

Since its first description in 1963, significant advances in lipoprotein(a) [Lp(a)] biology have taken place. Lp(a) is composed of a low-density lipoprotein (LDL)-like particle where apolipoprotein B-100 (apoB) is covalently attached to the hydrophilic, glycosylated apolipoprotein(a) [apo(a)]. Our understanding of Lp(a)’s pathophysiology has been enhanced by epidemiology studies, meta-analyses and genetic instrumental variable analyses that suggest Lp(a) is a causal mediator, and not merely a risk marker, of cardiovascular disease (CVD) and calcific aortic valve stenosis (CAVS).1 Elevated plasma Lp(a) levels, >30–50 mg/dL or >75–125 nmol/L, are present in 20–30% of the general population, and in even higher percentages in patients with established CVD and CAVS. However, one area of Lp(a) biology that remains incompletely understood and in need of further clarification is defining the mechanisms regulating its catabolism.
In this issue of the Circulation Research, Sharma et al.2 elegantly extend our understanding of hepatic Lp(a) catabolism. Using confocal microscopy to study exogenously added Lp(a) to cultured hepatocytes and fibroblasts, the authors describe different intracellular fates of the lipid components of Lp(a) and of the protein apo(a). After initial binding and uptake, the lipid components leave the Rab5+ early endosomes and undergo lysosomal degradation. In contrast, apo(a) dissociates from apoB and traffics to the Golgi network, and ultimately recycles via Rab11+ endosomes and is secreted back into the media. Via this process, the authors suggest that ~30% of apo(a) recycles back to the extracellular space over a 24-hr period. The fate of any such “free” apo(a) is unknown, but it could bind to LDL in the extracellular space or circulate free until it binds LDL to generate Lp(a), be cleared by other receptors, enter the vessel wall or kidney or be degraded by proteolytic enzymes.
Furthermore, Sharma et al. showed that apo(a) internalization and recycling was diminished by inhibition of the plasminogen receptor (KT) (PlgRKT). This novel finding implicates PlgRKT, a widely-expressed receptor that binds plasminogen in Lp(a) catabolism. However, binding of Lp(a) to PlgRKT was not fully evaluated and plasminogen did not compete for Lp(a) internalization, so whether PlgRKT is a de facto Lp(a) receptor, solely an apo(a) recycling receptor or both remains to be clarified (Figure). Moreover, PlgRKT knockout cells internalize apo(a), albeit much less than control cells, suggesting additional pathways for Lp(a) catabolism. The authors excluded a significant contribution of the LDLR to Lp(a) uptake since an LDLR antibody known to compete with LDL binding was unable to alter Lp(a) clearance.
Figure. Proposed Model for Hepatic Lp(a) Catabolism and Apo(a) Recycling.
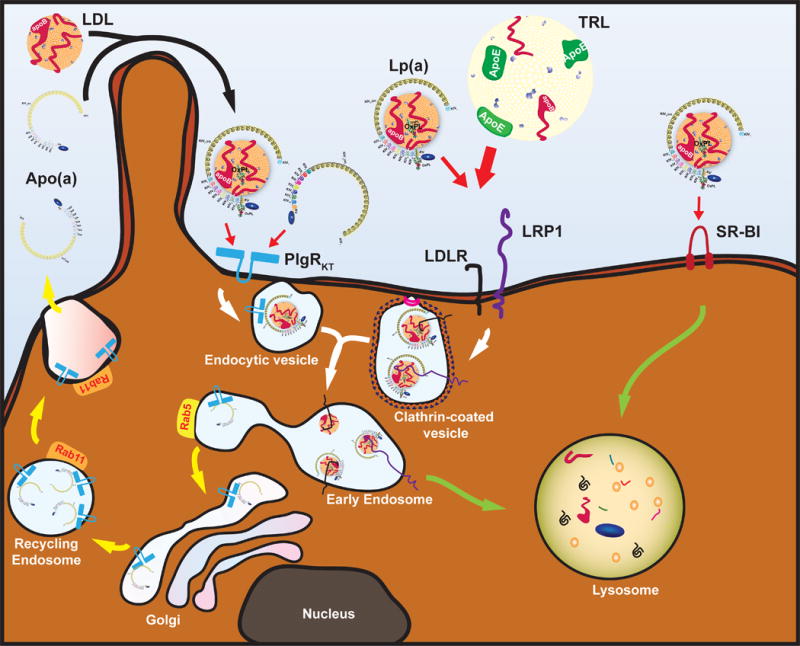
Several hepatic receptors are involved in Lp(a) catabolism. The plasminogen receptor PlgRKT is responsible for a futile cycle of Lp(a) and apo(a) uptake in Rab5 early endosomes with subsequent apo(a) recycling via Rab11 recycling endosomes. The secreted apo(a) can re-associate with newly-formed or circulating LDL to generate new Lp(a). LDLR and LRP1 are PCSK9-sensitive clearance receptors for which Lp(a) needs to compete with apoE-bearing TRLs, a process influenced by the apoE genotype. Uptake via this clathrin-mediated endocytosis pathway will lead to lysosomal degradation of the Lp(a)-lipid moiety (i.e. LDL) and associated apo(a). Finally, other receptors such as SR-BI have been implicated and result in lysosomal degradation of the whole particle or the lipid content.
