficacy of different chemotherapies or targeted therapies in a personalized manner. These models not only include rule-based decision-tree methods but are also more complex computational models. In addition to predicting the efficacy of combination therapies, computational methods for drug repositioning are also continuously gaining popularity and producing effective therapies.80
Because many of these drug development and prediction approaches rely on accurate and detailed patient stratification based on genomic data, clinical samples increasingly undergo whole genome, exome, and transcriptome sequencing, either at the time of collection for rapid turnaround or after the banking for future analysis. A vast amount of sequencing data has enabled better assessment of prognosis in many cases, although this is not new to the sequencing era. By 2000, microarrays were being used for molecular stratification of cancer samples through the identification of gene signatures defining differential survival.81 Unsurprisingly, the advent of NGS methods increased studies in this vein.
Even with applications to all aspects of human health and disease, cancer remains the one disease (really an innumerable collection of diseases) on which genomics has had the biggest impact. Cancer is genetic in nature; cancers arise from the accumulation of inherited and somatic genetic alterations.82 Heterogeneous subpopulations comprising tumors have been experimentally observed through cytogenetic, Sanger sequencing, and NGS experiments.83 As originally proposed by Nowell in 1976, these subpopulations compete with each other for space and resources, and the clones better equipped to survive and proliferate in the tumor microenvironment will progress.84 Genomics enables researchers to assess the compositions of tumors and infer the molecular characteristics of distinct subpopulations.
The main challenge in accurately inferring heterogeneity and clonal evolution is that most tumorprofiling methods involve a bulk sample of cells, effectively masking intratumoral variability. With novel technological developments in single-cell sequencing, we can now measure these subpopulations directly and at a previously unprecedented resolution. Single-cell sequencing will add a new level to clinical applications of tumor sequencing by increasing the resolution with which we can model complex tumor dynamics and incorporate that into prognosis assessment and drug efficacy prediction (Figure 3). The development of single-cell sequencing methods has addressed this issue, especially single cell RNA-seq, which researchers have used in immune cells,85 breast cancer,86 melanoma circulating tumor cells,87 and glioblastoma.88 Each of these cases revealed new levels of heterogeneity that are undetectable in bulk samples, suggesting that single-cell resolution is necessary to accurately characterize complex tissue samples.
FIG. 3.
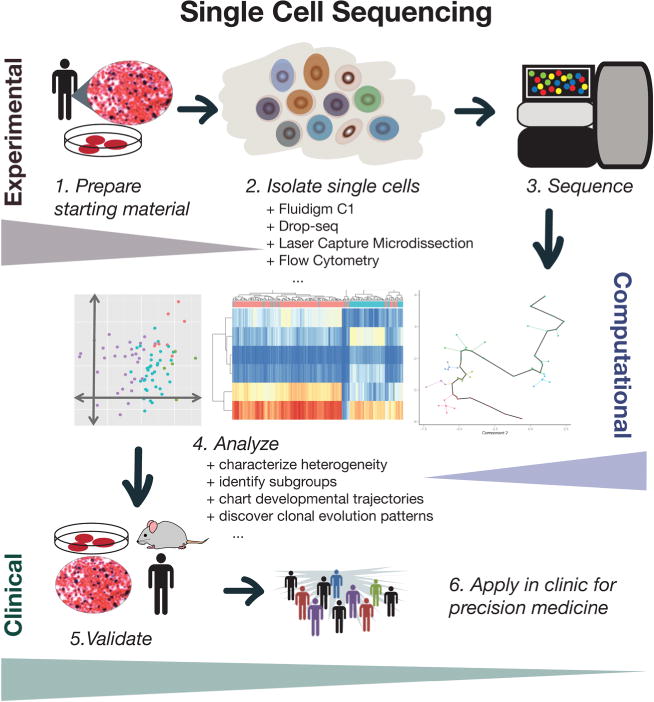
Conceptual overview of single-cell sequencing for clinical applications.
An added benefit is that all of these sequencing data are submitted to curated repositories with publication, such as the database of Genotypes and Phenotypes, the Sequencing Reads Archive, and the Gene Expression Omnibus. Public data help alleviate the problem of small sample sizes common in clinical settings and/or rare diseases. Researchers interested in any of these data can download them and apply their own analysis. For those unfamiliar with computational and bioinformatics methods, there are also pipelines with guided user interfaces that facilitate these steps, such as STORMseq,89 Genesifter, Ingenuity variant analysis software, and more. Currently research is also being conducted in software design for use by non-computational clinical scientists.90 Although the data repositories in place serve a much-needed purpose, there are opportunities for better infrastructure, support for IRB approvals, ease of submission, and ease of access.
E. Advances in Genomics Approaches for Neurobiology
The use of molecular stratification with genomic sequencing to guide patient therapy is not limited to cancer drugs. Although less understood, genomic approaches also apply to neurobiology, especially in the study of Alzheimer’s and autism spectrum disorders.91 With large-scale efforts in mapping the human brain using cutting edge brain imaging techniques, high-volume data approaches are becoming increasingly useful in understanding neurodegenerative diseases. Understanding mutations and predispositions to these diseases would allow for early intervention, which is often the only hope for therapy.
F. National and International Personalized Medicine Initiatives
Overall, clinical genomics pervasively affects human health and disease, especially in oncology. Federal policy changes mirror this evolution in our understanding and treatment of cancer, most notably through President Obama’s Precision Medicine Initiative, announced in his 2015 State of the Union Address. This initiative includes increased funding to the National Cancer Institute for researching genomic drivers in cancer and for streamlining the design and testing of targeted therapies based on genetics. Relatedly, the prototypical clinical trial is transforming to reflect a personalized medicine approach, as seen by the success of the IMPACT and following IMPACT2 studies. Importantly, these changes in clinical genomics are occurring on a global scale, inspiring international cooperation to advance medicine.92–93
Acknowledgments
We would like to thank the Epigenomics Core Facility at Weill Cornell Medicine. The authors would like to thank the following sources of financial support: Ty Louis Campbell Foundation, Elizabeth’s Hope and the Families of the Children’s Brain Tumor Project, WorldQuant Foundation, the Starr Cancer Consortium grants (I7-A765, I9-A9-071) and funding from the Irma T. Hirschl and Monique Weill-Caulier Charitable Trusts, Bert L and N Kuggie Vallee Foundation, the Pershing Square Sohn Cancer Research Alliance, NASA (NNX14AH50G, 15-15Omni2-0063), the National Institutes of Health (R25EB020393, R01NS076465, R01AI125416, R01ES021006), the Bill and Melinda Gates Foundation (OPP1151054), and the Alfred P. Sloan Foundation (G-2015-13964).
