

Abstract
Proteins composed entirely of unnatural D-amino acids and the achiral amino acid glycine are mirror image forms of their native L-protein counterparts. Recent advances in chemical protein synthesis afford unique and facile synthetic access to domain-sized mirror image D-proteins, enabling protein research to be conducted through “the looking glass” and in a way previously unattainable. D-proteins can facilitate structure determination of their native L-forms that are difficult to crystallize (racemic X-ray crystallography); D-proteins can serve as the bait for library screening to ultimately yield pharmacologically superior D-peptide/D-protein therapeutics (mirror image phage display); D-proteins can also be used as a powerful mechanistic tool for probing molecular events in biology. This review examines recent progress in the application of mirror image proteins to structural biology, drug discovery, and immunology.
Introduction
Alpha-amino acids – the basic building blocks of proteins – are chiral molecules that exist in two forms: L-enantiomer (“L” for levorotatory or left-handed) and D-enantiomer (“D” for dextrorotatory or right-handed). The two non-superimposable forms of amino acid differing in handedness or chirality are mirror images of one another and have otherwise identical physical and chemical properties. Life, however, uses only L-amino acids and the achiral amino acid glycine to construct proteins that perform a great variety of biological functions. Although present in nature [1], notably in the peptidoglycans of cell walls and in peptide antibiotics of bacterial origin, in proteins of lower animals such as insects, snails and amphibians, and even in the brain as neurotransmitters, D-amino acids in various organisms are thought to be converted from parent L-enantiomers through enzyme-catalyzed post-translational reactions [2,3]. The fascinating question of why and how life on Earth favors these left-handed molecules has been a subject of intense debate for decades among chemists, physicists, biologists, and even astronomers. While the origin of homochirality of alpha-amino acids continually remains a mystery [4], scientists have learned a great deal already from studying the physicochemical and biological properties of unnatural or artificial D-peptides and D-proteins that contain only chiral D-amino acids.
Mirror image proteins can only be made by chemistry. Peptides and small proteins are traditionally synthesized using stepwise solid phase peptide synthesis techniques [5], which limit the size of a polypeptide chain to be assembled to roughly 60 amino acid residues. Since the average size of eukaryotic protein domains is about 125 amino acid residues in length [6], synthetic peptide chemistry for decades was limited to studies of peptides and a few small proteins and failed to unlock its full potential for protein research. Kent and colleagues revolutionized peptide and protein research by developing a robust chemistry termed native chemical ligation [7-9], which enables the chemoselective ligation of fully unprotected peptides in aqueous solution, forming a product polypeptide linked by the native peptide bond. Numerous proteins have been chemically synthesized using the native chemical ligation technique or its varied forms, greatly advancing our understanding of the molecular basis of how proteins function in a way previously unattainable. Native chemical ligation also makes it now possible to routinely synthesize and study mirror image D-protein forms of domain-sized natural proteins, further expanding the capacity and augmenting the power of mirror image protein technology. This review summarizes important progress made during the past few years on research using mirror image proteins, with a focus on their applications in structural biology, drug discovery, and immunology.
Racemic X-ray crystallography for protein structural biology
Crystallization and phase determination are often two rate-limiting steps in X-ray crystallographic analysis of protein structure. Zawadzke & Berg pioneered racemic protein crystallography in 1993, where an equal molar mixture of the L- and D-enantiomers of a 45-residue iron-sulfur protein, rubredoxin, was crystallized in a centrosymmetric space group with two molecules in the unit cell, one of each enantiomer, related to each other by a center of inversion [10]. In these centrosymmetric crystals the phase of each reflection was restricted to either 0 or 180 degrees, as predicted [11]. The combination of a much simplified phase problem [11] and the greater ease with which racemic proteins crystallize [12] should facilitate, structure determination of moderately sized macromolecules, as anticipated by Berg and Zawadzke [13].
This expectation has largely been fulfilled by the work from the Kent laboratory at the University of Chicago [14]. Armed with their ability to create synthetic mirror image proteins and racemic crystallization, Kent and colleagues have determined the crystal structures of a variety of small proteins previously proven difficult to tackle, including the snow flea antifreeze protein [15], the scorpion toxin BmBKTx1 [16], the fungal defensin plectasin [17], the snake venom protein omwaprin [18], the scorpion toxin kaliotoxin [19], an engineered insulin molecule [20], and the peptide toxin ShK from sea anemones [21]. More recently, they used racemic crystallography to determine the crystal structure of the first heterochiral protein complex, in which a designed small D-protein antagonist, D-RFX001, of 56 amino acid residues bound to its target protein - the angiogenic protein vascular endothelial growth factor (VEGF-A, a covalent dimer of residues 8-109) [22]. Interestingly, the structure of a quasi-centrosymmetric crystal formed by two chemically non-identical enantiomers of a chemokine, N-glycosylated L-CCL1 protein and non-glycosylated D-CCL1 protein, was also determined [23], showcasing the power of quasiracemic crystallography in structure determination of glycoproteins that are difficult to crystallize.
