

Abstract
The tumor-suppressor protein p53 is tightly controlled in normal cells by its two negative regulators—the E3 ubiquitin ligase MDM2 and its homolog MDMX. Under stressed conditions such as DNA damage, p53 escapes MDM2- and MDMX-mediated functional inhibition and degradation, acting to prevent damaged cells from proliferating through induction of cell cycle arrest, DNA repair, senescence or apoptosis. Ample evidence suggests that stress signals induce phosphorylation of MDM2 and MDMX, leading to p53 activation. However, the structural basis of stress-induced p53 activation remains poorly understood because of the paucity of technical means to produce site-specifically phosphorylated MDM2 and MDMX proteins for biochemical and biophysical studies. Herein, we report total chemical synthesis, via native chemical ligation, and functional characterization of (24–108) MDMX and its Tyr99-phosphorylated analog with respect to their ability to interact with a panel of p53-derived peptide ligands and PMI, a p53-mimicking but more potent peptide antagonist of MDMX, using FP and surface plasmon resonance techniques. Phosphorylation of MDMX at Tyr99 weakens peptide binding by approximately two orders of magnitude. Comparative X-ray crystallographic analyses of MDMX and of pTyr99 MDMX in complex with PMI as well as modeling studies reveal that the phosphate group of pTyr99 imposes extensive steric clashes with the C-terminus of PMI or p53 peptide and induces a significant lateral shift of the peptide ligand, contributing to the dramatic decrease in the binding affinity of MDMX for p53. Because DNA damage activates c-Abl tyrosine kinase that phosphorylates MDMX at Tyr99, our findings afford a rare glimpse at the structural level of how stress-induced MDMX phosphorylation dislodges p53 from the inhibitory complex and activates it in response to DNA damage.
INTRODUCTION
The tumor-suppressor protein p53 is activated as a transcription factor to induce powerful growth-inhibitory and apoptotic responses to cellular stress, but is otherwise tightly controlled in normal cells by the E3 ubiquitin ligase MDM2 and its homolog MDMX.1–4 MDM2 primarily targets p53 for proteasomal degradation, and MDMX mainly functions as a transcriptional antagonist of p53. Growing evidence suggests that MDMX also heterodimerizes with MDM2 to enhance E2 recruitment and augment p53 degradation.5 Whereas deletion of MDM2 or MDMX is embryonic lethal owing to uncontrolled p53 activity,6–8 overexpression or amplification of either gene, as found in many tumors harboring wild-type TP53, is oncogenic owing to p53 inactivation. A double knockout of both the TP53 and MDM2 or MDMX genes, however, fully rescues embryos from lethality, demonstrating the critical and nonredundant regulatory roles of MDM2 and MDMX in the p53 pathway.6–8 Understanding the molecular mechanisms by which MDM2 and MDMX negatively regulate p53 activity and stability is of fundamental importance in cancer biology and therapy.9–11
MDM2 and MDMX phosphorylation, mediated by stress-induced protein kinases, is thought to be chiefly responsible for p53 activation and stabilization in response to DNA damage.5,12,13 In vitro and in vivo studies have pinpointed various phosphorylation sites in MDM2 and MDMX and provided ample evidence that phosphorylation debilitates MDM2 and MDMX to bind p53,14,15 accelerates MDM2 and MDMX degradation,16–19 and impairs the ability of MDM2 to target p53 for degradation.20 Despite significant progress in understanding stress-induced p53 activation at the cellular level, the precise molecular mechanisms of phosphorylation-induced p53 activation remain poorly understood and, in some cases, even controversial.14,21–24
The stress-activated, non-receptor tyrosine kinase, c-Abl, phosphorylates both MDM2 and MDMX to activate p53 in response to DNA damage.15,25,26 For MDMX, both Tyr55 and Tyr99 in the p53-binding domain (equivalent to Tyr56 and Tyr100 in MDM2) have been identified as the major sites of phosphorylation by c-Abl.15 In fact, Tyr99 phosphorylation is implicated in immunoblotting and immunoprecipitation assays as a detriment to MDMX binding to p53.15 Modeling studies of MDMX suggest that Tyr99 phosphorylation would create a steric clash between the phosphate group and Pro27 of p53, thus destabilizing the p53-MDMX complex.15,27 However, definitive and direct biochemical and structural evidence is lacking in the absence of studies of site-specifically phosphorylated MDMX analogs with respect to their ability to bind p53. This paucity is compounded by limited availability of state-of-the-art technologies in site-specific protein phosphorylation, a common deficiency in the field of signal transduction. As a consequence, it has been difficult to decipher the precise physiological role of MDM2/MDMX phosphorylation and how it switches on the p53 pathway in response to genotoxic stress.
We chemically synthesized the p53-binding domain of MDMX and its pTyr99 analog using the native chemical ligation technique,28,29 and comparatively quantified their interactions with a potent peptide antagonist of MDMX termed PMI30 and a panel of p53-derived peptide ligands using fluorescence polarization (FP) and surface plasmon resonance techniques. The crystal structure of pTyr99 MDMX in complex with PMI was also determined. Our data shed important light on the molecular basis of how MDMX phosphorylation at Tyr99 induces p53 activation in response to cellular stress.
RESULTS
Total chemical synthesis of MDMX proteins via native chemical ligation
Residues 24–108 of MDMX encompass its p53-binding domain.30 The total chemical synthesis of (24–108) MDMX was carried out by ligating (24–75) MDMX-αCOSR (R= CH2CH2CO-Leu) to (76–108) MDMX. Both peptide fragments were synthesized using Boc chemistry for solid phase peptide synthesis. To introduce pTyr99 to MDMX, however, pTyr99-(76–108) MDMX was prepared using standard Fmoc chemistry as the phosphate group is acid labile. The two ligation reactions went to completion in 6 h (Supplementary Figure S1), yielding (24–108) MDMX and pTyr99-(24–108) MDMX (referred to thereafter as MDMX and pTyr99 MDMX, respectively) purified to homogeneity and verified by mass spectrometry (Supplementary Figure S2).
Both MDMX and pTyr99 MDMX folded spontaneously in aqueous solution as the chemical denaturant GuHCl was stripped through dialysis. Circular dichroism spectroscopic analysis verified their correct folding. As shown in Supplementary Figure S3, both proteins displayed nearly identical circular dichroism spectra with double negative peaks at 208 and 222 nM and a single positive peak at 195 nM, characteristic of alpha-helical secondary structure and consistent with the known structural features of MDMX.27,30 As expected for single domain globular proteins, MDMX and pTyr99 MDMX unfolded in the presence of increasing concentrations of GuHCl, conforming to a two-state (single transition) cooperative denaturation process as monitored by circular dichroism measurements at 222 nM (Supplementary Figure S4). Not surprisingly, phosphorylation of MDMX at Tyr99 slightly stabilized the protein by 0.3 kcal/mol. Taken together, these data suggest that Tyr99 phosphorylation is well tolerated structurally with little impact on MDMX folding.