References
- 1.Mason CE, Porter SG, Smith TM. Characterizing multi-omic data in systems biology. Adv Exp Med Biol. 2014;799:15–38. doi: 10.1007/978-1-4614-8778-4_2. [DOI] [PubMed] [Google Scholar]
- 2.Chaisson MJ, Tesler G. Mapping single molecule sequencing reads using basic local alignment with successive refinement (BLASR): Theory and Application. BMC Bioinformatics. 2012;13:238. doi: 10.1186/1471-2105-13-238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jain M, Fiddes IT, Miga KH, Olsen HE, Paten B, Akeson M. Improved data analysis for the MinION nanopore sequencer. Nat Methods. 2015;12(4):351–356. doi: 10.1038/nmeth.3290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang J, Moore NE, Deng Y-M, Eccles DA, Hall RJ. MinION nanopore sequencing of an influenza genome. Front Microbiol. 2015 Aug 18;6:766. doi: 10.3389/fmicb.2015.00766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ashton PM, Nair S, Dallman T, Rubino S, Rabsch W, Mwaigwisya S, Wain J, O’Grady J. MinION nanopore sequencing identifies the position and structure of a bacterial antibiotic resistance island. Nat Biotechnol. 2014;33(3):296–300. doi: 10.1038/nbt.3103. [DOI] [PubMed] [Google Scholar]
- 6.Gardy J, Loman NJ, Rambaut A. Real-time digital pathogen surveillance — the time is now. Genome Biol. 2015;16(1):155. doi: 10.1186/s13059-015-0726-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bolisetty MT, Rajadinakaran G, Graveley BR. Determining exon connectivity in complex mRNAs by nanopore sequencing. Genome Biol. 2015 Sep;16(1):204. doi: 10.1186/s13059-015-0777-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lam HYK, Clark MJ, Chen R, Natsoulis G, Chen R, O’Huallachain M, Dewey FE, Habegger F, Ashley EA, Gerstein MB, Butte AJ, Ji HP, Snyder M. Performance comparison of whole-genome sequencing platforms. Nat Biotechnol. 2012 doi: 10.1038/nbt.2065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Loman NJ, Misra RV, Dallman TJ, Constantinidou C, Gharbia SE, Wain J, Pallen MJ. Performance comparison of benchtop high-throughput sequencing platforms. Nat Biotechnol. 2012 May;30(5):434–9. doi: 10.1038/nbt.2198. [DOI] [PubMed] [Google Scholar]
- 10.Tan R, Wang Y, Kleinstein SE, Liu Y, Zhu X, Guo H, Jiang Q, Allen AS, Zhu M. An evaluation of copy number variation detection tools from whole-exome sequencing data. Hum Mutat. 2014 Jul;35(7):899–907. doi: 10.1002/humu.22537. [DOI] [PubMed] [Google Scholar]
- 11.Retterer K, Scuffins J, Schmidt D, Lewis R, Pineda-Alvarez D, Stafford A, Schmidt L, Warren S, Gibellini F, Kondakova A, Blair A, Bale S, Matyakhina L, Meck J, Aradhya S, Haverfield E. Assessing copy number from exome sequencing and exome array CGH based on CNV spectrum in a large clinical cohort. Genet Med. 2015 Aug;17(8):623–9. doi: 10.1038/gim.2014.160. [DOI] [PubMed] [Google Scholar]
- 12.Clark MJ, Chen R, Lam HYK, Karczewski KJ, Chen R, Euskirchen G, Butte AJ, Snyder M. Performance comparison of exome DNA sequencing technologies. Nat Biotechnol. 2011 Oct;29(10):908–14. doi: 10.1038/nbt.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sulonen AM, Ellonen P, Almusa H, Lepistö M, Eldfors S, Hannula S, Miettinen T, Tyynismaa H, Salo P, Heckman C, Joensuu H, Raivio T, Suomalainen A, Saarela J. Comparison of solution-based exome capture methods for next generation sequencing. Genome Biol. 2011 Sep 28;12(9):R94. doi: 10.1186/gb-2011-12-9-r94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Clark MJ, Chen R, Lam HY, Karczewski KJ, Chen R, Euskirchen G, Butte AJ, Snyder M. Performance comparison of exome DNA sequencing technologies. Nat Biotechnol. 2011 Sep 25;29(10):908–14. doi: 10.1038/nbt.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ding L, Ley TJ, Larson DE, Miller CA, Koboldt DC, Welch JS, Ritchey JK, Young MA, Lamprecht T, McLellan MD, McMichael JF, Wallis JW, Lu C, Shen D, Harris CC, Dooling DJ, Fulton RS, Fulton LL, Chen K, Schmidt H, Kalicki-Veizer J, Magrini VJ, Cook L, McGrath SD, Vickery TL, Wendl MC, Heath S, Watson MA, Link DC, Tomasson MH, Shannon WD, Payton JE, Kulkarni S, Westervelt P, Walter MJ, Graubert TA, Mardis ER, Wilson RK, DiPersio JF. Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature. 2012 Jan 11;481(7382):506–10. doi: 10.1038/nature10738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Patel JP, Gönen M, Figueroa ME, Fernandez H, Sun Z, Racevskis J, Van Vlierberghe P, Dolgalev I, Thomas S, Aminova O, Huberman K, Cheng J, Viale A, Socci ND, Heguy A, Cherry A, Vance G, Higgins RR, Ketterling RP, Gallagher RE, Litzow M, van den Brink MR, Lazarus HM, Rowe JM, Luger S, Ferrando A, Paietta E, Tallman MS, Melnick A, Abdel-Wahab O, Levine RL. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N Engl J Med. 2012 Mar 22;366(12):1079–89. doi: 10.1056/NEJMoa1112304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Van Keuren-Jensen K, Keats JJ, Craig DW. Bringing RNA-seq closer to the clinic. Nat Biotechnol. 2014 Sep;32(9):884–5. doi: 10.1038/nbt.3017. [DOI] [PubMed] [Google Scholar]
