

Abstract
Mucosal melanomas are a rare subtype of melanoma, arising in mucosal tissues, which have a very poor prognosis due to the lack of effective targeted therapies. This study aimed to better understand the molecular landscape of these cancers and find potential new therapeutic targets. Whole-exome sequencing was performed on mucosal melanomas from 19 patients and 135 sun-exposed cutaneous melanomas, with matched peripheral blood samples when available. Mutational profiles were compared between mucosal subgroups and sun-exposed cutaneous melanomas. Comparisons of molecular profiles identified 161 genes enriched in mucosal melanoma (P<0.05). KIT and NF1 were frequently comutated (32%) in the mucosal subgroup, with a significantly higher incidence than that in cutaneous melanoma (4%). Recurrent SF3B1 R625H/S/C mutations were identified and validated in 7 of 19 (37%) mucosal melanoma patients. Mutations in the spliceosome pathway were found to be enriched in mucosal melanomas when compared with cutaneous melanomas. Alternative splicing in four genes were observed in SF3B1-mutant samples compared with the wild-type samples. This study identified potential new therapeutic targets for mucosal melanoma, including comutation of NF1 and KIT, and recurrent R625 mutations in SF3B1. This is the first report of SF3B1 R625 mutations in vulvovaginal mucosal melanoma, with the largest whole-exome sequencing project of mucosal melanomas to date. The results here also indicated that the mutations in SF3B1 lead to alternative splicing in multiple genes. These findings expand our knowledge of this rare disease.
Keywords: genomic landscape, mucosal melanoma, SF3B1 mutation, spliceosome, whole-exome sequencing
Introduction
Melanomas in mucosal tissue are uncommon and account for only 1% or less of all melanomas. They are not related to sun exposure or other known environmental factors and are not familial. Mucosal melanomas have been shown to have distinct molecular profiles compared with cutaneous melanoma arising on sun-exposed sites 1,2. Unfortunately, many of the advances made in the understanding and treatment of cutaneous melanoma in the past 5 years are not readily applicable to mucosal melanomas 3,4. Common molecular drivers identified in cutaneous melanoma, such as mutated BRAF V600E, have not been identified in mucosal melanoma. Oncogenic drivers of mucosal melanoma remain unclear as there have been no consistent molecular targets described 5. In addition, the current immunotherapies targeting CTLA4 and PD1 for cutaneous melanomas have had little success in mucosal melanomas 3.
In order to better understand the mutational profile of mucosal melanoma, several investigators have performed next-generation sequencing on a small number of mucosal melanomas and compared them with cutaneous melanoma. Furney et al. 1 examined the mutational spectra of 10 mucosal melanomas from the head and neck, rectum, penis, and vagina. These studies have provided the foundation for understanding the molecular profiles of mucosal melanoma. Our study expands this knowledge by contributing the largest whole-exome sequenced cohort of mucosal melanoma known to date with validated mutations and differentially spliced genes as a result of these mutations.
In the present study, we performed whole-exome sequencing (WES) on tissue samples from 19 patients with mucosal melanomas from various sites. The samples were from five anorectal melanomas, five nasopharyngeal melanomas, and nine vulvovaginal melanomas. We compared and contrasted mutational profiles from these with 135 cutaneous melanomas from sun-exposed sites from our in-house melanoma tissue bank [International Melanoma Biorepository and Research Laboratory (IMBRL)]. We identified comutations in NF1 and KIT in 32% of our patient samples. Discovery of these comutations could provide insight into the lack of response of mucosal melanoma to traditional melanoma treatments. Importantly, we discovered a recurrent R625 mutation in SF3B1, in vulvovaginal melanomas that was previously only described in anorectal and ocular melanomas 6,7. We confirmed that SF3B1-mutant samples have differential splicing of at least four genes found in previously described SF3B1-mutant cancers 6,8. This study provides important new clues into the molecular changes that occur in mucosal melanomas. These findings may lead to a better understanding of the development, molecular drivers, and hence improved therapeutics for these rare forms of melanoma.