Tyr99 phosphorylation is detrimental to MDMX interactions with peptide ligands
To evaluate the functional impact of Tyr99 phosphorylation on p53 binding to MDMX, we used two quantitative tools previously developed for MDM2/MDMX interactions with their peptide and small-molecule ligands: FP and surface plasmon resonance.24,30–32 The transactivation peptide of p53, that is, (15–29) p53 (SQETFSDLWKLLPEN), was fluorescently labeled with TAMRA at Lys24, a solvent-exposed non-contact residue with its lysyl side chain projecting outward and away from the p53-MDMX binding interface.27 Because peptide-protein complex has a higher FP than peptide alone, titration of an increasing amount of MDMX or pTyr99 MDMX to a fixed concentration of TAMRA-(15–29) p53 generated a dose-dependent increase in FP (Figure 1a), which formed the basis for quantification of an equilibrium dissociation constant, Kd, in solution. As shown in Figure 1a, the Kd value of MDMX for the p53 peptide increased from 119 nM to 9.3 μM—a 78-fold reduction in binding affinity as the result of MDMX phosphorylation at Tyr99.
Figure 1.
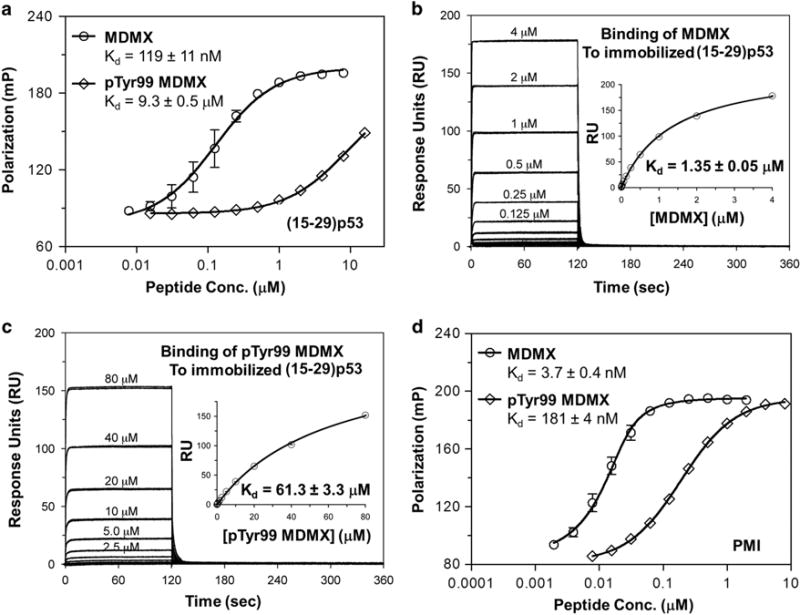
Binding affinities of (15–29) p53 (a–c) and PMI (d) for MDMX and pTyr99 MDMX as quantified by FP (a and d) and surface plasmon resonance (SPR) (b and c) techniques. For FP measurements at room temperature on a Tecan Infinite M1000 plate reader, succinimidyl ester-activated carboxytetramethylrhodamine (TAMRA-NHS) was covalently conjugated to the Lys24 side chain of an N-acetyl-(15–29) p53 and the N-terminus of PMI. Serially diluted MDMX protein was prepared in PBS (pH 7.4) in 96-well plates and incubated for 30 min with 20 nM TAMRA-labeled peptide in a total volume of 100 μl per well before readings were taken at λex =530 nM, λem= 580 nM. Non-linear regression analyses were performed as described to give rise to Kd values (mean ± s.e.m., n = 3); each curve is the mean of three independent measurements with the error bars denoting s.e.m. SPR-based steady-state binding assays were carried out at 25 °C on a Biacore T100 instrument using a CM5 sensor chip to which (15–29) p53 is covalently attached via its N-terminus or Lys24 side chain. The buffer (HBS-EP) was 10 mM HEPES, 150 mM NaCl and 0.005% surfactant P20, pH 7.4. MDMX proteins prepared in HBS-EP buffer in a twofold serial dilution were injected onto the p53 peptide-immobilized sensor chip at a flow rate of 20 μl/min for 2 min, followed by 4 min dissociation. Non-linear regression analyses were performed as described to yield Kd values (mean ±s.e.m., n =2); each curve is the mean of two independent measurements with the error bars denoting s.e.m.
To verify these results, we immobilized (15–29) p53 via the amino group of its N-terminus or Lys24 side chain on a CM5 biosensor chip, and quantified direct binding of the p53 peptide to MDMX and pTyr99 MDMX using the surface plasmon resonance technique. Shown in Figures 1b and c are steady-state binding isotherms of varying concentrations of MDMX protein, from which Kd values of 1.35 and 61.3 μM were derived for MDMX and pTyr99 MDMX, respectively. Apparently, Tyr99 phosphorylation weakened the p53-MDMX interaction by 45-fold, consistent with the results obtained from FP studies.
PMI is a dodecapeptide (TSFAEYWNLLSP) discovered via phage display that mimics p53 but binds to MDM2/MDMX approximately two orders of magnitude stronger than (15–29) p53.30 We N-terminally labeled PMI with TAMRA and quantified its interaction with MDMX and pTyr99 MDMX using the FP technique. As shown in Figure 1d, Tyr99 phosphorylation substantially reduced the binding affinity of PMI for MDMX by 49-fold, as evidenced by an increase in Kd from 3.7 to 181 nM. Taken together, these data from our direct binding assays using two different techniques clearly demonstrate that phosphorylation of MDMX at Tyr99 is functionally detrimental with respect to its binding to peptide ligands.
Deleterious functional effects of Tyr99 phosphorylation exacerbate as the p53 peptide lengthens
Recent experimental evidence indicates that the binding affinity of MDM2/MDMX for p53-derived peptides is dependent on the size of the ligand.24 We comparatively characterized MDMX and pTyr99 MDMX interactions with three p53-derived peptides differing in length: (17–28) p53, (15–29) p53 and (1–44) p53. To simplify quantification, we developed a FP-based competitive binding assay, where different concentrations of an unlabeled p53 peptide ligand were added to a preformed TAMRA-PMI-MDMX complex to compete off TAMRA-PMI. As expected, as the peptide concentration increased, FP progressively decreased (Figure 2), which, after a non-linear regression analysis, gave rise to IC50 values or peptide concentrations at which 50% of the TAMRA-PMI-MDMX complex dissociated (Table 1).