- 18.Li S, Tighe SW, Nicolet CM, Grove D, Levy S, Farmerie W, Viale A, Wright C, Schweitzer PA, Gao Y, Kim D, Boland J, Hicks B, Kim R, Chhangawala S, Jafari N, Raghavachari N, Gandara J, Garcia-Reyero N, Hendrickson C, Roberson D, Rosenfeld J, Smith T, Underwood JG, Wang M, Zumbo P, Baldwin DA, Grills GS, Mason CE. Multi-platform assessment of transcriptome profiling using RNA-seq in the ABRF next-generation sequencing study. Nat Biotechnol. 2014 Sep;32(9):915–25. doi: 10.1038/nbt.2972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li S, Labaj PP, Zumbo P, Sykacek P, Shi W, Shi L, Phan J, Wu PY, Wang M, Wang C, Thierry-Mieg D, Thierry-Mieg J, Kreil DP, Mason CE. Detecting and correcting systematic variation in large-scale RNA sequencing data. Nat Biotechnol. 2014 Sep;32(9):888–95. doi: 10.1038/nbt.3000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Risso D, Schwartz K, Sherlock G, Dudoit S. Normalization of RNA-seq data using factor analysis of control genes or samples. Nat Biotechnol. 2011 Dec 17;12:480. doi: 10.1038/nbt.2931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Risso D1, Schwartz K, Sherlock G, Dudoit S. GC-content normalization for RNA-Seq data. BMC Bioinformatics. 2011 Dec 17;12:480. doi: 10.1186/1471-2105-12-480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stegle O, Parts L, Piipari M, Winn J, Durbin R. Using probabilistic estimation of expression residuals (PEER) to obtain increased power and interpretability of gene expression analyses. Nat Protoc. 2012 Feb 16;7(3):500–7. doi: 10.1038/nprot.2011.457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Su Z, Łabaj PP, Li S, Thierry-Mieg J, Thierry-Mieg D, Shi W, Wang C, Schroth GP, Setterquist RA, Thompson JF, Jones WD, Xiao W, Xu W, Jensen RV, Kelly R, Xu J, Conesa A, Furlanello C, Gao H, Hong H, Jafari N, Letovsky S, Liao Y, Lu F, Oakeley EJ, Peng Z, Praul CA, Santoyo-Lopez J, Scherer A, Shi T, Smyth GK, Staedtler F, Sykacek P, Tan XX, Thompson EA, Vandesompele J, Wang MD, Wang J, Wolfinger RD, Zavadil J, Auerbach SS, Bao W, Binder H, Blomquist T, Brilliant MH, Bushel PR, Cai W, Catalano JG, Chang CW, Chen T, Chen G, Chen R, Chierici M, Chu TM, Clevert DA, Deng Y, Derti A, Devanarayan V, Dong Z, Dopazo J, Du T, Fang H, Fang Y, Fasold M, Fernandez A, Fischer M, Furió-Tari P, Fuscoe JC, Caimet F, Gaj S, Gandara J, Gao H, Ge W, Gondo Y, Gong B, Gong M, Gong Z, Green B, Guo C, Guo L, Guo LW, Hadfield J, Hellemans J, Hochreiter S, Jia M, Jian M, Johnson CD, Kay S, Kleinjans J, Lababidi S, Levy S, Li QZ, Li L, Li L, Li P, Li Y, Li H, Li J, Li S, Lin SM, López FJ, Lu X, Luo H, Ma X, Meehan J, Megherbi DB, Mei N, Mu B, Ning B, Pandey A, Pérez-Florido J, Perkins RG, Peters R, Phan JH, Pirooznia M, Qian F, Qing T, Rainbow L, Rocca-Serra P, Sambourg L, Sansone SA, Schwartz S, Shah R, Shen J, Smith TM, Stegle O, Stralis-Pavese N, Stupka E, Suzuki Y, Szkotnicki LT, Tinning M, Tu B, van Delft J, Vela-Boza A, Venturini E, Walker SJ, Wan L, Wang W, Wang J, Wang J, Wieben ED, Willey JC, Wu PY, Xuan J, Yang Y, Ye Z, Yin Y, Yu Y, Yuan YC, Zhang J, Zhang KK, Zhang W, Zhang W, Zhang Y, Zhao C, Zheng Y, Zhou Y, Zumbo P, Tong W, Kreil DP, Mason CE, Shi L. A comprehensive assessment of RNA-seq accuracy, reproducibility and information content by the Sequencing Quality Control Consortium. Nat Biotechnol. 2014 Sep;32(9):903–14. doi: 10.1038/nbt.2957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Toung JM1, Morley M, Li M, Cheung VG. RNA-sequence analysis of human B-cells. Genome Res. 2011 Jun;21(6):991–8. doi: 10.1101/gr.116335.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rapaport F, Khanin R, Liang Y, Pirun M, Krek A, Zumbo P, Mason CE, Socci ND, Betel D. Comprehensive evaluation of differential gene expression analysis methods for RNA-seq data. Genome Biol. 2013;14(9):R95. doi: 10.1186/gb-2013-14-9-r95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Teschendorff AE1, Menon U, Gentry-Maharaj A, Ramus SJ, Weisenberger DJ, Shen H, Campan M, Noushmehr H, Bell CG, Maxwell AP, Savage DA, Mueller-Holzner E, Marth C, Kocjan G, Gayther SA, Jones A, Beck S, Wagner W, Laird PW, Jacobs IJ, Widschwendter M. Age-dependent DNA methylation of genes that are suppressed in stem cells is a hallmark of cancer. Genome Res. 2010 Apr;20(4):440–6. doi: 10.1101/gr.103606.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rodríguez-Paredes M1, Esteller M. Cancer epigenetics reaches mainstream oncology. Nat Med. 2011 Mar;17(3):330–9. doi: 10.1038/nm.2305. [DOI] [PubMed] [Google Scholar]
- 28.Noushmehr H, Weisenberger DJ, Diefes K, Phillips HS, Pujara K, Berman BP, Pan F, Pelloski CE, Sulman EP, Bhat KP, Verhaak RG, Hoadley KA, Hayes DN, Perou CM, Schmidt HK, Ding L, Wilson RK, Van Den Berg D, Shen H, Bengtsson H, Neuvial P, Cope LM, Buckley J, Herman JG, Baylin SB, Laird PW, Aldape K, Cancer Genome Atlas Research Network Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell. 2010 May 18;17(5):510–22. doi: 10.1016/j.ccr.2010.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Figueroa ME, Lugthart S, Li Y, Erpelinck-Verschueren C, Deng X, Christos PJ, Schifano E, Booth J, van Putten W, Skrabanek L, Campagne F, Mazumdar M, Greally JM, Valk PJM, Löwenberg B, Delwel R, Melnick A. DNA methylation signatures identify biologically distinct subtypes in acute myeloid leukemia. Cancer Cell. 2010 Apr;17(1):13–27. doi: 10.1016/j.ccr.2009.