Methods
Clinical review
The IMBRL at the University of Colorado Cancer Center currently contains over 3900 melanoma patient tissue and peripheral blood samples. Nineteen mucosal melanoma patients had formalin-fixed paraffin-embedded, and/or fresh frozen tissues meeting quality standards for mutational analysis by means of WES and had (when available) normal peripheral blood DNA in our biorepository. More than one tissue sample was available for some patients. WES was performed on 40 tissue samples from 19 patients.
Sample collection and genomic DNA isolation
Tissue and peripheral blood samples were collected between 2008 and 2016 from melanoma patients and stored in the IMBRL at the University of Colorado Cancer Center. All collections were made in accordance with review board approval and with the patient’s written consent (COMIRB-05-0309). Many of these patients had multiple tissue or peripheral blood samples collected at different times, and all samples from which adequate DNA could be extracted were submitted for WES. Blood samples were collected in PAXGene (Qiagen & BD, Feldbachstrasse, Switzerland) DNA tubes and stored at 4°C until processed. Depending on material, tissue genomic DNA was isolated using the DNeasy Blood and Tissue kit (Qiagen, Valencia, California, USA) or the QiaAmp DNA formalin-fixed paraffin-embedded kit (Qiagen). The concentration of DNA was determined using a UV spectrophotometer and stored at 4°C.
Next-generation whole-exome sequencing
DNA concentration and purity were determined using Qubit (Thermo Fisher Scientific, Waltham, Massachusetts, USA) and Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, California, USA) analysis. Genomic DNA (200 ng) was sheared using Covaris (Woburn, Massachusetts, USA) S220 at 150 bp. Sheared DNA was used to construct the exome library following Agilent SureSelect XT Target Enrichment System (Agilent Technologies) for Illumina Paired End Multiplexed Sequencing Library (catalog #G9641B; Illumina, San Diego, California, USA). Sheared DNA was end repaired, followed by addition of adapter tags to construct DNA libraries through PCR amplification. Exome capture was performed through hybridization using XT5 probe. Resulting captured libraries were indexed and purified. The cDNA library was validated on the Agilent 2100 Bioanalyzer using DNA-1000 chip. Libraries were sequenced on the Illumina HiSeq 2000 with 125 bp paired-end reads. We obtained an average of 400× and 200× sequencing coverage for the cancer and normal exomes, respectively.
Whole-exome sequencing pipeline and bioinformatics analysis
In order to establish the mutational profiles in mucosal melanomas, we used WES to analyze tissue and matched peripheral blood (when available) from 19 patients with mucosal melanomas. For comparison, we used WES data from 135 primary and metastatic cutaneous sun-exposed melanoma samples from our biorepository.
All samples from both cutaneous and mucosal patients were analyzed for variants and copy number alterations using IMPACT, our recently published WES analysis pipeline 9. In brief, IMPACT is a discovery-oriented WES analysis pipeline designed to detect variant reads found in at least 10% of sequencing reads (minimum of four variant reads out of 20 mapped reads). To eliminate potential false positives, IMPACT performs a stringent filtering step, keeping only nonsynonymous somatic variants that are predicted to be deleterious by SIFT 10 and PolyPhen2 11 or were present in the COSMIC database 12. By doing this, IMPACT is able to detect more subclonal variants but only keeps those variants that are predicted to be the most deleterious. To remove germline variants, a normal blood DNA sample from the same patient was used as control. Somatic variants found in each patient are listed in Supplementary Table 1 (http://tanlab.ucdenver.edu/Mucosal_Melanoma/MucosalData/).
Somatic variants in genes were compared between mucosal and cutaneous groups. A Fisher’s exact test was performed for each gene found in at least 30% of the mucosal samples. False discovery rate was applied to the gene list. Genes with P value less than 0.05 and false discovery rate less than 20% were considered as mutated genes enriched in the mucosal samples. The BAM files from our mucosal cohort can be downloaded at http://tanlab.ucdenver.edu/Mucosal_Melanoma/MucosalData/. We queried the mutated genes with the KEGG pathway database to identify enriched pathways in the mucosal samples 13. For mucosal samples in which we had more than one sample, we combined all samples into one profile for each patient. A total of 161 genes were found to be enriched in mucosal melanoma (Supplementary Table 2, http://tanlab.ucdenver.edu/Mucosal_Melanoma/MucosalData/).