Figure 2.
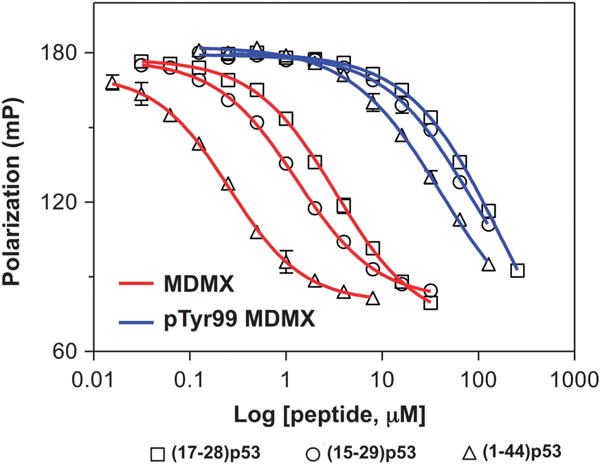
Competitive binding of p53 peptides of different lengths to MDMX and pTyr99 MDMX as measured by FP techniques. Serially diluted p53 peptide was prepared in PBS (pH 7.4), to which 10 nM TAMRA-PMI and 50 nM MDMX or 1 μM pTyr99 MDMX were added in a total volume of 120 μl per well. After a 30-min incubation at room temperature, FP was measured at λex = 530 nM, λem = 580 nM. Nonlinear regression analyses were performed as described to give rise to half maximal inhibitory concentration (IC50) values from three independent experiments. IC50 values are tabulated in Table 1.
Table 1.
Half maximal inhibitory concentrations of p53 peptides (IC50 ± s.d.) for MDMX and pTyr99 MDMX as determined by a fluorescence polarization-based competitive binding assay
| Peptide | IC50 (nM) MDMX | IC50 (μM) pTyr99 MDMX | IC50 ratio |
|---|---|---|---|
| Effects of p53 peptide length | |||
| (17–28) p53 | 3186 ± 520 | 118 ± 25 | 37 |
| (15–29) p53 | 1193 ± 354 | 90.3 ± 15.0 | 76 |
| (1–44) p53 | 260 ± 57 | 41.6 ± 5.3 | 160 |
| Effects of P27A mutation | |||
| (17–28) p53 | 3186 ± 520 | 118 ± 25 | 37 |
| P27A-(17–28) p53 | 1193 ± 52 | 20.1 ± 3.1 | 17 |
Abbreviation: IC, inhibitory concentration.
As expected, all three p53 peptides bound substantially weaker to pTyr99 MDMX than to MDMX. An increase in the size of the p53 peptide ligand invariably improved its binding to both MDMX proteins. The improvement was more pronounced for MDMX than for pTyr99 MDMX. For (15–29) p53, the ratio of IC50 of pTyr99 MDMX to MDMX was found to be 76, nearly identical to the 78-fold reduction in Kd measured by FP techniques for the two MDMX proteins. Truncation of (15–29) p53 by three amino acids to (17–28) p53 lowered the ratio of IC50 to 37. By sharp contrast, lengthening (15–29) p53 to (1–44) p53 augmented the ratio of IC50 to 160. These data demonstrate that the longer the peptide ligand is, the greater the deleterious functional effect of Tyr99 phosphorylation will become. Because full-length p53 is a protein of 393 amino acid residues, an immediate implication of these findings is that in vivo phosphorylation of MDMX at Tyr99 may be far more effective in dislodging and activating p53 than indicated biochemically with these small peptide ligands.
Pro27 of p53 is partially responsible for the deleterious functional effect of Tyr99 phosphorylation
Pro27 is highly conserved in p53. However, Pro27 as a known helix breaker debilitates the p53 transactivation peptide from adopting a more extended and stable helical conformation favorable for MDM2/MDMX binding. In fact, we previously found that the P27A mutation enhanced (17–28) p53 binding affinity by 10-fold for MDM2 and 3-fold for MDMX.31 Structural studies of MDMX in complex with (17–37) p53 suggested that phosphorylation of Tyr99 would create a steric clash between the phosphate group and the side chain of Pro27 of p53.27 To verify this functionally, we determined the IC50 values of P27A-(17–28) p53 in the FP-based competitive binding assay (Figure 3 and Table 1). Consistent with our previous finding, the P27A mutation decreased the IC50 value of (17–28) p53 for MDMX by threefold. By contrast, replacement of Pro27 by Ala lowered the IC50 value of the p53 peptide for pTyr99 MDMX by sixfold. The more pronounced enhancement in binding seen with P27A-(17–28) p53 and pTyr99 MDMX is indicative of the existence of a steric clash between Pro27 of p53 and pTyr99 of MDMX in the complex. However, on the basis of the ratios of IC50 of MDMX to pTyr99 MDMX, 17-fold for P27A-(17–28) p53 is only modestly lower than 37-fold for (17–28) p53, suggesting that Pro27 is only partially responsible for the deleterious functional effect of Tyr99 phosphorylation. This is understandable as the IC50 ratio for P27A-(17–28) p53 would have approached 1 if Pro27 were the sole culprit. Further, the amino acid residue in PMI structurally equivalent to Pro27 of p53 is Ser11. Yet, the binding affinity of PMI for pTyr99 MDMX is 49-fold weaker than that for MDMX (Figure 1d), in support of the above conclusion that Pro is only partially responsible. It is plausible that a global conformational change in p53 or PMI not restricted to local clashes involving Pro27 or Ser11 side chains may have occurred to justify the huge deleterious effect of Tyr99 phosphorylation.
Figure 3.