The LPA gene on chromosome 6, evolutionarily derived from the neighboring plasminogen gene, encodes kringle domains KIV, KV and an inactive protease domain. KIV of LPA contains 10 subtypes, of which KIV2 is composed of a variable number of tandem repeats, ranging from 1 to >40. For this reason, the apo(a) protein is highly polymorphic among human populations. Large apo(a) isoforms with high number of repeats are synthesized slower in molar quantities than smaller apo(a) isoforms. Therefore, apo(a) size is inversely correlated with plasma Lp(a) levels, with small isoforms being highly overrepresented in patients with elevated Lp(a) levels. Statins, the mainstay of preventative therapy for CVD, reduce circulating LDL-C via upregulation of the LDL receptor (LDLR). Interestingly, statins do not lower plasma Lp(a), and may in fact raise them 10–20%.3 Therefore further increases in Lp(a) with statins may represent a significant component of the “residual risk” not addressed by current medical management. This generates a clinical need to understand the mechanisms involved in Lp(a) catabolism and to identify novel approaches to lower Lp(a).
The significance of these findings is that apo(a) is trafficked via a pathway distinct from the well described endo-lysosomal circuit for LDLR mediated LDL internalization. The role of LDLR in Lp(a) catabolism is controversial with divergent findings in cell culture.4,5 Multiple cohort studies with familial hypercholesterolemia (FH) patients show an LDLR gene-dose effect on Lp(a) levels with highest levels in those with homozygous FH (HoFH) and lowest in non-FH subjects. Consistent with this, mice that overexpress human LDLR show accelerated Lp(a) clearance.6 Experiments in human cell lines demonstrate that Lp(a) can bind the LDLR and that Lp(a) internalization is reduced by adding the proprotein convertase subtilisin/kexin type 9 (PCSK9) which shunts the LDLR for lysosomal degradation,4,7 PCSK9 inhibition also lowers plasma Lp(a) levels in all patient subsets and increases the fractional catabolic rate (FCR) of apo(a) in healthy volunteers.8 However, the FCR of Lp(a) in LDLR-null mice is not different from wild-type controls9 or between HoFH patients and their heterozygous relatives, albeit in very small studies, and other cells culture studies have suggested no role of the LDLR in PCSK9 mediated Lp(a) reduction.5 In further support of an LDLR independent clearance pathway, evolocumab treated LDLR-null HoFH subjects (n=2) experienced a 16–20% decrease in Lp(a) whereas their LDL-C remained unchanged,10 but a larger study that included 8 LDLR-null HoFH patients suggested much smaller reductions of <10%.11 The mechanisms of PCSK9-mediated Lp(a) reduction need to be reconciled with more definitive studies on Lp(a) catabolism. Additional receptors in Lp(a) clearance have also been suggested by preclinical models reflecting human disease including the VLDLR, LRP1 and SR-BI.
More recently, a study of 431,239 patients found a strong correlation between APOE genotype and plasma Lp(a) levels. Individuals with APOE e4/e4 genotype had 65% higher Lp(a) levels compared to those with e2/e2 genotype.12 ApoE4 has high affinity for large lipoproteins and facilitates triglyceride-rich lipoprotein (TRL) clearance via high affinity binding to LDLR, LRP-1 and syndecan-1. In contrast, apoE2 binds to LDLR and LRP1, but not syndecan-1, with lower affinity, implying that apoE on TRLs competes with Lp(a) for LDLR and LRP1-mediated hepatic clearance. This could mean that the apoE-sensitive clearance receptors either precede the apo(a) recycling pathway or bypass the recycling pathway and set up Lp(a) directly for lysosomal degradation (Figure). The kidney may also play a role in the relationship between Lp(a) and apoE since it expresses high levels of LRP1 and megalin/LRP2, both of which bind apoE and Lp(a), and apo(a) fragments are also found in the urine, even in patients with very low Lp(a) levels.