Direct Sanger sequencing of SF3B1 R625 variants
Exon 14 of the SF3B1 gene was amplified from ∼100 to 300 ng of genomic DNA. Previously reported forward and reverse primers were used 14. PCR was carried out using GoTaq Master Mix (Promega, Madison, Wisconsin, USA) with a final primer concentration of 0.4 mmol/l and a final reaction volume of 50 μl. Thermocycling was carried out on the GeneAmp PCR System (Applied Biosystems, Carlsbad, California, USA). PCR products were visualized on a 1.5% agarose gel. PCR products were purified using the ExoSap IT enzyme mix (USB Corporation, Santa Clara, California, USA). Approximately 15 ng/ml of the PCR product was sent for direct sequence analysis at the Barbara Davis Center Molecular Biology Core Facility.
Splice variant analysis using quantitative reverse-transcription polymerase chain reaction
Previously reported forward and reverse primers were retrieved from Furney et al.
6 and Maguire et al. 8 for predicted splice variants in SF3B1-mutant and wild-type samples (Supplementary Table 2, http://tanlab.ucdenver.edu/Mucosal_Melanoma/MucosalData/). cDNA (10 ng) was analyzed in triplicate to quantify spliced and unspliced forms using real-time quantitative PCR, and repeated three times for an n=3. Briefly, 45 cycles of quantitative reverse-transcription PCR (qRT-PCR) were conducted in 96-well plates using PowerUp SYBR Green reagents (Invitrogen, Thermo Fisher Scientific) on the StepOnePlus instrument (Applied Biosystems). To perform splice variant analysis for each gene of interest was calculated by averaging the Ct values per sample, and calculating the ratio of spliced form using the formula 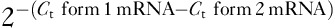 . For each splicing event, a two-tailed unpaired t-test (using GraphPad Prism software; GraphPad Software Inc., La Jolla, California, USA) was applied between SF3B1 mutated (n=3) and wild-type (n=4) cases. Differences were considered significant at P value less than 0.05.
. For each splicing event, a two-tailed unpaired t-test (using GraphPad Prism software; GraphPad Software Inc., La Jolla, California, USA) was applied between SF3B1 mutated (n=3) and wild-type (n=4) cases. Differences were considered significant at P value less than 0.05.
Patient-derived tumor xenograft and cell line models
The human mucosal melanoma explant xenografts were generated according to previously published methods 15,16. Female athymic nude (nu/nu) mice were purchased form Harlan Labs at age 4–6 weeks. Briefly, surgical specimens from consented patients undergoing removal of primary or metastatic tumor at the University of Colorado Hospital were reimplanted subcutaneously into five mice for each patient. Tumors were allowed to grow to a size of 1000–1500 mm3 (F1), at which point they were harvested, divided, and transplanted to another five mice (F2) to maintain the tumor bank. Tumor samples were collected from F1 and F2 for RNA isolation. Animal studies were performed in a facility accredited by the American Association for Accreditation of Laboratory Animal Care in accordance with the NIH guidelines for care and use of laboratory animals and approved by the University of Colorado Institutional Animal Care and Use Committee before initiation. Cell lines Mel202 (ECACC) and MP41 (ATCC) were maintained according to manufacturer’s instructions. The SF3B1 status was verified by means of Sanger sequencing using the methods and primers (Supplementary Table 3, http://tanlab.ucdenver.edu/Mucosal_Melanoma/MucosalData/) described above.
Results
Mucosal melanoma patient information
To define the mutational landscape of mucosal melanoma patients, we performed WES on 19 patients with paired tumor and normal samples. On average, we obtained 400× and 200× coverage for the cancer and normal exomes (when available), respectively. The clinical details of the patients included in this study are shown in Table 1. There were five patients each with anorectal and nasopharyngeal melanoma and nine patients with vulvovaginal melanomas. The median age was 64 years across all subtypes. The tissue source includes a combination of both primary and metastatic lesions (Fig. 1 and Table 1).