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mack SC, Witt H, Piro RM, Gu L, Zuyderduyn S, Stütz AM, Wang X, Gallo M, Garzia L, Zayne K, Zhang X, Ramaswamy V, Jäger N, Jones DT, Sill M, Pugh TJ, Ryzhova M, Wani KM, Shih DJ, Head R, Remke M, Bailey SD, Zichner T, Faria CC, Barszczyk M, Stark S, Seker-Cin H, Hutter S, Johann P, Bender S, Hovestadt V, Tzaridis T, Dubuc AM, Northcott PA, Peacock J, Bertrand KC, Agnihotri S, Cavalli FM, Clarke I, Nethery-Brokx K, Creasy CL, Verma SK, Koster J, Wu X, Yao Y, Milde T, Sin-Chan P, Zuccaro J, Lau L, Pereira S, Castelo-Branco P, Hirst M, Marra MA, Roberts SS, Fults D, Massimi L, Cho YJ, Van Meter T, Grajkowska W, Lach B, Kulozik AE, von Deimling A, Witt O, Scherer SW, Fan X, Muraszko KM, Kool M, Pomeroy SL, Gupta N, Phillips J, Huang A, Tabori U, Hawkins C, Malkin D, Kongkham PN, Weiss WA, Jabado N, Rutka JT, Bouffet E, Korbel JO, Lupien M, Aldape KD, Bader GD, Eils R, Lichter P, Dirks PB, Pfister SM, Korshunov A, Taylor MD. Epigenomic alterations define lethal CIMP-positive ependymomas of infancy. Nature. 2014 Feb 27;506(7489):445–50. doi: 10.1038/nature13108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sandoval J, Mendez-Gonzalez J, Nadal E, Chen G, Carmona FJ, Sayols S, Moran S, Heyn H, Vizoso M, Gomez A, Sanchez-Cespedes M, Assenov Y, Müller F, Bock C, Taron M, Mora J, Muscarella LA, Liloglou T, Davies M, Pollan M, Pajares MJ, Torre W, Montuenga LM, Brambilla E, Field JK, Roz L, Lo Iacono M, Scagliotti GV, Rosell R, Beer DG, Esteller M. A prognostic DNA methylation signature for stage I non-small-cell lung cancer. J Clin Oncol. 2013 Nov 10;31(32):4140–7. doi: 10.1200/JCO.2012.48.5516. [DOI] [PubMed] [Google Scholar]
- 32.Weisenberger DJ. Characterizing DNA methylation alterations from The Cancer Genome Atlas. J Clin Invest. 2014 Jan;124(1):17–23. doi: 10.1172/JCI69740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fang F1, Turcan S, Rimner A, Kaufman A, Giri D, Morris LG, Shen R, Seshan V, Mo Q, Heguy A, Baylin SB, Ahuja N, Viale A, Massague J, Norton L, Vahdat LT, Moynahan ME, Chan TA. Breast cancer methylomes establish an epigenomic foundation for metastasis. Sci Trans Med. 2011 Mar 23;3(75):75ra25. doi: 10.1126/scitranslmed.3001875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pajtler KW, Witt H, Sill M, Jones DT, Hovestadt V, Kratochwil F, Wani K, Tatevossian R, Punchihewa C, Johann P, Reimand J, Wamatz HJ, Ryzhova M, Mack S, Ramaswamy V, Capper D, Schweizer L, Sieber L, Wittmann A, Huang Z, van Sluis P, Volckmann R, Koster J, Versteeg R, Fults D, Toledano H, Avigad S, Hoffman LM, Donson AM, Foreman N, Hewer E, Zitterbart K, Gilbert M, Armstrong TS, Gupta N, Allen JC, Karajannis MA, Zagzag D, Hasselblatt M, Kulozik AE, Witt O, Collins VP, von Hoff K, Rutkowski S, Pietsch T, Bader G, Yaspo ML, von Deimling A, Lichter P, Taylor MD, Gilbertson R, Ellison DW, Aldape K, Korshunov A, Kool M, Pfister SM. Molecular Classification of Ependymal Tumors across All CNS Compartments, Histopathological Grades, and Age Groups. Cancer Cell. 2015 May 11;27(5):728–43. doi: 10.1016/j.ccell.2015.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bass AJ, Thorsson V, Shmulevich I, Reynolds SM, Miller M, Bernard B, Hinoue T, Laird PW, Curtis C, Shen H, Weisenberger DJ, Schultz N, Shen R, Weinhold N, Kelsen DP, Bowlby R, Chu A, Kasaian K, Mungall AJ, Robertson AG, Sipahimalani P, Cherniack AD, Getz G, Liu Y, Noble MS, Pedamallu C, Sougnez C, Taylor-Weiner A, Akbani R, Lee JS, Liu W, Mills GB, Yang D, Zhang W, Pantazi A, Parfenov M, Gulley M, Piazuelo MB, Schneider BG, Kim J, Boussioutas A, Sheth M, Demchok JA, Rabkin CS, Willis JE, Ng S, Garman K, Beer DG, Pennathur A, Raphael BJ, Wu HT, Odze R, Kim HK, Bowen J, Leraas KM, Lichtenberg TM, Weaver S, McLellan M, Wiznerowicz M, Sakai R, Getz G, Sougnez C, Lawrence MS, Cibulskis K, Lichtenstein L, Fisher S, Gabriel SB, Lander ES, Ding L, Niu B, Ally A, Balasundaram M, Birol I, Bowlby R, Brooks D, Butterfield YS, Carlsen R, Chu A, Chu J, Chuah E, Chun HJ, Clarke A, Dhalla N, Guin R, Holt RA, Jones SJ, Kasaian K, Lee D, Li HA, Lim E, Ma Y, Marra MA, Mayo M, Moore RA, Mungall AJ, Mungall KL, Ming Nip K, Robertson AG, Schein JE, Sipahimalani P, Tam A, Thiessen N, Beroukhim R, Carter SL, Cherniack AD, Cho J, Cibulskis K, DiCara D, Frazer S, Fisher S, Gabriel SB, Gehlenborg N, Heiman DI, Jung J, Kim J, Lander ES, Lawrence MS, Lichtenstein L, Lin P, Meyerson M, Ojesina AI, Sekhar Pedamallu C, Saksena G, Schumacher SE, Sougnez C, Stojanov P, Tabak B, Taylor-Weiner A, Voet D, Rosenberg M, Zack TI, Zhang H, Zou L, Protopopov A, Santoso N, Parfenov M, Lee S, Zhang J, Mahadeshwar HS, Tang J, Ren X, Seth S, Yang L, Xu AW, Song X, Pantazi A, Xi R, Bristow CA, Hadjipanayis A, Seidman J, Chin L, Park PJ, Kucherlapati R, Akbani R, Ling S, Liu W, Rao A, Weinstein JN, Kim SB, Lee JS, Lu Y, Mills G, Laird PW, Hinoue T, Weisenberger DJ, Bootwalla MS, Lai PH, Shen H, Triche T, Jr, Van Den Berg DJ, Baylin SB, Herman JG, Getz G, Chin L, Liu Y, Murray BA, Noble MS, Askoy BA, Ciriello G, Dresdner G, Gao J, Gross B, Jacobsen A, Lee W, Ramirez R, Sander C, Schultz N, Senbabaoglu Y, Sinha R, Sumer SO, Sun Y, Weinhold N, Thorsson V, Bernard B, Iype L, Kramer RW, Kreisberg R, Miller M, Reynolds SM, Rovira H, Tasman N, Shmulevich I, Ng S, Haussler D, Stuart JM, Akbani R, Ling S, Liu W, Rao A, Weinstein JN, Verhaak RG, Mills GB, Leiserson MD, Raphael BJ, Wu HT, Taylor BS, Black AD, Bowen J, Carney JA, Gastier-Foster JM, Helsel C, Leraas KM, Lichtenberg TM, McAllister C, Ramirez NC, Tabler TR, Wise L, Zmuda E, Penny R, Crain D, Gardner J, Lau K, Curely E, Mallery D, Morris S, Paulauskis J, Shelton T, Shelton C, Sherman M, Benz C, Lee JH, Fedosenko K, Manikhas G, Potapova O, Voronina O, Belyaev D, Dolzhansky O, Rathmell WK, Brzezinski J, Ibbs M, Korski K, Kycler W, Laźniak R, Leporowska E, Mackiewicz A, Murawa D, Murawa P, Spychala A, Suchorska WM, Tatka H, Teresiak M, Wiznerowicz M, Abdel-Misih R, Bennett J, Brown J, Iacocca M, Rabeno B, Kwon SY, Penny R, Gardner J, Kemkes A, Mallery D, Morris S, Shelton T, Shelton C, Curley E, Alexopoulou I, Engel J, Bartlett J, Albert M, Park DY, Dhir R, Luketich J, Landreneau R, Janjigian YY, Kelsen DP, Cho E, Ladanyi M, Tang L, McCall SJ, Park YS, Cheong JH, Ajani J, Camargo MC, Alonso S, Ayala B, Jensen MA, Pihl T, Raman R, Walton J, Wan Y, Demchok JA, Eley G, Mills Shaw KR, Sheth M, Tarnuzzer R, Wang Z, Yang L, Zenklusen JC, Davidsen T, Hutter CM, Sofia HJ, Burton R, Chudamani S, Liu J. Comprehensive molecular characterization of gastric adenocarcinoma. Nature. 