There are a few limitations to consider when interpreting these studies and similar studies in the literature. First, the translatability of in vitro cell culture studies to in vivo effects in humans remains a potential limitation. Second, isolation of “pure” Lp(a) has not been perfected, and most preparations have variable amounts of LDL and HDL when analyzed by sensitive ELISA techniques, which may impact interpretation of Lp(a) binding to LDLR, LRP1 and SRB1 in setting of small amounts of contaminating lipoproteins. Third, the authors evaluated the effects of the PlgRKT on apo(a) 6 hours into the experiment, where most Lp(a) was internalized within 1 hour. This suggests that apo(a) may not be internalized by PlgRKT but rather resorted to PlgRKT in the early endosome that allows slow recycling and prevention of rapid lysosomal degradation. Consistent with this concept is the observation that PlgRKT knockdown or overexpression affected the cellular apo(a) content and not apoB or plasminogen uptake. It remains unclear if Lp(a)-associated apoB uptake was affected in the PlgRKT-deficient cells when challenged with Lp(a), which may have clarified this issue. Finally, the relative quantitative contribution and clinical relevance of the PlgRKT pathway compared to other pathways needs to be defined in future studies.
Overall, the study by Sharma et al. provides valuable insights into Lp(a) catabolism and suggests a novel physiological pathway for apo(a) catabolism and apo(a) recycling. Additionally, whether this thrifty conservation pathway of apo(a) protein had a primordial beneficial function that no longer seems to be needed, as lack of Lp(a) does not apparently lead to any known pathophysiological consequences,1 remains to be determined. It was also intriguing that the authors suggested that apo(a) may mediate the degradation within lysosomes of accumulated oxidized phospholipids on apo(a) from cell membranes. Oxidized phospholipids on apoB (OxPL-apoB), mainly reflecting OxPL on Lp(a), have been shown to be integral in the clinical prediction of Lp(a)-mediated CVD risk.13 This may represent a putative, long-sought physiological function of Lp(a) that may explain its evolutionary arrival as a remodeled duplication from plasminogen, which also carries oxidized phospholipids.14 This hypothesis can be tested by quantitating OxPL, either with mass spectroscopy or immunostaining with antibody E06, in apo(a) before and after secretion to the media.
Going forward, it will be important to systematically characterize the interaction between Lp(a) and its various components in vitro and in vivo to optimally define the physiological contribution of each receptor and pathway to Lp(a) catabolism. Ultimately, with such an understanding, novel means to decrease circulating plasma Lp(a) levels by accelerating catabolism of Lp(a) or apo(a) may be developed, in parallel with the recent successes in reducing apo(a) synthesis with antisense oligonucleotides.15
Acknowledgments
Dr Tsimikas is supported by NHLBI grants NIH HL119828, HL055798, HL088093, HL106579, HL078610, HL124174. Dr Gordts is supported by AHA grant 15BGIA25550111.
Footnotes
Disclosures
Dr Tsimikas is a co-inventor of and receives royalties from patents or patent applications owned by the University of California San Diego on antibodies used in biotheranostic applications and has a dual appointment at Ionis Pharmaceuticals, Inc. and UCSD. The other authors have no conflicts to disclose.
References
- 1.Tsimikas S. A Test in Context: Lipoprotein(a): Diagnosis, Prognosis, Controversies, and Emerging Therapies. J Am Coll Cardiol. 2017;69:692–711. doi: 10.1016/j.jacc.2016.11.042. [DOI] [PubMed] [Google Scholar]
- 2.Sharma M, Redpath GM, Williams MJ, McCormick SP. Recycling of apolipoprotein(a) after PlgRKT-mediated endocytosis of lipoprotein(a) Circ Res. 2016 doi: 10.1161/CIRCRESAHA.116.310272. [DOI] [PubMed] [Google Scholar]