Table 1.
Cumulative clinical data and mutational status for 19 patients with mucosal melanoma
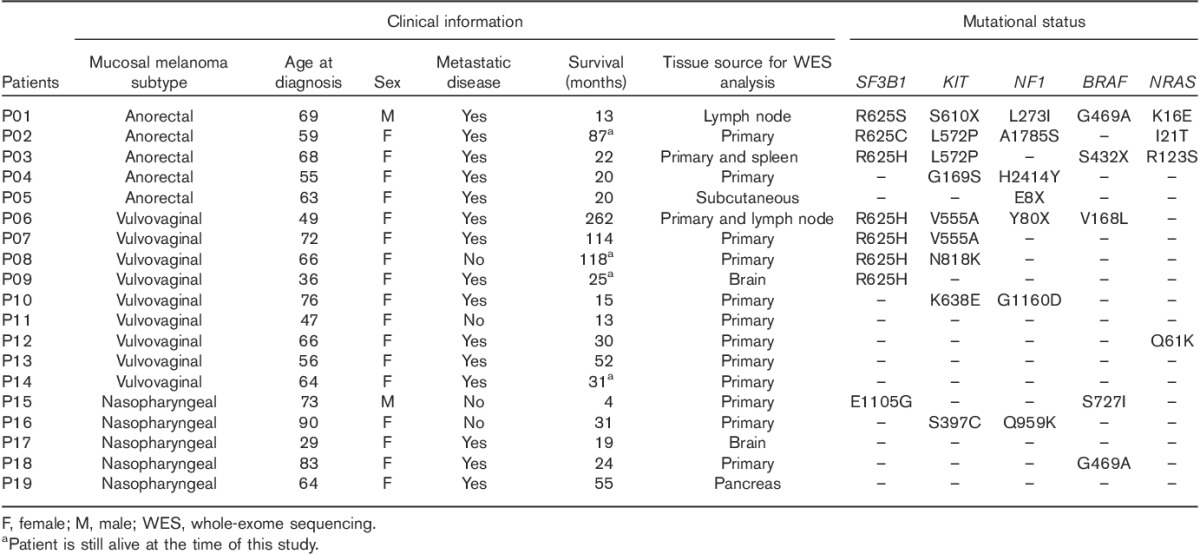
Fig. 1.
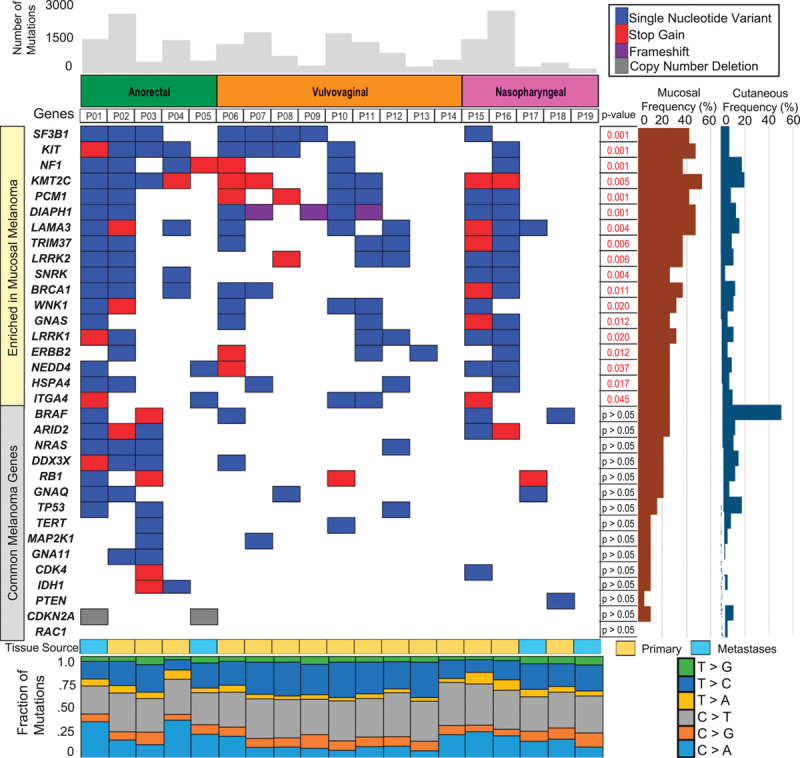
Mutational landscape of 19 mucosal melanomas compared with cutaneous melanomas. Important differences in individual genes are shown along with P values. Note in particular the significantly higher frequency of mutations in KIT, NF1, and SF3B1 in mucosal melanomas. Common melanoma genes occur in low frequency as highlighted within the figure. Tissue source and mutational signature for individual samples are presented in the lower panel.