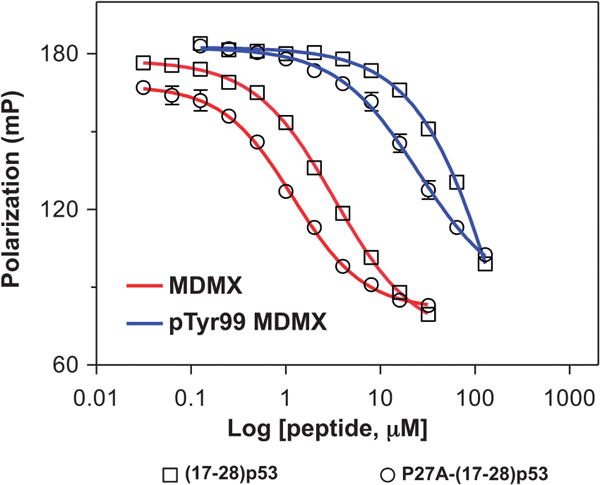
Competitive binding of P27A-(17–28) p53 peptide to MDMX and pTyr99 MDMX as measured by FP techniques. The experimental parameters were described in Figure 2 caption. For comparison and clarity, the competition curves of (17–28) p53 from Figure 2 were re-plotted in this figure.
Structural basis of how stress-induced MDMX phosphorylation activates p53
We previously determined the crystal structure of (24–108) MDMX in complex with PMI.30 To better understand why phosphorylation of Tyr99 reduced the binding affinity of PMI for MDMX by nearly two orders of magnitude, we solved the crystal structure of the PMI-pTyr99 MDMX complex at 1.55 Å resolution. As shown in Figure 4, the PMI peptide binds within the hydrophobic p53-binding cleft of pTyr99 MDMX in a mode closely resembling its binding to the wild-type MDMX, where the interaction is energetically dominated by the three non-polar residues, Phe3, Trp7 and Leu10.
Figure 4.
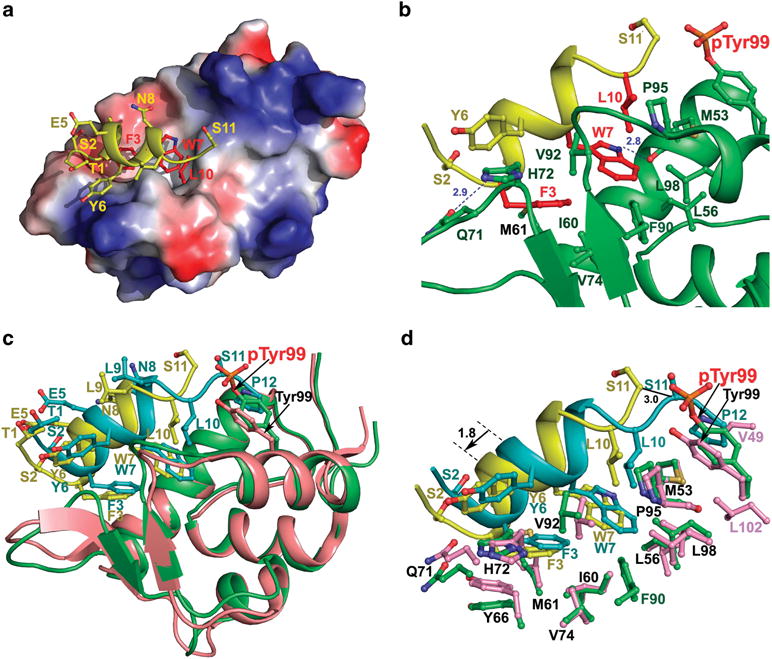
PMI in complex with pTyr99 MDMX (a and b) and structural comparison of PMI-pTyr99 MDMX and PMI-MDMX complexes (c and d). (a) Electrostatic potential is displayed over the molecular surface of pTyr99 MDMX to show the hydrophobic pocket accommodating the most critical residues for binding, Phe3, Trp7 and Leu10 of PMI (shown in red). The electrostatic potential surface is colored red for negative, blue for positive and white for apolar. The PMI peptide is shown as a ribbon and stick representation, where nitrogen atoms are colored blue and oxygen atoms red. (b) Interface of the PMI-pTyr99 MDMX complex. The PMI peptide and pTyr99 MDMX are shown as ribbons, and the side chains at the interface are shown as sticks. Hydrogen bonds are shown as blue dashes. PMI anchors Phe3, Trp7 and Leu10 within the hydrophobic p53-binding pocket lined with Met53, Leu56, Val74, Met61, Phe90, Pro95 and Leu98. The predominant hydrophobic interactions of Phe3, Trp7 and Leu10 are supplemented with two hydrogen bonds involving Phe3 and Trp7 and formed between Phe3 N and Gln71 Oε1 (2.9Å) and between Trp7 Nε1 and Met53 O (2.8Å). (c) PMI-pTyr99 MDMX (yellow/green) and PMI-MDMX (cyan/pink) complexes are aligned based on MDMX molecule from the PMI-MDMX complex (PDB code 3EQY). Residues of PMI and pTyr99/Tyr99 are shown as sticks. (d) Close-up view into the interfaces of PMI-pTyr99 MDMX and PMI-MDMX complexes. Ser2, Phe3, Tyr6, Trp7 and Leu10, Ser11 and Pro12 of PMI, and residues of pTyr99 MDMX and MDMX contributing to the binding are shown as sticks.
Phosphorylation of Tyr99 of MDMX, however, induces a significant change to the position of PMI in its binding cavity. In the PMI-pTyr99 MDMX complex, the bulky side chain of pTyr99 forms a rim that blocks binding of the two C-terminal residues of the peptide, that is, Ser11 and Pro12, and pushes the C-terminus of PMI outside of the binding pocket (Figures 4a and b). In fact, no electron density was observed for Pro12 of PMI in the complex and poorly defined density for Ser11 was observed in one of the two copies of complex present in the asymmetric unit (Supplementary Figure S5), indicating that this region is largely disordered. By contrast, when MDMX is non-phosphorylated, the C-terminus of PMI is structurally ordered (Figures 4c and d), with Ser11 N forming an H-bond to Tyr99 Oη and Pro12 side chain situated in a shallow hydrophobic cleft formed by Val49, Met53, Tyr99 and Leu102.30
Ala-scan mutagenesis studies indicated that the side chains of Ser11 and Pro12, together, contributed only marginally to the binding of PMI to MDMX.31 Despite severe steric clashes imposed by the phosphate group of pTyr99 that result in Ser11 moving away by 3 Å (Figure 4d), loss of the Ser11-Tyr99 H-bonding and side chain interactions involving Pro12 of PMI, alone, is unlikely to account for the two orders of magnitude reduction in binding affinity as the consequence of Tyr99 phosphorylation. A substantial structural change also comes from a lateral shift of PMI upon Tyr99 phosphorylation, by approximately 1.8 Å from its original position in relation to the p53-binding pocket of MDMX (Figures 4c and d); the side chains of Phe3, Trp7 and Leu10 all recess from their respective binding cavities. Compensatory conformational changes resulting from Tyr99 phosphorylation were observed to MDMX that accommodated, to some extent, Phe3, Trp7 and Leu10 side chains in their new positions. For example, although Tyr99 phosphorylation breaks off hydrophobic interactions with PMI afforded by Leu102 and Val49 of MDMX (Figure 4d), Phe90 side chain of MDMX shifts to form a new apolar interaction with a recessed Trp7 of PMI (Figure 4d). Further, the two H-bonds formed between Phe3 N and Gln71 Oε1 and between Trp7 Nε1 and Met53 O are largely maintained, independent of Tyr99 phosphorylation, in both the PMI-pTyr99 MDMX and PMI-MDMX complexes. However, calculations using PISA Interface indicate that the lateral shift of PMI reduces the buried surface areas of Phe3 and Trp7 by approximately 10%, sufficient to cause detrimental effects on binding energetics of PMI–MDMX interactions.