2014 Sep 11;513(7517):202–9. doi: 10.1038/nature13480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shen L. DNA Methylation and Environmental Exposures in Human Hepatocellular Carcinoma. Cancer Spectrum Knowl Environ. 2002 May;94(10):755–761. doi: 10.1093/jnci/94.10.755. [DOI] [PubMed] [Google Scholar]
- 37.Pan H, Chen L, Dogra S, Teh AL, Tan JH, Lim YI, Lim YC, Jin S, Lee YK, Ng PY, Ong ML, Barton S, Chong YS, Meaney MJ, Gluckman PD, Stunkel W, Ding C, Holbrook JD. Measuring the methylome in clinical samples: improved processing of the Infinium Human Methylation450 BeadChip Array. Epigenetics. 2012 Oct;7(10):1173–87. doi: 10.4161/epi.22102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lowe R, Rakyan VK. Marmal-aid—a database for Infinium Human Methylation 450. BMC Bioinformatics. 2013 Jan;14:359. doi: 10.1186/1471-2105-14-359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Frommer M1, McDonald LE, Millar DS, Collis CM, Watt F, Grigg GW, Molloy PL, Paul CL. A genomic sequencing protocol that yields a positive display of 5-methylcytosine residues in individual DNA strands. Proc Natl Acad Sci U S A. 1992 Mar 1;89(5):1827–31. doi: 10.1073/pnas.89.5.1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Meissner A. Reduced representation bisulfite sequencing for comparative high-resolution DNA methylation analysis. Nucleic Acids Res. 2005 Oct;33(18):5868–5877. doi: 10.1093/nar/gki901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Down TA, Rakyan VK, Turner DJ, Flicek P, Li H, Kulesha E, Gräf S, Johnson N, Herrero J, Tomazou EM, Thorne NP, Bäckdahl L, Heiberth M, Howe KL, Jackson DK, Miretti MM, Marioni JC, Birney E, Hubbard TJ, Durbin R, Tavaré S, Beck S. A Bayesian deconvolution strategy for immunoprecipitation-based DNA methylome analysis. Nat Biotechnol. 2008 Jul;26(7):779–85. doi: 10.1038/nbt1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Deng J, Shoemaker R, Xie B, Gore A, LeProust EM, Antosiewicz-Bourget J, Egli D, Maherali N, Park IH, Yu J, Daley GQ, Eggan K, Hochedlinger K, Thomson J, Wang W, Gao Y, Zhang K. Targeted bisulfite sequencing reveals changes in DNA methylation associated with nuclear reprogramming. Nat Biotechnol. 2009 Apr;27(4):353–60. doi: 10.1038/nbt.1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Clark C, Palta P, Joyce CJ, Scott C, Grundberg E, Deloukas P, Palotie A, Coffey AJ. A comparison of the whole genome approach of MeDIP-seq to the targeted approach of the Infinium Human Methylation450 BeadChip(®) for methylome profiling. PLoS One. 2012;7(11):e50233. doi: 10.1371/journal.pone.0050233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29(1):15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Krueger F, Andrews SR. Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics. 2011 Jun;27(11):1571–2. doi: 10.1093/bioinformatics/btr167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Xi Y, Li W. BSMAP: Whole genome bisulfite sequence MAPping program. BMC Bioinformatics. 2009 Jan;10(1):232. doi: 10.1186/1471-2105-10-232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chatterjee A, Stockwell PA, Rodger EJ, Morison IM. Comparison of alignment software for genomewide bisulphite sequence data. Nucleic Acids Res. 2012 May;40(10):e79. doi: 10.1093/nar/gks150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mielczarek M, Szyda J. Review of alignment and SNP calling algorithms for next-generation sequencing data. J Appl Genet. 2016 Feb;57(1):71–9. doi: 10.1007/s13353-015-0292-7. [DOI] [PubMed] [Google Scholar]
- 50.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kemytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010 Sep;20(9):1297–303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cingolani P, Platts A, Wang le L, Coon M, Nguyen T, Wang L, Land SJ, Lu X, Ruden DM. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 2012 Apr-Jun;6(2):80–92. doi: 10.4161/fly.19695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ramos AH, Lichtenstein L, Gupta M, Lawrence MS, Pugh TJ, Saksena G, Meyerson M, Getz G. Oncotator: cancer variant annotation tool. Hum Mutat. 2015 Apr;36(4):E2423–9. doi: 10.1002/humu.22771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Roth A, Khattra J, Yap D, Wan A, Laks E, Biele J, Ha G, Aparicio S, Bouchard-Côté A, Shah SP. PyClone: statistical inference of clonal population structure in cancer. Nat Methods. 