Mutated genes enriched in mucosal melanomas
Overall, 161 genes were found to be enriched in the mucosal patient samples (Supplementary Table 2, http://tanlab.ucdenver.edu/Mucosal_Melanoma/MucosalData/) when compared with our cutaneous melanoma cohort. KM2TC was the most frequently mutated gene (52%) in the cohort, followed by KIT, DIAPH1, and LAMA3 (50%). NF1 was found mutated in seven patient samples; six of these patient samples (32% of all samples) also had a co-occurring KIT mutation. Figure 1 summarizes the genes significantly enriched for mutations in mucosal versus cutaneous melanomas.
We identified SF3B1, with a recurrent somatic mutation at codon R625 in 37% (7/19) of the mucosal melanomas. Notably, the recurrent SF3B1 mutation was only found to be mutated in anorectal and vulvovaginal patients but not in nasopharyngeal melanoma samples. There was one nasopharyngeal patient with a previously undescribed SF3B1 E1105G mutation. Table 1 describes the specific mutations found in each patient sample.
In contrast, oncogenic driver mutations in BRAF were not detected in mucosal melanomas, and the mutations found were not commonly identified in cutaneous melanoma (Fig. 1 and Table 1). Other commonly mutated genes (e.g. NRAS, GNA11, GNAS) in cutaneous and/or uveal melanoma are not enriched in mucosal melanomas (Fig. 1). Figure 2 molecularly classifies the patient samples with the common mutations of BRAF (V600), NRAS (G12, G13, Q61), NF1, and KIT mutations in our mucosal and cutaneous cohorts, as well as those samples that lacked these gene mutations (pan negative). The most striking observation is the significantly higher occurrence of comutation of KIT and NF1 in mucosal melanoma (32%), compared with cutaneous melanoma (1%) (Fig. 2b). This occurrence was observed in 22% in the vulvovaginal, 20% in the nasopharyngeal, and 60% in the anorectal subtype (Fig. 2b). We found that the nasopharyngeal mucosal melanomas are highly enriched (80%) in the pan-negative cluster (Fig. 2b). This supports the hypothesis that mucosal melanomas have a different mutational landscape compared with the cutaneous melanomas.
Fig. 2.
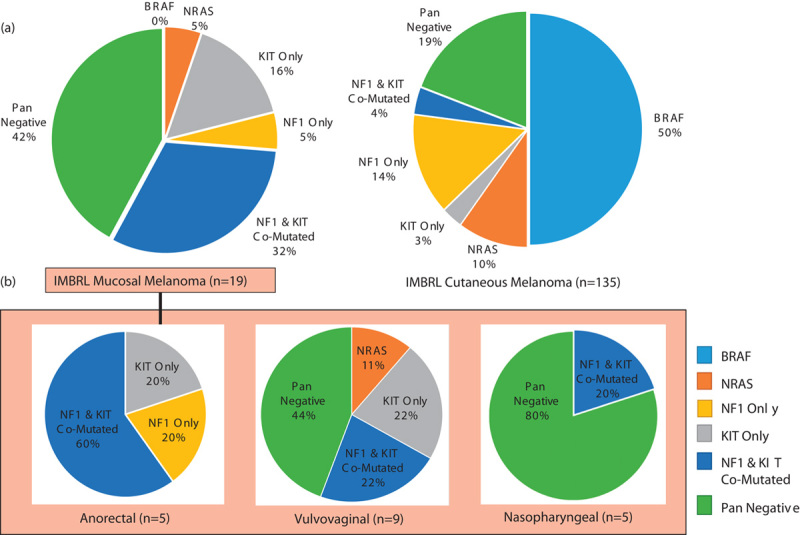
Molecular classification of melanoma with BRAF (V600), NRAS (G12, G13, Q61), NF1, and KIT mutations. (a) Comparison of all 19 mucosal melanomas with our 135 cutaneous melanomas. (b) The difference in molecular classifications between mucosal melanomas arising in different anatomic sites.