DISCUSSION
Site-specifically phosphorylated proteins are difficult to attain for functional and structural studies. To elucidate the importance of protein phosphorylation at sites of Ser and Thr, biochemists often resort to Ser/Thr-to-Asp/Glu mutations to mimic phosphorylation. However, because a phosphorylated side chain of Ser/Thr differs considerably in chemical structure from the carboxylate group of Asp/Glu, these ‘knock-in’ mutations cannot faithfully recapitulate phospho-Ser/Thr functionality.33 On the other hand, no natural amino acid mimicking phospho-Tyr exists in the genetic code. These dilemmas often limit scientists to the use of ‘knock-out’ mutations such as Ser/Thr-to-Ala and Tyr-to-Phe to prevent phosphorylation.
Several powerful technologies are now available that allow for site-specific incorporation of phospho-amino acids or mimetics into proteins, including total chemical protein synthesis via native chemical ligation,28,29,34 protein semisynthesis via expressed protein ligation33,35,36 and protein in vitro translation via nonsense codon suppression.37 For example, expressed protein ligation was used to introduce phosphonomethylene-phenylalanine as a nonhydrolyzable phosphotyrosine (pTyr) mimetic into the C-terminus of SHP-2, a multi-domain protein tyrosine phosphatase that has an important role in cell signaling.38,39 Kent and colleagues28,29 pioneered native chemical ligation—a revolutionary synthetic methodology that enables two or more fully unprotected synthetic peptides to react chemoselectively in aqueous solution, yielding a longer polypeptide chain linked in native peptide bonds. Since its debut, native chemical ligation has been widely used for total synthesis of many domain-sized proteins with high efficiency.34 Using native chemical ligation, we have recently synthesized a phosphorylated MDM2 protein, pSer17-(1–109) MDM2, helping clarify a controversial role of MDM2 phosphorylation at Ser17 in stress-induced p53 activation.24 This work further demonstrates total chemical protein synthesis aided by native chemical ligation as a powerful tool for site-specific incorporation of phospho-amino acids or mimetics into proteins.
MDM2 and MDMX phosphorylation induced by stress signals is known to be important at the cellular level for p53 activation. However, the structural basis for stress-induced p53 activation remains less well understood. By chemically synthesizing MDMX and pTyr99 MDMX via native chemical ligation, comparatively characterizing their functionality with respect to peptide ligand binding, we have shown biochemically that MDMX phosphorylation at Tyr99 is functionally detrimental to MDMX interactions with PMI and p53-derived peptide ligands.
PMI is significantly stronger than p53-derived peptides with respect to MDM2/MDMX binding despite the conservation of the three critical hydrophobic residues Phe, Trp and Leu.30,31 For example, (17–28)p53 (ETFSDLWKLLPE) binds to MDMX two orders of magnitude weaker than PMI (TSFAEYWNLLSP).31 We failed to crystallize the complex between pTyr99 MDMX and p53 peptides presumably because of low binding affinity. The crystal structure of a recombinant human MDMX complexed with (17–37)p53 was previously reported,27 in which only residues 17–29 of p53 are ordered and visible. A superposition of PMI-MDMX and (17–37) p53-MDMX complexes reveals that the p53 peptide also shifts laterally by 1.8 Å with respect to PMI (Supplementary Figure S6). Not surprisingly, when the PMI-pTyr99 MDMX and (17–37) p53-MDMX structures are superimposed, a laterally shifted PMI well-overlaps with the p53 peptide in the N-terminal region (Figure 5). As shown in Figure 5, the two structures of MDMX are similar with an root mean sqaure deviation of 0.54 Å; however, they differ in binding of the C-terminal portion of their respective peptide ligands (Figure 5a and Supplementary Figure S7). Tyr99 Oη forms a side chain-main chain hydrogen bond to Pro27 O of p53 and this interaction contributes significantly to p53-MDMX complex stability (Figure 5b).27 Corresponding to Pro27 is Ser11 of PMI, which in the PMI-pTyr99 MDMX complex is pushed away from the binding interface by the protruding phosphate group of pTyr99. Phosphorylation of MDMX at Tyr99 not only breaks the H-bond between Tyr99 Oη and Pro27 O of p53, but also induces steric clashes with the side chains of Pro27 and Asn29, thus hindering p53 binding. Perhaps more importantly, these local conformational changes on p53 imposed by the phosphate group of pTyr99 will propagate into a lateral shift of the peptide ligand as seen with PMI-pTyr99 MDMX, further weakening the p53-MDMX interaction. Of note, it was previously reported that Tyr99 could flip away from the binding pocket of MDMX it constitutes upon binding of a p53 peptidomimetic containing a bulky 6-Cl-Trp residue.40 However, such a large conformational change surrounding Tyr99 has not been observed in MDMX complexed with natural peptide or small-molecule ligands, nor can it be induced by Tyr99 phosphorylation.
Figure 5.
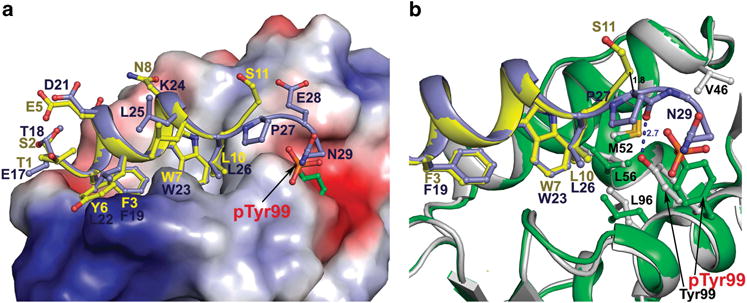
Structural comparison of PMI-pTyr99 MDMX and p53-MDMX complexes. The structure of the PMI-pTyr99 MDMX complex (green/yellow) was superimposed onto the p53-MDMX complex (blue/grey) (PDB code 3DAC) based on MDMX molecules. (a) Electrostatic potential is displayed over the molecular surface of MDMX. PMI and p53 peptides are shown as ribbons and colored yellow and blue, respectively. pTyr99/Tyr99 and residues of PMI and p53 are shown as sticks. (b) The PMI-pTyr99 MDMX and p53-MDMX binding interfaces. Contact residues of pTyr99 MDMX and MDMX contributing to the binding of the C-terminal portion of PMI or p53 peptide are shown as sticks. The hydrogen bond formed between Pro27 O and Tyr99 Oη in the p53-MDMX complex is shown as blue dashes.