Mirror image phage display for D-peptide/D-protein drug discovery
Developing small molecules to target enzymes and receptors has been a traditional drug discovery approach widely adopted by the pharmaceutical industry. Despite an exponential growth over the past several decades in the expenditures for drug research and development, however, the number of annual new drug approvals by FDA has remained stagnant, or even has declined in some years. The exhaustion of “low-hanging fruit” in drug discovery further exacerbates the growing disparity between investment and outcome, underscoring an urgent need to explore new classes of drugs beyond small molecules such as peptides and proteins known as biologics [24,25], and new drug targets such as protein-protein interactions [26,27].
Small peptide inhibitors of protein-protein interactions, due to their high affinity and superb specificity, show promise as an important class of therapeutic agents for the treatment of various human diseases. However, peptide drugs suffer poor bioavailability as they are highly susceptibility to proteolytic degradation in vivo, severely limiting their systemic use and therapeutic potential. Different chemistries that aim to modify side chains and/or the backbone have been used to improve peptide resistance to proteolysis [28-30], among which the use of D-peptides remains one of the most promising approaches to overcoming the pharmacologic hurdle in peptide drug discovery. D-peptides are resistant to proteolytic degradation due to an exceedingly high free energy barrier, afforded by steric incompatibility, to the transition state of the enzyme-substrate complex [31]. This intrinsic pharmacologic property endows D-peptide therapeutics with the ability to circulate longer in vivo, and may render them orally available and less immunogenic due to poor efficiency in antigen presentation.
Kim and colleagues pioneered an elegant technique for D-peptide drug discovery, termed ‘mirror image phage display’ [32], where a phage-expressed peptide library was screened against the D-enantiomer of an SH3 domain of the Src tyrosine kinase family, yielding a consensus L-peptide ligand for the chemically synthesized D-protein; when inverted and prepared by chemical synthesis, the resultant D-peptide ligand specifically bound, for reasons of symmetry, recombinant L-SH3 domain at the binding site of physiological ligands on natural SH3 domains. This powerful tool enabled the subsequent identification of several classes of proteolysis-resistant D-peptide inhibitors with therapeutic potential [31], including antagonists that target the HIV-1 envelope glycoprotein gp41 to block viral fusion/entry [33-35], and the aggregation-prone amyloid peptide Aβ to inhibit amyloid plaque formation in Alzheimer's disease [36-38].
Mirror image phage display necessitates the use of enantiomeric D-protein targets as the bait for library screening. Such D-protein molecules can only be synthesized chemically. Early practitioners of the mirror image phage display technique relied on peptide or small protein targets such as SH3 domain, HIV-1 gp41 peptide, and amyloid peptide Aβ, chemically accessible via stepwise solid phase peptide synthesis. The native chemical ligation technique developed by Kent and colleagues [7,9] enables facile synthetic access to domain-sized proteins, greatly expanding the breadth, depth and power of mirror image phage display in peptide drug discovery.
The master tumor suppressor protein p53 is functionally inhibited in many tumors by the two overexpressed oncogenic proteins MDM2 and MDMX, which interplay to inhibit p53 transactivation activity and target p53 for degradation [39]. MDM2 and MDMX contain an N-terminal p53-binding domain of ∼100 amino acid residues that forms a tight complex with p53 through extensive hydrophobic interactions with p53 transactivation peptide of ∼15 amino acid residues. MDM2 and/or MDMX antagonists that disrupt the p53-MDM2/MDMX interaction can rescue p53 function and kill tumor cells in vitro and in vivo [40], promising a novel class of anticancer therapeutics. Lu and colleagues screened a phage-expressed peptide library against both L- and D-enantiomers of MDM2 synthesized via native chemical ligation, and discovered an array of L- and D-peptide ligands that bound to both MDM2 and MDMX with high affinity [41-45]. Three D-peptide ligands, TNWYANLEKLLR (DPMI-α), TAWYANFEKLLR (DPMI-β), and DWWPLAFEALLR (DPMI-γ), bound to synthetic MDM2 and MDMX with affinities of 219 nM and 18 μM, 34.5 nM and 2.4 μM, 52.8 nM and 4.9 μM, respectively. Structural studies validated the productive binding mode of these D-peptide antagonists in complex with MDM2/MDMX, where they adopt a left-handed alpha-helical conformation, docking their hydrophobic residues inside the p53-binding pocket of MDM2/MDMX (Figure 1). DPMI-α activated the p53 pathway in human glioblastoma cell lines harboring wild type p53 and elevated MDM2, inhibited tumor growth in vitro and in vivo, and prolonged the survival of nude mice bearing intracranial glioblastoma. More recently, Lu and colleagues successfully designed, on the basis of dPMI-β, a superactive D-peptide antagonist of MDM2, termed DPMI-δ, which bound to MDM2 with an affinity of 220 pM, promising a highly attractive lead drug candidate for anticancer therapy [46].