2014 Apr;11(4):396–8. doi: 10.1038/nmeth.2883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Robinson MD, McCarthy DJ, Smyth GK. EdgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010 Jan;26(1):139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Akalin A, Kormaksson M, Li S, Garrett-Bakelman FE, Figueroa ME, Melnick A, Mason CE. MethylKit: a comprehensive R package for the analysis of genome-wide DNA methylation profiles. Genome Biol. 2012 Oct;13(10):R87. doi: 10.1186/gb-2012-13-10-r87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Li S, Garrett-Bakelman FE, Akalin A, Zumbo P, Levine R, To BL, Lewis ID, Brown AL, D’Andrea RJ, Melnick A, Mason CE. An optimized algorithm for detecting and annotating regional differential methylation. BMC Bioinformatics. 2013;14(Suppl 5):S10. doi: 10.1186/1471-2105-14-S5-S10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Li S, Garrett-Bakelman F, Perl AE, Luger SM, Zhang C, To BL, Lewis ID, Brown AL, D’Andrea RJ, Ross ME, Levine R, Carroll M, Melnick A, Mason CE. Dynamic evolution of clonal epialleles revealed by methclone. Genome Biol. 2014 Sep 27;15(9):472. doi: 10.1186/s13059-014-0472-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shen R, Olshen AB, Ladanyi M. Integrative clustering of multiple genomic data types using a joint latent variable model with application to breast and lung cancer subtype analysis. Bioinformatics. 2009 Nov;25(22):2906–12. doi: 10.1093/bioinformatics/btp543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shannon P1, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003 Nov;12(11):2498–504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mo Q, Wang S, Seshan VE, Olshen AB, Schultz N, Sander C, Powers RS, Ladanyi M, Shen R. Pattern discovery and cancer gene identification in integrated cancer genomic data. Proc Natl Acad Sci U S A. 2013 Mar 12;110(11):4245–50. doi: 10.1073/pnas.1208949110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Su M, Dou X, Cheng H, Han J-DJ. In: Integrative Epigenomics. Computational and Statistical Epigenomics, Media Dordrecht 2015. Teschendorff AE, editor. Vol. 7. Springer; Netherlands: pp. 127–139. [Google Scholar]
- 62.Wang G, Jung K, Winnenburg R, Shah NH. A method for systematic discovery of adverse drug events from clinical notes. J Am Med Inform Assoc. 2015 Nov;22(6):1196–204. doi: 10.1093/jamia/ocv102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Boland MR, Tatonetti NP, Hripcsak G. Development and validation of a classification approach for extracting severity automatically from electronic health records. J Biomed Semantics. 2015 Apr 6;6:14. doi: 10.1186/s13326-015-0010-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Marsolo K, Spooner SA. Clinical genomics in the world of the electronic health record. Genet Med. 2013 Oct;15(10):786–91. doi: 10.1038/gim.2013.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mandal RS, Saha S, Das S. Metagenomic Surveys of Gut Microbiota. Genomics Proteomics Bioinformatics. 2015 Jun;13(3):148–58. doi: 10.1016/j.gpb.2015.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Drekonja D, Reich J, Gezahegn S, Greer N, Shaukat A, MacDonald R, Rutks I, Wilt TJ. Fecal microbiota transplantation for clostridium difficile infection: a systematic review. Ann Intern Med. 2015 May 5;162(9):630–8. doi: 10.7326/M14-2693. [DOI] [PubMed] [Google Scholar]
- 67.Patel CJ, Sivadas A, Tabassum R, Preeprem T, Zhao J, Arafat D, Chen R, Morgan AA, Martin GS, Brigham KL, Butte AJ, Gibson G. Whole genome sequencing in support of wellness and health maintenance. Genome Med. 2013 Jun 27;5(6):58. doi: 10.1186/gm462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Li L, Ruau DJ, Patel CJ, Weber SC, Chen R, Tatonetti NP, Dudley JT, Butte AJ. Disease risk factors identified through shared genetic architecture and electronic medical records. Sci Transl Med. 2014 Apr 30;6(234):234ra57. doi: 10.1126/scitranslmed.3007191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Rosenfeld JA, Mason CE. Pervasive sequence patents cover the entire human genome. Genome Med. 2013 Mar 25;5(3):27. doi: 10.1186/gm431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Botkin JR, Belmont JW, Berg JS, Berkman BE, Bombard Y, Holm IA, Levy HP, Ormond KE, Saal HM, Spinner NB, Wilfond BS, McInerney JD. Points to consider: ethical, legal, and psychosocial implications of genetic testing in children and adolescents. Am J Hum Genet. 2015 Jul 2;97(1):6–21. doi: 10.1016/j.ajhg.2015.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ciriello G, Miller ML, Aksoy BA, Senbabaoglu Y, Schultz N, Sander C. Emerging landscape of oncogenic signatures across human cancers. Nat Genet. 2013 Oct;45(10):1127–33. doi: 10.1038/ng.2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.T International Cancer Genome Consortium. Hudson TJ, Anderson W, Artez A, Barker AD, Bell C, Bernabé RR, Bhan MK, Calvo F, Eerola I, Gerhard DS, Guttmacher A, Guyer M, Hemsley FM, Jennings JL, Kerr D, Klatt P, Kolar P, Kusada J, Lane DP, Laplace F, Youyong L, Nettekoven G, Ozenberger B, Peterson J, Rao TS, Remacle J, Schafer AJ, Shibata T, Stratton MR, Vockley JG, Watanabe K, Yang H, Yuen MM, Knoppers BM, Bobrow M, Cambon-Thomsen A, Dressler LG, Dyke SO, Joly Y, Kato K, Kennedy KL, Nicolás P, Parker MJ, Rial-Sebbag E, Romeo-Casabona CM, Shaw KM, Wallace S, Wiesner GL, Zeps N, Lichter P, Biankin AV, Chabannon C, Chin L, Clément B, de Alava E, Degos F, Ferguson ML, Geary P, Hayes DN, Hudson TJ, Johns AL, Kasprzyk A, Nakagawa H, Penny R, Piris MA, Sarin R, Scarpa A, Shibata T, van de Vijver M, Futreal PA, Aburatani H, Bayés M, Botwell DD, Campbell PJ, Estivill X, Gerhard DS, Grimmond SM, Gut I, Hirst M, López-Otín C, Majumder P, Marra M, McPherson