It is worth pointing out that the p53 transactivation peptide contains multiple acidic residues including Glu17, Asp21 and Glu28, which could potentially contribute to a reduced binding of p53 to Tyr99-phosphorylated MDMX due, presumably, to electrostatic repulsion. However, none of the three solvent-exposed acidic residues are involved in direct interactions with MDM2 or MDMX, as they project away from the binding interface, consistent with the fact that the p53-MDM2/MDMX interaction is energetically dictated by hydrophobic force. Structural analysis further indicates that Glu17 and Asp21 of p53 are ∼18 Å away from Tyr99 of MDMX and the side chain of Glu28 points away from Tyr99 at a distance of 8 Å.27 The distance and geometry of Glu28 in relation to Tyr99 is such that an electrostatic repulsion created by Tyr99 phosphorylation, if any, is unlikely to be energetically significant.
In conclusion, structural studies of the PMI-pTyr99 MDMX complex strongly suggest that phosphorylation of Tyr99 creates a steric clash between the phosphate group and neighboring contact residues of the p53 transactivation domain, resulting in a displacement of the p53 backbone in the vicinity of Tyr99. This local structural change is accompanied by a lateral shift of p53 along the hydrophobic binding pocket of MDMX, attributing to a substantial reduction in the binding affinity of p53 for MDMX that ultimately ensues the dislodging of p53 from the p53-MDMX inhibitory complex. As DNA damage activates c-Abl tyrosine kinase that phosphorylates MDMX at Tyr99, our findings afford a rare glimpse at the structural level of how stress-induced MDMX phosphorylation activates p53 in response to DNA damage.
MATERIALS AND METHODS
Solid phase peptide synthesis
Boc-amino acids were obtained from Peptides International (Louisville, KY, USA); Fmoc-amino acids, Fmoc-Tyr(PO(OBzl)OH)-OH were purchased from Novabiochem (San Diego, CA, USA); p-methyl-BHA resin and Boc-Leu-OCH2 – PAM resin were purchased from Applied Biosystems (Foster City, CA, USA); Tris-(2-carboxyethyl) phosphine, dichloromethane, N,N-dimethyl-formamide and high performance liquid chromatography grade acetonitrile were obtained from Thermo-Fisher Scientific (Pittsburgh, PA, USA), and 2-(1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluroniumhexa-fluoropho-sphate was acquired from Oakwood Products, Inc (West Columbia, SC, USA). Trifluoroacetic acid was purchased from Halocarbon and hydrogen fluoride was from Matheson Trigas (Basking Ridge, NJ, USA). N,N-diisopropylethylamine and p-cresol were from Sigma-Aldrich (St Louis, MO, USA), and ultrapure guanidine hydrochloride was obtained from ICN Biochemicals (Irvine, CA, USA). 5-(and-6)-Carboxytetramethylrhodamine succinimidyl ester was purchased from Life Technologies (Grand Island, NY, USA); BIAcore series S sensor chips CM5 and HBS-EP buffer were purchased from GE Healthcare Bio-Sciences AB (Pittsburgh, PA, USA).
The amino acid sequence of (24–108) MDMX is as follows: INQVRPK30 LPLLKILHAA40 GAQGEMFTVK50 EVMHYLGQYI60 MVKQLYDQQE70 QHMVYCGGDL80 LGELLGRQSF90 SVKDPSPLYD100 MLRKNLVT108, whe re the ligation site Tyr75-Cys76 is underlined. The two peptide segments of MDMX, that is, (24–75) MDMX-αCOSCH2CH2COLeu and (76–108)MDMX, were individually synthesized on appropriate resins on an ABI 433A automated peptide synthesizer using the optimized 2-(1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluroniumhexa-fluorophosphate activation/N,N-diisopropylethylamine in situ neutralization protocol developed by Kent and colleagues41 for Boc-chemistry solid phase peptide synthesis or an ABI-supplied 2-(1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluroniumhexa-fluoro-phosphate/HOBt protocol for Fmoc-chemistry solid phase peptide synthesis. After cleavage and deprotection in hydrogen fluoride (Boc-chemistry) or in a reagent cocktail containing 95% trifluoroacetic acid, 2.5% TIS and 2.5% H2O (Fmoc-chemistry), crude products were precipitated with cold ether and purified to homogeneity by preparative C18 reversed-phase high performance liquid chromatography. The molecular masses were ascertained by electrospray ionization mass spectrometry.
Crystallization
The initial screening for crystals was carried out with the Art Robinson crystallization robot with various crystal screens available commercially from Hampton Research (Aliso Viejo, CA, USA) and Qiagen (Valencia, CA, USA). All crystallization experiments were performed with PMI-pTyr99 MDMX complex at 8.2 mg/ml in 20 mM Tris pH 7.4. Conditions that produced micro crystals were optimized with respect to the protein concentration, precipitant concentration and pH using the hanging drop vapor diffusion method. Diffraction quality crystals were obtained from the drops containing 0.8 μl of protein mixed with equal volume of the reservoir solution containing 12% isopropanol, 0.1M 2-(N-morpholino)ethanesulfonic acid pH 6.5 and 10% PEG5000 monomethyl ether.
Data collection, structure solution and refinement
Crystals were flash-frozen in liquid nitrogen after briefly soaking in the crystallization condition plus 10% 2-Methyl-2,4-pentanediol prior to data collection. Diffraction data for PMI-pTyr99 MDMX complex was collected at the Stanford Synchrotron Radiation Light Source (SSRL) BL7-1 beam line on an ADSC Quantum 315 area detector. The crystals belong to a space group P1 with the unit-cell parameters a = 28.6, b = 41.4, c = 45.6 Å and α = 103.3°, β = 103.7°, γ = 102.4° and two PMI-pTyr99 MDMX complexes present in the asymmetric unit (Supplementary Table S1).