Figure 1.
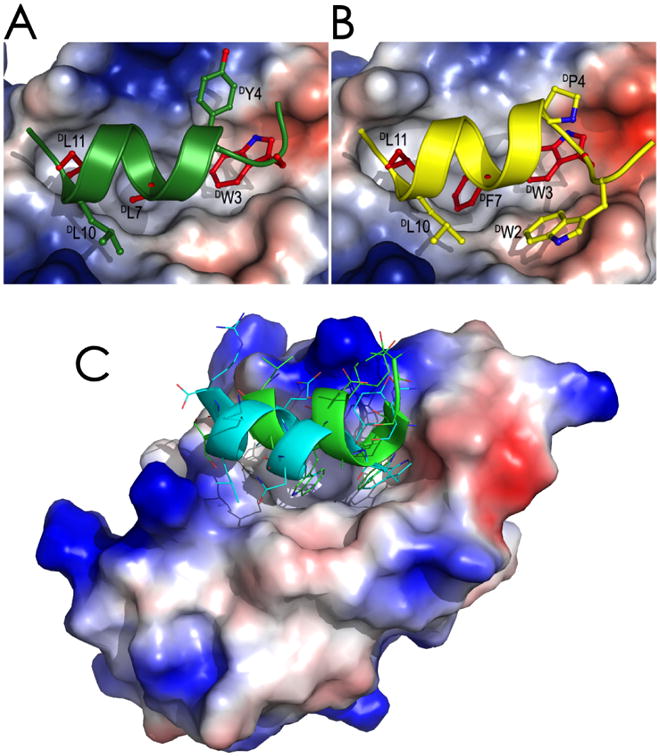
Crystal structures of synthetic MDM2 in complex with D-peptide antagonists identified by mirror image phage display. (A) TNWYANLEKLLR (DPMI-α) in complex with MDM2; (B) TAWYANFEKLLR (DPMI-β) in complex with MDM2; (C) Comparison of DPMI-α in a left-handed alpha-helical conformation (cyan) with an L-peptide antagonist in a right-handed alpha-helical conformation (green) bound to the same p53-binding pocket of MDM2.
A notable recent development in mirror image phage display is the emerging application of this technology to the development of D-protein therapeutics [22], which are expected to possess enhanced binding affinity and specificity compared with small peptide ligands. The enhanced affinity and specificity is understandable, as a structurally stable protein scaffold generally loses less conformational entropy in the course of target binding than a flexible and disordered peptide ligand. The Sachdev Sidhu group at the University of Toronto constructed a phage-displayed protein library using the 56-residue B1 domain of streptococcal protein G as a scaffold, screened the library against the mirror image form of VEGF-A of 102 amino acid residues chemically synthesized using a three-segment ligation strategy by the Kent lab at the University of Chicago, and identified the D-protein ligand D-RFX001 that bound to native L-VEGF-A at an affinity of 82 nM and competitively blocked VEGF interactions with its cognate receptor [22]. This D-protein antagonist of VEGF, mechanistically similar to therapeutic monoclonal antibodies, may have the potential to be developed as a novel class of angiogenesis inhibitors for anticancer therapy.
A powerful mechanistic tool for biology
Unlike chiral peptides and proteins, lipids that constitute plasma membranes are generally achiral molecules. It is therefore expected that interactions of peptides/proteins with achiral lipid bilayer membranes are functionally independent of the chirality of the (peptide/protein) ligand. Merrifield and colleagues first demonstrated that the L- and D-enantiomers of alpha-helical antimicrobial peptides (AMPs) such as cecropin, magainin, and melittin were equally active in killing Gram-positive and -negative strains of bacteria, polarizing planar lipid bilayers, and lysing erythrocytes [47]. Lehrer and coworkers made similar observations with the L- and D-enantiomers of protegrins, Cys-rich and beta-sheet AMPs from porcine leukocytes, in their killing of bacteria and Candida albicans [48,49]. These findings immediately suggested that chiral target molecules were not functionally involved in the action of cationic AMPs. Despite a rare exception reported on the L- and D-enantiomers of apidaecin, a small insect cationic AMP rich in prolines [50], membrane disruption had long been thought to be an overriding mechanism of action by which most, if not all, classes of cationic AMPs kill bacteria, including defensins – a major family of disulfide bridged AMPs found in mammals [51].