JD, Nakagawa H, Ning Z, Puente XS, Ruan Y, Shibata T, Stratton MR, Stunnenberg HG, Swerdlow H, Velculescu VE, Wilson RK, Xue HH, Yang L, Spellman PT, Bader GD, Boutros PC, Campbell PJ, Flicek P, Getz G, Guigó R, Guo G, Haussler D, Heath S, Hubbard TJ, Jiang T, Jones SM, Li Q, López-Bigas N, Luo R, Muthuswamy L, Ouellette BF, Pearson JV, Puente XS, Quesada V, Raphael BJ, Sander C, Shibata T, Speed TP, Stein LD, Stuart JM, Teague JW, Totoki Y, Tsunoda T, Valencia A, Wheeler DA, Wu H, Zhao S, Zhou G, Stein LD, Guigó R, Hubbard TJ, Joly Y, Jones SM, Kasprzyk A, Lathrop M, López-Bigas N, Ouellette BF, Spellman PT, Teague JW, Thomas G, Valencia A, Yoshida T, Kennedy KL, Axton M, Dyke SO, Futreal PA, Gerhard DS, Gunter C, Guyer M, Hudson TJ, McPherson JD, Miller LJ, Ozenberger B, Shaw KM, Kasprzyk A, Stein LD, Zhang J, Haider SA, Wang J, Yung CK, Cros A, Liang Y, Gnaneshan S, Guberman J, Hsu J, Bobrow M, Chalmers DR, Hasel KW, Joly Y, Kaan TS, Kennedy KL, Knoppers BM, Lowrance WW, Masui T, Nicolás P, Rial-Sebbag E, Rodriguez LL, Vergely C, Yoshida T, Grimmond SM, Biankin AV, Bowtell DD, Cloonan N, deFazio A, Eshleman JR, Etemadmoghadam D, Gardiner BB, Kench JG, Scarpa A, Sutherland RL, Tempero MA, Waddell NJ, Wilson PJ, McPherson JD, Gallinger S, Tsao MS, Shaw PA, Petersen GM, Mukhopadhyay D, Chin L, DePinho RA, Thayer S, Muthuswamy L, Shazand K, Beck T, Sam M, Timms L, Ballin V, Lu Y, Ji J, Zhang X, Chen F, Hu X, Zhou G, Yang Q, Tian G, Zhang L, Xing X, Li X, Zhu Z, Yu Y, Yu J, Yang H, Lathrop M, Tost J, Brennan P, Holcatova I, Zaridze D, Brazma A, Egevard L, Prokhortchouk E, Banks RE, Uhlén M, Cambon-Thomsen A, Viksna J, Ponten F, Skryabin K, Stratton MR, Futreal PA, Birney E, Borg A, Børresen-Dale AL, Caldas C, Foekens JA, Martin S, Reis-Filho JS, Richardson AL, Sotiriou C, Stunnenberg HG, Thoms G, van de Vijver M, van’t Veer L, Calvo F, Birnbaum D, Blanche H, Boucher P, Boyault S, Chabannon C, Gut I, Masson-Jacquemier JD, Lathrop M, Pauporté I, Pivot X, Vincent-Salomon A, Tabone E, Theillet C, Thomas G, Tost J, Treilleux I, Calvo F, Bioulac-Sage P, Clément B, Decaens T, Degos F, Franco D, Gut I, Gut M, Heath S, Lathrop M, Samuel D, Thomas G, Zucman-Rossi J, Lichter P, Eils R, Brors B, Korbel JO, Korshunov A, Landgraf P, Lehrach H, Pfister S, Radlwimmer B, Reifenberger G, Taylor MD, von Kalle C, Majumder PP, Sarin R, Rao TS, Bhan MK, Scarpa A, Pederzoli P, Lawlor RA, Delledonne M, Bardelli A, Biankin AV, Grimmond SM, Gress T, Klimstra D, Zamboni G, Shibata T, Nakamura Y, Nakagawa H, Kusada J, Tsunoda T, Miyano S, Aburatani H, Kato K, Fujimoto A, Yoshida T, Campo E, López-Otín C, Estivill X, Guigó R, de Sanjosé S, Piris MA, Montserrat E, González-Díaz M, Puente XS, Jares P, Valencia A, Himmelbauer H, Quesada V, Bea S, Stratton MR, Futreal PA, Campbell PJ, Vincent-Salomon A, Richardson AL, Reis-Filho JS, van de Vijver M, Thomas G, Masson-Jacquemier JD, Aparicio S, Borg A, Borresen-Dale AL, Caldas C, Foekens JA, Stunnenberg HG, van’t Veer L, Easton DF, Spellman PT, Martin S, Barker AD, Chin L, Collins FS, Compton CC, Ferguson ML, Gerhard DS, Getz G, Gunter C, Guttmacher A, Guyer M, Hayes DN, Lander ES, Ozenberger B, Penny R, Peterson J, Sander C, Shaw KM, Speed TP, Spellman PT, Vockley JG, Wheeler DA, Wilson RK, Hudson TJ, Chin L, Knoppers BM, Lander ES, Lichter P, Stein LD, Stratton MR, Anderson W, Barker AD, Bell C, Bobrow M, Burke W, Collins FS, Compton CC, DePinho RA, Easton DF, Futreal PA, Gerhard DS, Green AR, Guyer M, Hamilton SR, Hubbard TJ, Kallioniemi OP, Kennedy KL, Ley TJ, Liu ET, Lu Y, Majumder P, Marra M, Ozenberger B, Peterson J, Schafer AJ, Spellman PT, Stunnenberg HG, Wainwright BJ, Wilson RK, Yang H. International network of cancer genome projects. Nature. 2010 Apr 15;464(7291):993–8. doi: 10.1038/nature08987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, Antipin Y, Reva B, Goldberg AP, Sander C, Schultz N. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012 May;2(5):401–4. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Chiron D, Di Liberto M, Martin P, Huang X, Sharman J, Blecua P, Mathew S, Vijay P, Eng K, Ali S, Johnson A, Chang B, Ely S, Elemento O, Mason CE, Leonard JP, Chen-Kiang S. Cell-cycle reprogramming for PI3K inhibition overrides a relapse-specific C481S BTK mutation revealed by longitudinal functional genomics in mantle cell lymphoma. Cancer Discov. 2014 Sep;4(9):1022–35. doi: 10.1158/2159-8290.CD-14-0098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, Dummer R, Garbe C, Testori A, Maio M, Hogg D, Lorigan P, Lebbe C, Jouary T, Schadendorf D, Ribas A, O’Day SJ, Sosman JA, Kirkwood JM, Eggermont AM, Dreno B, Nolop K, Li J, Nelson B, Hou J, Lee RJ, Flaherty KT, McArthur GA, BRIM-3 Study Group Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011 Jun 30;364(26):2507–16. doi: 10.1056/NEJMoa1103782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Robert C, Karaszewska B, Schachter J, Rutkowski P, Mackiewicz A, Stroiakovski D, Lichinitser M, Dummer R, Grange F, Mortier L, Chiarion-Sileni V, Drucis K, Krajsova I, Hauschild A, Lorigan P, Wolter P, Long GV, Flaherty K, Nathan P, Ribas A, Martin AM, Sun P, Crist W, Legos J, Rubin SD, Little SM, Schadendorf D. Improved Overall Survival in Melanoma with Combined Dabrafenib and Trametinib. N Engl J Med. 2015 Jan 1;372(1):30–9. doi: 10.1056/NEJMoa1412690. [DOI] [PubMed] [Google Scholar]