The data were processed and scaled with HKL2000 package.42 Structure was solved by molecular replacement with Phaser43 from the CCP4i suite based on the coordinates extracted from the structure of PMI-MDMX complex (PDB code: 3EQY30). The model was refined using Refmac and the structure was completed manually using COOT.44 For both the copies of PMI-pTyr99 MDMX present in the asymmetric unit, the electron density map showed no density for the last residue Pro12 of the PMI peptide and only one copy of complex had defined Ser11 (Supplementary Figure S1). The final model with the resolution of 1.55 Å was refined to R-factor of 0.183 and Rfree of 0.224. The Ramachandran plot obtained by the MolProbity validation tool45 shows 92.8% of the total amino acids in the most favored region 7.2% and no residues in the generously allowed and disallowed regions, respectively. The coordinates and structure factors have been deposited in the PDB with accession code 4RXZ.
Acknowledgments
This work was partially supported by the US National Institutes of Health grant CA167296 and the Overseas Scholars Collaborative Research grant 81128015 from the National Natural Science Foundation of China (WL), and by the National Basic Research Program of China (973 Program, 2013CB932500) (W-YL). XC was the recipient of a scholarship from the China Scholarship Council. Portions of this research were carried out at the University of Maryland X-ray Crystallography Shared Service and the Stanford Synchrotron Radiation Lightsource, a Directorate of SLAC National Accelerator Laboratory and an Office of Science User Facility operated for the U.S. Department of Energy Office of Science by Stanford University. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research and by the National Institutes of Health, National Center for Research Resources, Biomedical Technology Program (P41RR001209), and the National Institute of General Medical Sciences.
Footnotes
CONFLICT OF INTEREST
The authors declare no conflict of interest.
Supplementary Information accompanies this paper on the Oncogene website (http://www.nature.com/onc)
References
- 1.Wade M, Li YC, Wahl GM. MDM2, MDMX and p53 in oncogenesis and cancer therapy. Nat Rev Cancer. 2013;13:83–96. doi: 10.1038/nrc3430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Levine AJ, Oren M. The first 30 years of p53: growing ever more complex. Nat Rev Cancer. 2009;9:749–758. doi: 10.1038/nrc2723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature. 2000;408:307–310. doi: 10.1038/35042675. [DOI] [PubMed] [Google Scholar]
- 4.Vousden KH, Lane DP. p53 in health and disease. Nat Rev Mol Cell Biol. 2007;8:275–283. doi: 10.1038/nrm2147. [DOI] [PubMed] [Google Scholar]
- 5.Wade M, Wang YV, Wahl GM. The p53 orchestra: Mdm2 and Mdmx set the tone. Trends Cell Biol. 2010;20:299–309. doi: 10.1016/j.tcb.2010.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jones SN, Roe AE, Donehower LA, Bradley A. Rescue of embryonic lethality in Mdm2-deficient mice by absence of p53. Nature. 1995;378:206–208. doi: 10.1038/378206a0. [DOI] [PubMed] [Google Scholar]
- 7.Montes de Oca Luna R, Wagner DS, Lozano G. Rescue of early embryonic lethality in mdm2-deficient mice by deletion of p53. Nature. 1995;378:203–206. doi: 10.1038/378203a0. [DOI] [PubMed] [Google Scholar]
- 8.Parant J, Chavez-Reyes A, Little NA, Yan W, Reinke V, Jochemsen AG, et al. Rescue of embryonic lethality in Mdm4-null mice by loss of Trp53 suggests a non-overlapping pathway with MDM2 to regulate p53. Nat Genet. 2001;29:92–95. doi: 10.1038/ng714. [DOI] [PubMed] [Google Scholar]
- 9.Brown CJ, Lain S, Verma CS, Fersht AR, Lane DP. Awakening guardian angels: drugging the p53 pathway. Nat Rev Cancer. 2009;9:862–873. doi: 10.1038/nrc2763. [DOI] [PubMed] [Google Scholar]
- 10.Khoo KH, Verma CS, Lane DP. Drugging the p53 pathway: understanding the route to clinical efficacy. Nat Rev Drug Discov. 2014;13:217–236. doi: 10.1038/nrd4236. [DOI] [PubMed] [Google Scholar]
- 11.Popowicz GM, Domling A, Holak TA. The structure-based design of Mdm2/ Mdmx-p53 inhibitors gets serious. Angew Chem Int Ed Engl. 2011;50:2680–2688. doi: 10.1002/anie.201003863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kruse JP, Gu W. Modes of p53 regulation. Cell. 2009;137:609–622. doi: 10.1016/j.cell.2009.04.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Meek DW. Tumour suppression by p53: a role for the DNA damage response? Nat Rev Cancer. 2009;9:714–723. doi: 10.1038/nrc2716. [DOI] [PubMed] [Google Scholar]
- 14.Mayo LD, Turchi JJ, Berberich SJ. Mdm-2 phosphorylation by DNA-dependent protein kinase prevents interaction with p53. Cancer Res. 1997;57:5013–5016. [PubMed] [Google Scholar]
- 15.Zuckerman V, Lenos K, Popowicz GM, Silberman I, Grossman T, Marine JC, et al. c-Abl phosphorylates Hdmx and regulates its interaction with p53. J Biol Chem. 2009;284:4031–4039. doi: 10.1074/jbc.M809211200. [DOI] [PubMed] [Google Scholar]
- 16.Chen L, Gilkes DM, Pan Y, Lane WS, Chen J. ATM and Chk2-dependent phosphorylation of MDMX contribute to p53 activation after DNA damage. EMBO J. 2005;24:3411–3422. doi: 10.1038/sj.emboj.7600812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Maya R, Balass M, Kim ST, Shkedy D, Leal JF, Shifman O, et al. ATM-dependent phosphorylation of Mdm2 on serine 395: role in p53 activation by DNA damage. Genes Dev. 2001;15:1067–1077. doi: 10.1101/gad.886901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Okamoto K, Kashima K, Pereg Y, Ishida M, Yamazaki S, Nota A, et al. DNA damage-induced phosphorylation of MdmX at serine 367 activates p53 by targeting MdmX for Mdm2-dependent degradation. Mol Cell Biol. 2005;25:9608–9620. doi: 10.1128/MCB.25.21.9608-9620.