The use of D-enantiomeric defensins in functional studies, however, called this mechanism of action into question [52]. Shown in Figure 2 are the crystal structures of the L- and D-enantiomers of human neutrophil alpha-defensin 1 or HNP1, both adopting the canonical three-stranded beta-sheet fold stabilized by three intra-molecular disulfides and arranged in a dimeric form; a nearly perfect plane of symmetry relates the two mirror image defensin dimers to one another. While L- and D-HNP1 showed no functional difference in killing E. coli, consistent with the membrane-centric mechanism of action of HNP1 against Gram-negative bacteria [53], the D-enantiomer exhibited a substantially reduced bactericidal activity against the Gram-positive bacterium S. aureus compared with its L-counterpart [52]. Similar results were reproduced with the L- and D-enantiomers of the human enteric alpha-defensin 5 or HD5 [52]. By contrast, both L- and D-HNP1 displayed identical membranolytic activity in inducing the leakage of fluorescent dyes from large unilamellar phospholipid vesicles.
Figure 2.
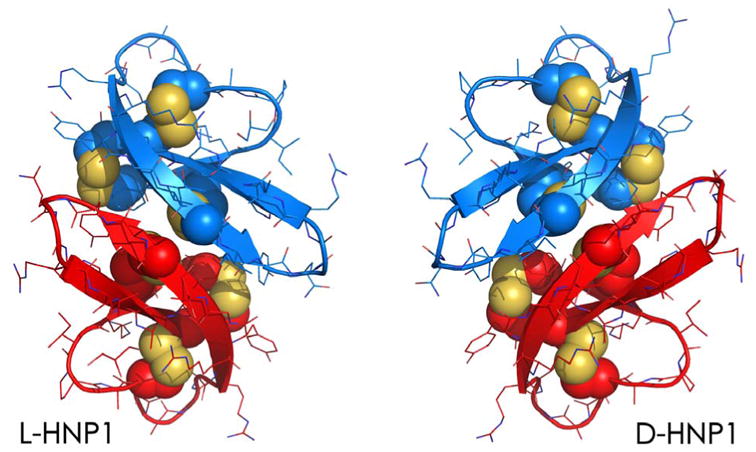
Crystal structures of dimeric HNP1 made up of L-amino acids (left) and D-amino acids (right). The side chains of the six Cys residues of HNP1 that form three intramolecular disulfide bridges are depicted in spheres.
These findings led to the suggestion that the lethal event in the killing of Gram-positive bacteria by human alpha-defensins is not dominated by membrane disruption and likely involves a chiral molecular target that is preferentially recognized by native defensins [52]. Since both HNP1 and HD5 were found capable of binding peptidoglycans that constitute the thick bacterial cell wall in Gram-positive bacteria, the authors further speculated that human alpha-defensins kill S. aureus by engaging the cell wall precursor lipid II to inhibit bacterial cell wall synthesis [52]. This mode of bacterial killing by human alpha-defensins, reminiscent of nisin and vancomysin [54-58], was subsequently confirmed experimentally [59,60]. To date, several more defensins from different origins have been reported to kill Gram-positive bacteria via sequestration of lipid II and inhibition of bacterial cell wall synthesis [61-65], completing the paradigm-shifting discovery of a novel mechanism of action of AMPs exposed by mirror image proteins.
Conclusion
Mirror image proteins are powerful tools with a wide range of applications in structural biology, peptide/protein drug design, and mechanistic studies of biological processes. As chemical protein synthesis techniques become more robust and readily available to scientists from different disciplines, the huge potential of mirror image proteins in chemical, biological, and biomedical research will be fully unlocked. The two enabling technologies – native chemical ligation and mirror image phage display are particularly attractive, and will have a profound impact on the discovery of novel classes of pharmacologically superior peptide and protein therapeutics for the treatment of a variety of human diseases.
Highlights.
Native chemical ligation enables facile synthetic access to mirror image proteins
Mirror image proteins facilitate protein crystallization and structure solution
Mirror image proteins aid drug discovery of novel classes of therapeutics
Mirror image proteins can be a powerful mechanistic tool for biology
Acknowledgments
W.L. has been supported by NIH grants for the past decade and L.Z. was a Guanghua Scholar supported by Xi'an Jiaotong University School of Medicine.