- 77.Costello JC, Heiser LM, Georgii E, Gönen M, Menden MP, Wang NJ, Bansal M, Ammad-ud-din M, Hintsanen P, Khan SA, Mpindi JP, Kallioniemi O, Honkela A, Aittokallio T, Wennerberg K, NCI DREAM Community. Collins JJ, Gallahan D, Singer D, Saez-Rodriguez J, Kaski S, Gray JW, Stolovitzky G. A community effort to assess and improve drug sensitivity prediction algorithms. Nat Biotechnol. 2014 Dec;32(12):1202–12. doi: 10.1038/nbt.2877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Shoemaker RH. The NCI60 human tumour cell line anticancer drug screen. Nat Rev Cancer. 2006 Oct;6(10):813–23. doi: 10.1038/nrc1951. [DOI] [PubMed] [Google Scholar]
- 79.Lamb J, Crawford ED, Peck D, Modell JW, Blat IC, Wrobel MJ, Lerner J, Brunet JP, Subramanian A, Ross KN, Reich M, Hieronymus H, Wei G, Armstrong SA, Haggarty SJ, Clemons PA, Wei R, Carr SA, Lander ES, Golub TR. The Connectivity Map: using gene-expression signatures to connect small molecules, genes, and disease. Science. 2006 Sep 29;313(5795):1929–35. doi: 10.1126/science.1132939. [DOI] [PubMed] [Google Scholar]
- 80.Shameer K, Readhead B, Dudley JT. Computational and experimental advances in drug repositioning for accelerated therapeutic stratification. Curr Top Med Chem. 2015;15(1):5–20. doi: 10.2174/1568026615666150112103510. [DOI] [PubMed] [Google Scholar]
- 81.Perez-Diez A, Morgun A, Shulzhenko N. Microarrays for Cancer Diagnosis and Classification. Austin TX: Landes Bioscience; 2000. pp. 2–13. [DOI] [PubMed] [Google Scholar]
- 82.Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, Jr, Kinzler KW. Cancer genome landscapes. Science. 2013 Mar 29;339(6127):1546–58. doi: 10.1126/science.1235122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Landau DA, Carter SL, Getz G, Wu CJ. Clonal evolution in hematological malignancies and therapeutic implications. Leukemia. 2014 Jan;28(1):34–43. doi: 10.1038/leu.2013.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Nowell PC. The clonal evolution of tumor cell populations. Science. 1976;194:23–28. doi: 10.1126/science.959840. [DOI] [PubMed] [Google Scholar]
- 85.Shalek AK, Satija R, Adiconis X, Gertner RS, Gaublomme JT, Raychowdhury R, Schwartz S, Yosef N, Malboeuf C, Lu D, Trombetta JJ, Gennert D, Gnirke A, Goren A, Hacohen N, Levin JZ, Park H, Regev A. Single-cell transcriptomics reveals bimodality in expression and splicing in immune cells. Nature. 2013 Jun 13;498(7453):236–40. doi: 10.1038/nature12172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Navin N, Kendall J, Troge J, Andrews P, Rodgers L, McIndoo J, Cook K, Stepansky A, Levy D, Esposito D, Muthuswamy L, Krasnitz A, McCombie WR, Hicks J, Wigler M. Tumour evolution inferred by single-cell sequencing. Nature. 2011 Apr 7;472(7341):90–94. doi: 10.1038/nature09807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Hou Y, Fan W, Yan L, Li R, Lian Y, Huang J, Li J, Xu L, Tang F, Xie XS, Qiao J. Genome analyses of single human oocytes. Cell. 2013 Dec 19;155(7):1492–506. doi: 10.1016/j.cell.2013.11.040. [DOI] [PubMed] [Google Scholar]
- 88.Patel AP, Tirosh I, Trombetta JJ, Shalek AK, Gillespie SM, Wakimoto H, Cahill DP, Nahed BV, Curry WT, Martuza RL, Louis DN, Rozenblatt-Rosen O, Suvà ML, Regev A, Bernstein BE. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science. 2014 Jun 20;344(6190):1396–401. doi: 10.1126/science.1254257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Karczewski KJ, Fernald GH, Martin AR, Snyder M, Tatonetti NP, Dudley JT. STORMSeq: an open-source, user-friendly pipeline for processing personal genomics data in the cloud. PLoS One. 2014 Jan 15;9(1):e84860. doi: 10.1371/journal.pone.0084860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Shyr C, Kushniruk A, van Karnebeek CD, Wasserman WW. Dynamic software design for clinical exome and genome analyses: insights from bioinformaticians, clinical geneticists, and genetic counselors. J Am Med Inform Assoc. 2016 Mar;23(2):257–68. doi: 10.1093/jamia/ocv053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Han G, Sun J, Wang J, Bai Z, Song F, Lei H. Genomics in neurological disorders. Genomics Proteomics Bioinformatic. 2014 Aug;12(4):156–63. doi: 10.1016/j.gpb.2014.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Manolio TA, Abramowicz M, Al-Mulla F, Anderson W, Balling R, Berger AC, Bleyl S, Chakravarti A, Chantratita W, Chisholm RL, Dissanayake VH, Dunn M, Dzau VJ, Han BG, Hubbard T, Kolbe A, Korf B, Kubo M, Lasko P, Leego E, Mahasirimongkol S, Majumdar PP, Matthijs G, McLeod HL, Metspalu A, Meulien P, Miyano S, Naparstek Y, O’Rourke PP, Patrinos GP, Rehm HL, Relling MV, Rennert G, Rodriguez LL, Roden DM, Shuldiner AR, Sinha S, Tan P, Ulfendahl M, Ward R, Williams MS, Wong JE, Green ED, Ginsburg GS. Global implementation of genomic medicine: we are not alone. Sci Transl Med. 2015 Jun 3;7(290):290ps13. doi: 10.1126/scitranslmed.aab0194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Li S, Garrett-Bakelman FE, Chung SS, Sanders MA, Hricik T, Rapaport F, Patel J, Dillon R, Vijay P, Brown AL, Perl AE, Cannon J, Bullinger L, Luger S, Becker M, Lewis ID, To LB, Delwel R, Löwenberg B, Döhner H, Döhner K, Guzman ML, Hassane DC, Roboz GJ, Grimwade D, Valk PJ, D’Andrea RJ, Carroll M, Park CY, Neuberg D, Levine R, Melnick AM, Mason CE. Distinct evolution and dynamics of epigenetic and genetic heterogeneity in acute myeloid leukemia. Nature Medicine. 2016 Jul;22(7):792–9. doi: 10.1038/nm.4125. [DOI] [PMC free article] [PubMed] [Google Scholar]