- 3.Yeang C, Wilkinson MJ, Tsimikas S. Lipoprotein(a) and oxidized phospholipids in calcific aortic valve stenosis. Current opinion in cardiology. 2016;31:440–450. doi: 10.1097/HCO.0000000000000300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Romagnuolo R, Scipione CA, Boffa MB, Marcovina SM, Seidah NG, Koschinsky ML. Lipoprotein(a) catabolism is regulated by proprotein convertase subtilisin/kexin type 9 through the low density lipoprotein receptor. J Biol Chem. 2015;290:11649–11662. doi: 10.1074/jbc.M114.611988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Villard EF, Thedrez A, Blankenstein J, Croyal M, Tran T-T-T, Poirier B, Le Bail J-C, Illiano S, Nobécourt E, Krempf M, Blom DJ, Marais AD, Janiak P, Muslin AJ, Guillot E, Lambert G. PCSK9 Modulates the secretion but not the cellular uptake of lipoprotein(a) ex vivo. JACC: Basic to Translational Science. 2016;1:419. doi: 10.1016/j.jacbts.2016.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hofmann SL, Eaton DL, Brown MS, McConathy WJ, Goldstein JL, Hammer RE. Overexpression of human low density lipoprotein receptors leads to accelerated catabolism of Lp(a) lipoprotein in transgenic mice. J Clin Invest. 1990;85:1542–1547. doi: 10.1172/JCI114602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Raal FJ, Giugliano RP, Sabatine MS, Koren MJ, Blom D, Seidah NG, Honarpour N, Lira A, Xue A, Chiruvolu P, Jackson S, Di M, Peach M, Somaratne R, Wasserman SM, Scott R, Stein EA. PCSK9 inhibition-mediated reduction in Lp(a) with evolocumab: an analysis of 10 clinical trials and the LDL receptor’s role. J Lipid Res. 2016;57:1086–1096. doi: 10.1194/jlr.P065334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Reyes-Soffer G, Pavlyha M, Ngai C, Thomas T, Holleran S, Ramakrishnan R, Karmally W, Nandakumar R, Fontanez N, Obunike J, Marcovina SM, Lichtenstein AH, Matthan NR, Matta J, Maroccia M, Becue F, Poitiers F, Swanson B, Cowan L, Sasiela WJ, Surks HK, Ginsberg HN. Effects of PCSK9 Inhibition with alirocumab on lipoprotein metabolism in healthy humans. Circulation. 2017;135:352–362. doi: 10.1161/CIRCULATIONAHA.116.025253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cain WJ, Millar JS, Himebauch AS, Tietge UJF, Maugeais C, Usher D, Rader DJ. Lipoprotein [a] is cleared from the plasma primarily by the liver in a process mediated by apolipoprotein [a] J Lipid Res. 2005;46:2681–2691. doi: 10.1194/jlr.M500249-JLR200. [DOI] [PubMed] [Google Scholar]
- 10.Stein EA, Honarpour N, Wasserman SM, Xu F, Scott R, Raal FJ. Effect of the proprotein convertase subtilisin/kexin 9 monoclonal antibody, AMG 145, in homozygous familial hypercholesterolemia. Circulation. 2013;128:2113–2120. doi: 10.1161/CIRCULATIONAHA.113.004678. [DOI] [PubMed] [Google Scholar]
- 11.Raal FJ, Hovingh GK, Blom D, Santos RD, Harada-Shiba M, Bruckert E, Couture P, Soran H, Watts GF, Kurtz C, Honarpour N, Tang L, Kasichayanula S, Wasserman SM, Stein EA. Long-term treatment with evolocumab added to conventional drug therapy, with or without apheresis, in patients with homozygous familial hypercholesterolaemia: an interim subset analysis of the open-label TAUSSIG study. Lancet Diabetes Endocrinol. 2017 doi: 10.1016/S2213-8587(17)30044-X. [DOI] [PubMed] [Google Scholar]
- 12.Moriarty PM, Varvel SA, Gordts PL, McConnell JP, Tsimikas S. Lipoprotein(a) mass levels increase significantly according to APOE Genotype: an analysis of 431 239 patients. Arterioscler Thromb Vasc Biol. 2017 doi: 10.1161/ATVBAHA.116.308704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Byun YS, Yang X, Bao W, DeMicco D, Laskey R, Witztum JL, Tsimikas S, Investigators ST Oxidized phospholipids on apolipoprotein B-100 and recurrent ischemic events following stroke or transient ischemic attack. J Am Coll Cardiol. 2017;69:147–158. doi: 10.1016/j.jacc.2016.10.057. [DOI] [PubMed] [Google Scholar]
- 14.Leibundgut G, Arai K, Orsoni A, Yin H, Scipione C, Miller ER, Koschinsky ML, Chapman MJ, Witztum JL, Tsimikas S. Oxidized phospholipids are present on plasminogen, affect fibrinolysis, and increase following acute myocardial infarction. J Am Coll Cardiol. 2012;59:1426–1437. doi: 10.1016/j.jacc.2011.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Viney NJ, van Capelleveen JC, Geary RS, Xia S, Tami JA, Yu RZ, Marcovina SM, Hughes SG, Graham MJ, Crooke RM, Crooke ST, Witztum JL, Stroes ES, Tsimikas S. Antisense oligonucleotides targeting apolipoprotein(a) in people with raised lipoprotein(a): two randomised, double-blind, placebo-controlled, dose-ranging trials. Lancet. 2016;388:2239–2253. doi: 10.1016/S0140-6736(16)31009-1. [DOI] [PubMed] [Google Scholar]