Footnotes
Conflicts of Interest: There are no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
Papers of particular interest have been highlighted as:
■ of special interest
■■ of outstanding interest
- 1.Kreil G. D-amino acids in animal peptides. Annu Rev Biochem. 1997;66:337–345. doi: 10.1146/annurev.biochem.66.1.337. [DOI] [PubMed] [Google Scholar]
- 2.Kreil G. Conversion of L- to D-amino acids: a posttranslational reaction. Science. 1994;266:996–997. doi: 10.1126/science.7973683. [DOI] [PubMed] [Google Scholar]
- 3.Heck SD, Siok CJ, Krapcho KJ, Kelbaugh PR, Thadeio PF, Welch MJ, Williams RD, Ganong AH, Kelly ME, Lanzetti AJ. Functional consequences of posttranslational isomerization of Ser46 in a calcium channel toxin. Science. 1994;266:1065–1068. doi: 10.1126/science.7973665. [DOI] [PubMed] [Google Scholar]
- 4.Blackmond DG. The origin of biological homochirality. Cold Spring Harb Perspect Biol. 2010;2:a002147. doi: 10.1101/cshperspect.a002147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kent S. Chemical synthesis of peptides and proteins. Annu Rev Biochem. 1988;57:957–989. doi: 10.1146/annurev.bi.57.070188.004521. [DOI] [PubMed] [Google Scholar]
- 6.Berman AL, Kolker E, Trifonov EN. Underlying order in protein sequence organization. Proc Natl Acad Sci USA. 1994;91:4044–4047. doi: 10.1073/pnas.91.9.4044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dawson PE, Kent SB. Synthesis of native proteins by chemical ligation. Annu Rev Biochem. 2000;69:923–960. doi: 10.1146/annurev.biochem.69.1.923. [DOI] [PubMed] [Google Scholar]
- 8.Kent S. Total chemical synthesis of proteins. Chem Soc Rev. 2009;38:338–351. doi: 10.1039/b700141j. [DOI] [PubMed] [Google Scholar]
- 9■■.Dawson PE, Muir TW, Clark-Lewis I, Kent SB. Synthesis of proteins by native chemical ligation. Science. 1994;266:776–779. doi: 10.1126/science.7973629. This is the original report that describes the native chemical ligation technique developed by Kent and colleagues for the chemical synthesis of proteins. [DOI] [PubMed] [Google Scholar]
- 10■■.Zawadzke LE, Berg JM. The structure of a centrosymmetric protein crystal. Proteins. 1993;16:301–305. doi: 10.1002/prot.340160308. The first description of racemic protein crystallography pioneered by Berg and colleagues. [DOI] [PubMed] [Google Scholar]
- 11.Mackay AL. Crystal enigma. Nature. 1989;342:133. [Google Scholar]
- 12.Wukovitz SW, Yeates TO. Why protein crystals favour some space-groups over others. Nat Struct Mol Biol. 1995;2:1062–1067. doi: 10.1038/nsb1295-1062. [DOI] [PubMed] [Google Scholar]
- 13.Berg JM, Zawadzke LE. Racemic macromolecules for use in X-ray crystallography. Curr Opin Biotechnol. 1994;5:343–345. doi: 10.1016/0958-1669(94)90041-8. [DOI] [PubMed] [Google Scholar]
- 14■.Yeates TO, Kent S. Racemic protein crystallography. Annu Rev Biophys. 2012;41:41–61. doi: 10.1146/annurev-biophys-050511-102333. This is a comprehensive review on racemic protein crystallography by the two mostauthoritative experts in the field. [DOI] [PubMed] [Google Scholar]
- 15.Pentelute BL, Gates ZP, Tereshko V, Dashnau JL, Vanderkooi JM, Kossiakoff AA, Kent SBH. X-ray structure of snow flea antifreeze protein determined by racemic crystallization of synthetic protein enantiomers. J Am Chem Soc. 2008;130:9695–9701. doi: 10.1021/ja8013538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mandal K, Pentelute BL, Tereshko V, Kossiakoff AA, Kent SBH. X-ray structure of native scorpion toxin BmBKTx1 by racemic protein crystallography using direct methods. J Am Chem Soc. 2009;131:1362–1363. doi: 10.1021/ja8077973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mandal K, Pentelute BL, Tereshko V, Thammavongsa V, Schneewind O, Kossiakoff AA, Kent SBH. Racemic crystallography of synthetic protein enantiomers used to determine the X-ray structure of plectasin by direct methods. Protein Sci. 2009;18:1146–1154. doi: 10.1002/pro.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Banigan JR, Mandal K, Sawaya MR, Thammavongsa V, Hendrickx APA, Schneewind O, Yeates TO, Kent SBH. Determination of the X-ray structure of the snake venom protein omwaprin by total chemical synthesis and racemic protein crystallography. Protein Sci. 2010;19:1840–1849. doi: 10.1002/pro.468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pentelute BL, Mandal K, Gates ZP, Sawaya MR, Yeates TO, Kent SBH. Total chemicalsynthesis and X-ray structure of kaliotoxin by racemic protein crystallography. Chem Commun (Camb) 2010;46:8174–8176. doi: 10.1039/c0cc03148h. [DOI] [PubMed] [Google Scholar]