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pereg Y, Shkedy D, de Graaf P, Meulmeester E, Edelson-Averbukh M, Salek M, et al. Phosphorylation of Hdmx mediates its Hdm2- and ATM-dependent degradation in response to DNA damage. Proc Natl Acad Sci USA. 2005;102:5056–5061. doi: 10.1073/pnas.0408595102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cheng Q, Chen L, Li Z, Lane WS, Chen J. ATM activates p53 by regulating MDM2 oligomerization and E3 processivity. EMBO J. 2009;28:3857–3867. doi: 10.1038/emboj.2009.294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McCoy MA, Gesell JJ, Senior MM, Wyss DF. Flexible lid to the p53-binding domain of human Mdm2: implications for p53 regulation. Proc Natl Acad Sci USA. 2003;100:1645–1648. doi: 10.1073/pnas.0334477100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Showalter SA, Bruschweiler-Li L, Johnson E, Zhang F, Bruschweiler R. Quantitative lid dynamics of MDM2 reveals differential ligand binding modes of the p53-binding cleft. J Am Chem Soc. 2008;130:6472–6478. doi: 10.1021/ja800201j. [DOI] [PubMed] [Google Scholar]
- 23.Worrall EG, Worrall L, Blackburn E, Walkinshaw M, Hupp TR. The effects of phosphomimetic lid mutation on the thermostability of the N-terminal domain of MDM2. J Mol Biol. 2010;398:414–428. doi: 10.1016/j.jmb.2010.03.023. [DOI] [PubMed] [Google Scholar]
- 24.Zhan C, Varney K, Yuan W, Zhao L, Lu W. Interrogation of MDM2 phosphorylation in p53 activation using native chemical ligation: the functional role of Ser17 phosphorylation in MDM2 reexamined. J Am Chem Soc. 2012;134:6855–6864. doi: 10.1021/ja301255n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Goldberg Z, Vogt Sionov R, Berger M, Zwang Y, Perets R, Van Etten RA, et al. Tyrosine phosphorylation of Mdm2 by c-Abl: implications for p53 regulation. EMBO J. 2002;21:3715–3727. doi: 10.1093/emboj/cdf384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Waning DL, Lehman JA, Batuello CN, Mayo LD. c-Abl phosphorylation of Mdm2 facilitates Mdm2-Mdmx complex formation. J Biol Chem. 2011;286:216–222. doi: 10.1074/jbc.M110.183012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Popowicz GM, Czarna A, Holak TA. Structure of the human Mdmx protein bound to the p53 tumor suppressor transactivation domain. Cell Cycle. 2008;7:2441–2443. doi: 10.4161/cc.6365. [DOI] [PubMed] [Google Scholar]
- 28.Dawson PE, Kent SB. Synthesis of native proteins by chemical ligation. Annu Rev Biochem. 2000;69:923–960. doi: 10.1146/annurev.biochem.69.1.923. [DOI] [PubMed] [Google Scholar]
- 29.Dawson PE, Muir TW, Clark-Lewis I, Kent SB. Synthesis of proteins by native chemical ligation. Science. 1994;266:776–779. doi: 10.1126/science.7973629. [DOI] [PubMed] [Google Scholar]
- 30.Pazgier M, Liu M, Zou G, Yuan W, Li C, Li C, et al. Structural basis for high-affinity peptide inhibition of p53 interactions with MDM2 and MDMX. Proc Natl Acad Sci USA. 2009;106:4665–4670. doi: 10.1073/pnas.0900947106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li C, Pazgier M, Li C, Yuan W, Liu M, Wei G, et al. Systematic mutational analysis of peptide inhibition of the p53-MDM2/MDMX interactions. J Mol Biol. 2010;398:200–213. doi: 10.1016/j.jmb.2010.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhan C, Zhao L, Wei X, Wu X, Chen X, Yuan W, et al. An ultrahigh affinity d-peptide antagonist Of MDM2. J Med Chem. 2012;55:6237–6241. doi: 10.1021/jm3005465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tarrant MK, Cole PA. The chemical biology of protein phosphorylation. Annu Rev Biochem. 2009;78:797–825. doi: 10.1146/annurev.biochem.78.070907.103047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kent SB. Total chemical synthesis of proteins. Chem Soc Rev. 2009;38:338–351. doi: 10.1039/b700141j. [DOI] [PubMed] [Google Scholar]
- 35.Muir TW, Sondhi D, Cole PA. Expressed protein ligation: a general method for protein engineering. Proc Natl Acad Sci USA. 1998;95:6705–6710. doi: 10.1073/pnas.95.12.6705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schwarzer D, Cole PA. Protein semisynthesis and expressed protein ligation: chasing a protein’s tail. Curr Opin Chem Biol. 2005;9:561–569. doi: 10.1016/j.cbpa.2005.09.018. [DOI] [PubMed] [Google Scholar]
- 37.Noren CJ, Anthony-Cahill SJ, Griffith MC, Schultz PG. A general method for site-specific incorporation of unnatural amino acids into proteins. Science. 1989;244:182–188. doi: 10.1126/science.2649980. [DOI] [PubMed] [Google Scholar]
- 38.Lu W, Gong D, Bar-Sagi D, Cole PA. Site-specific incorporation of a phosphotyrosine mimetic reveals a role for tyrosine phosphorylation of SHP-2 in cell signaling. Mol Cell. 2001;8:759–769. doi: 10.1016/s1097-2765(01)00369-0. [DOI] [PubMed] [Google Scholar]
- 39.Lu W, Shen K, Cole PA. Chemical dissection of the effects of tyrosine phosphorylation of SHP-2. Biochemistry. 2003;42:5461–5468. doi: 10.1021/bi0340144. [DOI] [PubMed] [Google Scholar]
- 40.Kallen J, Goepfert A, Blechschmidt A, Izaac A, Geiser M, Tavares G, et al. Crystal structures of human MdmX (HdmX) in complex with p53 peptide analogues reveal surprising conformational changes. J Biol Chem. 2009;284:8812–8821. doi: 10.1074/jbc.M809096200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schnolzer M, Alewood P, Jones A, Alewood D, Kent SB. In situ neutralization in Boc-chemistry solid phase peptide synthesis. Rapid, high yield assembly of difficult sequences. Int J Pept Protein Res. 1992;40:180–193. doi: 10.1111/j.1399-3011.1992.tb00291.x. [DOI] [PubMed] [Google Scholar]
- 42.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. In: Carter CW Jr, Sweet RM, editors. Methods in Enzymology: Macromolecylar Crystallography, Part A. Vol. 276. Academic Press; New York: 1997. pp. 307–326. [DOI] [PubMed] [Google Scholar]
- 43.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 44.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 45.Chen VB, Arendall WB, 3rd, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]