- 20.Avital-Shmilovici M, Mandal K, Gates ZP, Phillips NB, Weiss MA, Kent SBH. Fully convergent chemical synthesis of ester insulin: determination of the high resolution X-ray structure by racemic protein crystallography. J Am Chem Soc. 2013;135:3173–3185. doi: 10.1021/ja311408y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dang B, Kubota T, Mandal K, Bezanilla F, Kent SBH. Native chemical ligation at Asx-Cys, Glx-Cys: chemical synthesis and high-resolution X-ray structure of ShK toxin by racemic protein crystallography. J Am Chem Soc. 2013;135:11911–11919. doi: 10.1021/ja4046795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22■.Mandal K, Uppalapati M, Ault-Riché D, Kenney J, Lowitz J, Sidhu SS, Kent SB. Chemical synthesis and X-ray structure of a heterochiral {D-protein antagonist plus vascular endothelial growth factor} protein complex by racemic crystallography. 2012:14779–14784. doi: 10.1073/pnas.1210483109. The first crystal structure of a heterochiral complex formed by D- and L-proteinssolved by racemic crystallography and the first D-protein antagonist withtherapeutic potential developed via mirror image phage display. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23■.Okamoto R, Mandal K, Sawaya MR, Kajihara Y, Yeates TO, Kent SBH. (Quasi-)racemic X-ray structures of glycosylated and non-glycosylated forms of thechemokine Ser-CCL1 prepared by total chemical synthesis. Angew Chem Int Ed Engl. 2014;53:5194–5198. doi: 10.1002/anie.201400679. A powerful example of racemic crystallography in structure determination of glycoproteins that are difficult to crystalize. [DOI] [PubMed] [Google Scholar]
- 24.Projan SJ, Gill D, Lu Z, Herrmann SH. Small molecules for small minds? The casefor biologic pharmaceuticals. Expert Opin Biol Ther. 2004;4:1345–1350. doi: 10.1517/14712598.4.8.1345. [DOI] [PubMed] [Google Scholar]
- 25.Leader B, Baca QJ, Golan DE. Protein therapeutics: a summary and pharmacological classification. Nat Rev Drug Discov. 2008;7:21–39. doi: 10.1038/nrd2399. [DOI] [PubMed] [Google Scholar]
- 26.Wells JA, McClendon CL. Reaching for high-hanging fruit in drug discovery at protein-protein interfaces. Nature. 2007;450:1001–1009. doi: 10.1038/nature06526. [DOI] [PubMed] [Google Scholar]
- 27.Arkin MR, Wells JA. Small-molecule inhibitors of protein-protein interactions: progressing towards the dream. Nat Rev Drug Discov. 2004;3:301–317. doi: 10.1038/nrd1343. [DOI] [PubMed] [Google Scholar]
- 28.Vagner J, Qu H, Hruby VJ. Peptidomimetics, a synthetic tool of drug discovery. Curr Opin Chem Biol. 2008;12:292–296. doi: 10.1016/j.cbpa.2008.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Walensky LD, Kung AL, Escher I, Malia TJ, Barbuto S, Wright RD, Wagner G, Verdine GL, Korsmeyer SJ. Activation of apoptosis in vivo by a hydrocarbon-stapled BH3 helix. Science. 2004;305:1466–1470. doi: 10.1126/science.1099191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Moellering RE, Cornejo M, Davis TN, Del Bianco C, Aster JC, Blacklow SC, Kung AL, Gilliland DG, Verdine GL, Bradner JE. Direct inhibition of the NOTCH transcription factor complex. Nature. 2009;462:182–188. doi: 10.1038/nature08543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhao L, Lu W. D-Peptide-Based Drug Discovery Aided by Chemical Protein Synthesis. Isr J Chem. 2011;51:868–875. [Google Scholar]
- 32■■.Schumacher TN, Mayr LM, Minor DL, Milhollen MA, Burgess MW, Kim PS. Identification of D-peptide ligands through mirror-image phage display. Science. 1996;271:1854–1857. doi: 10.1126/science.271.5257.1854. This is the original report the describes the mirror-image phage display technique developed by Kim and colleagues. [DOI] [PubMed] [Google Scholar]
- 33.Eckert DM, Malashkevich VN, Hong LH, Carr PA, Kim PS. Inhibiting HIV-1 entry: discovery of D-peptide inhibitors that target the gp41 coiled-coil pocket. Cell. 1999;99:103–115. doi: 10.1016/s0092-8674(00)80066-5. [DOI] [PubMed] [Google Scholar]
- 34■.Welch BD, VanDemark AP, Heroux A, Hill CP, Kay MS. Potent D-peptide inhibitors of HIV-1 entry. Proc Natl Acad Sci USA. 2007;104:16828–16833. doi: 10.1073/pnas.0708109104. This exceedingly potent D-peptide inhibitor of HIV-1 entry discovered via mirror-image phage display has a significant therapeutic potential. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Welch BD, Francis JN, Redman JS, Paul S, Weinstock MT, Reeves JD, Lie YS, Whitby FG, Eckert DM, Hill CP, et al. Design of a Potent D-Peptide HIV-1 Entry Inhibitor witha Strong Barrier to Resistance. J Virol. 2010;84:11235–11244. doi: 10.1128/JVI.01339-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wiesehan K, Buder K, Linke RP, Patt S, Stoldt M, Unger E, Schmitt B, Bucci E, Willbold D. Selection of D-amino-acid peptides that bind to Alzheimer's disease amyloid peptide abeta1-42 by mirror image phage display. Chembiochem. 2003;4:748–753. doi: 10.1002/cbic.200300631. [DOI] [PubMed] [Google Scholar]
- 37.van Groen T, Wiesehan K, Funke SA, Kadish I, Nagel-Steger L, Willbold D. Reduction of Alzheimer's disease amyloid plaque load in transgenic mice by D3, A D-enantiomeric peptide identified by mirror image phage display. ChemMedChem. 2008;3:1848–1852. doi: 10.1002/cmdc.200800273. [DOI] [PubMed] [Google Scholar]
- 38.Aileen Funke S, van Groen T, Kadish I, Bartnik D, Nagel-Steger L, Brener O, Sehl T, Batra-Safferling R, Moriscot C, Schoehn G, et al. Oral Treatment with the d-Enantiomeric Peptide D3 Improves the Pathology and Behavior of Alzheimer's Disease Transgenic Mice. ACS Chem Neurosci. 2010;1:639–648. doi: 10.1021/cn100057j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wade M, Li YC, Wahl GM. MDM2, MDMX and p53 in oncogenesis and cancer therapy. Nat Rev Cancer. 2012;13:83–96. doi: 10.1038/nrc3430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brown CJ, Lain S, Verma CS, Fersht AR, Lane DP. Awakening guardian angels: drugging the p53 pathway. Nat Rev Cancer. 2009;9:862–873. doi: 10.1038/nrc2763. [DOI] [PubMed] [Google Scholar]
- 41.Zhan C, Lu W. Peptide activators of the p53 tumor suppressor. Curr Pharm Des. 2011;17:603–609. doi: 10.2174/138161211795222577. [DOI] [PubMed] [Google Scholar]
- 42■.Liu M, Li C, Pazgier M, Li C, Mao Y, Lv Y, Gu B, Wei G, Yuan W, Zhan C, et al. D-peptide inhibitors of the p53-MDM2 interaction for targeted molecular therapy of malignant neoplasms. Proc Natl Acad Sci USA. 2010;107:14321–14326. doi: 10.1073/pnas.1008930107. The first example of the combined use of native chemical ligation and mirror image phage display for the discovery of potent D-peptide antagonists for anticancer therapy. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pazgier M, Liu M, Zou G, Yuan W, Li C, Li C, Li J, Monbo J, Zella D, Tarasov SG, et al. Structural basis for high-affinity peptide inhibition of p53 interactions with MDM2 and MDMX. Proc Natl Acad Sci USA. 2009;106:4665–4670. doi: 10.1073/pnas.0900947106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li C, Pazgier M, Li C, Yuan W, Liu M, Wei G, Lu WY, Lu W. Systematic mutational analysis of peptide inhibition of the p53-MDM2/MDMX interactions. J Mol Biol. 2010;398:200–213. doi: 10.1016/j.jmb.2010.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liu M, Pazgier M, Li C, Yuan W, Li C, Lu W. A left-handed solution to peptide inhibition of the p53-MDM2 interaction. Angew Chem Int Ed Engl. 2010;49:3649–3652. doi: 10.1002/anie.201000329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhan C, Zhao L, Wei X, Wu X, Chen X, Yuan W, Lu WY, Pazgier M, Lu W. An ultrahigh affinity d-peptide antagonist Of MDM2. J Med Chem. 2012;55:6237–6241. doi: 10.1021/jm3005465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wade D, Boman A, Wåhlin B, Drain CM, Andreu D, Boman HG, Merrifield RB. All-D amino acid-containing channel-forming antibiotic peptides. Proc Natl Acad Sci USA. 1990;87:4761–4765. doi: 10.1073/pnas.87.12.4761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cho Y, Turner JS, Dinh NN, Lehrer RI. Activity of protegrins against yeast-phase Candida albicans. Infect Immun. 1998;66:2486–2493. doi: 10.1128/iai.66.6.2486-2493.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Miyasaki KT, Iofel R, Oren A, Huynh T, Lehrer RI. Killing of Fusobacterium nucleatum, Porphyromonas gingivalis and Prevotella intermedia by protegrins. J Periodont Res. 1998;33:91–98. doi: 10.1111/j.1600-0765.1998.tb02297.x. [DOI] [PubMed] [Google Scholar]
- 50.Castle M, Nazarian A, Yi SS, Tempst P. Lethal effects of apidaecin on Escherichia coli involve sequential molecular interactions with diverse targets. J Biol Chem. 1999;274:32555–32564. doi: 10.1074/jbc.274.46.32555. [DOI] [PubMed] [Google Scholar]
- 51.Lehrer RI, Lu W. α-Defensins in human innate immunity. Immunol Rev. 2012;245:84–112. doi: 10.1111/j.1600-065X.2011.01082.x. [DOI] [PubMed] [Google Scholar]
- 52■.Wei G, de Leeuw E, Pazgier M, Yuan W, Zou G, Wang J, Ericksen B, Lu WY, Lehrer RI, Lu W. Through the looking glass, mechanistic insights from enantiomeric human defensins. J Biol Chem. 2009;284:29180–29192. doi: 10.1074/jbc.M109.018085. An elegant example of using mirror image proteins to probe the mechanisms of action of macromolecules in various biological processes. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kagan BL, Selsted ME, Ganz T, Lehrer RI. Antimicrobial defensin peptides formvoltage-dependent ion-permeable channels in planar lipid bilayer membranes. Proc Natl Acad Sci USA. 1990;87:210–214. doi: 10.1073/pnas.87.1.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Breukink E. Use of the Cell Wall Precursor Lipid II by a Pore-Forming Peptide Antibiotic. Science. 1999;286:2361–2364. doi: 10.1126/science.286.5448.2361. [DOI] [PubMed] [Google Scholar]
- 55.Hasper HE, Kramer NE, Smith JL, Hillman JD, Zachariah C, Kuipers OP, De Kruijff B, Breukink E. An Alternative Bactericidal Mechanism of Action for Lantibiotic Peptides That Target Lipid II. Science. 2006;313:1636–1637. doi: 10.1126/science.1129818. [DOI] [PubMed] [Google Scholar]
- 56.Hsu STD, Breukink E, Tischenko E, Lutters MAG, de Kruijff B, Kaptein R, Bonvin AMJJ, van Nuland NAJ. The nisin–lipid II complex reveals a pyrophosphate cage that provides a blueprint for novel antibiotics. Nat Struct Mol Biol. 2004;11:963–967. doi: 10.1038/nsmb830. [DOI] [PubMed] [Google Scholar]
- 57.Walsh C. MICROBIOLOGY:Deconstructing Vancomycin. Science. 1999;284:442–443. doi: 10.1126/science.284.5413.442. [DOI] [PubMed] [Google Scholar]
- 58.Breukink E, de Kruijff B. Lipid II as a target for antibiotics. Nat Rev Drug Discov. 2006;5:321–323. doi: 10.1038/nrd2004. [DOI] [PubMed] [Google Scholar]
- 59■.de Leeuw E, Li C, Zeng P, Li C, Diepeveen-de Buin M, Lu WY, Breukink E, Lu W. Functional interaction of human neutrophil peptide-1 with the cell wall precursor lipid II. FEBS Lett. 2010;584:1543–1548. doi: 10.1016/j.febslet.2010.03.004. The first demonstration that human alpha-defensins kill Gram-positive bacteriathrough lipid II sequestration. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Varney KM, Bonvin AMJJ, Pazgier M, Malin J, Yu W, Ateh E, Oashi T, Lu W, Huang J, Diepeveen-de Buin M, et al. Turning defense into offense: defensin mimetics as novel antibiotics targeting lipid II. PLoS Pathog. 2013;9:e1003732. doi: 10.1371/journal.ppat.1003732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Schmitt P, Wilmes M, Pugnière M, Aumelas A, Bachère E, Sahl HG, Schneider T, Destoumieux-Garzón D. Insight into invertebrate defensin mechanism of action: oyster defensins inhibit peptidoglycan biosynthesis by binding to lipid II. J Biol Chem. 2010;285:29208–29216. doi: 10.1074/jbc.M110.143388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62■■.Schneider T, Kruse T, Wimmer R, Wiedemann I, Sass V, Pag U, Jansen A, Nielsen AK, Mygind PH, Raventós DS, et al. Plectasin, a fungal defensin, targets the bacterial cell wall precursor Lipid II. Science. 2010;328:1168–1172. doi: 10.1126/science.1185723. This paper describes mechanistically how the fungal defensin plectasin kills Gram-positive bacteria through inhibition of bacterial cell wall synthesis and definitively identifies lipid II as the target molecule of defensins. [DOI] [PubMed] [Google Scholar]
- 63.Oeemig JS, Lynggaard C, Knudsen DH, Hansen FT, Nørgaard KD, Schneider T, Vad BS, Sandvang DH, Nielsen LA, Neve S, et al. Eurocin, a new fungal defensin: structure, lipid binding, and its mode of action. J Biol Chem. 2012;287:42361–42372. doi: 10.1074/jbc.M112.382028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sass V, Schneider T, Wilmes M, Körner C, Tossi A, Novikova N, Shamova O, Sahl HG. Human beta-defensin 3 inhibits cell wall biosynthesis in Staphylococci. Infect Immun. 2010;78:2793–2800. doi: 10.1128/IAI.00688-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wilmes M, Cammue BPA, Sahl HG, Thevissen K. Antibiotic activities of host defense peptides: more to it than lipid bilayer perturbation. Nat Prod Rep. 2011;28:1350. doi: 10.1039/c1np00022e. [DOI] [PubMed] [Google Scholar]