Pathways enriched in mucosal melanomas versus cutaneous melanomas
We next investigated these enriched genes mutated in mucosal melanomas by querying KEGG pathway database. We found that these enriched genes in mucosal melanomas were involved in focal adhesion, cell adhesion, the RAS, MAPK, and PI3K-AKT signaling pathways, as well as the spliceosome pathway (Fig. 3a). SF3B1 was the most enriched gene in the mucosal cohort, and the pathway analysis led to the identification of another spliceosome gene, EFTUD2. We expanded our investigation to include gene members described in the KEGG spliceosome pathway and published papers 8. We found that 68% of mucosal melanoma patients had one or more of the spliceosome genes mutated (Fig. 3b). The number of genes enriched in this pathway in the mucosal patient samples was significant when compared with the cutaneous cohort (P=0.0097). The occurrence of these mutations within the spliceosome pathway is depicted in Fig. 3c.
Fig. 3.
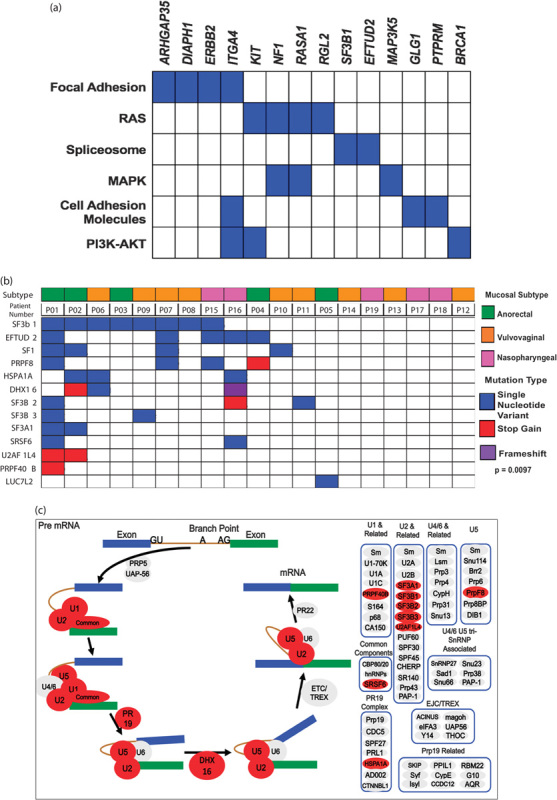
(a) Pathway analysis of genes enriched in mucosal melanoma. (b) Spliceosome pathway genes found in mucosal melanoma were enriched when compared with cutaneous melanomas. (c) Pathway visualization of the spliceosome pathway with mutated genes shown in red.
Recurrent mutation in SF3B1 in anorectal and vulvovaginal melanomas
From the WES analysis, we identified a total of seven vulvovaginal and anorectal mucosal melanomas harboring the SF3B1 recurrent mutations (R625H/S/C) and one nasopharyngeal mucosal melanoma mutated at E1105G. Within anorectal and vulvovaginal melanomas, SF3B1 R625 mutations occurred in 50% (7/14) of all samples. The recurrent mutation at codon R625 in SF3B1 is similar to the previously reported mutation in uveal melanoma 17. To validate this recurrent R625 mutation in mucosal melanoma identified by means of WES, we performed direct Sanger sequencing on exon 14 of SF3B1 in all of our SF3B1-mutant mucosal patient samples, and a subset of SF3B1 wild-type samples. The Sanger sequencing results validated the WES findings of SF3B1 R625 mutations in these samples (Supplementary Fig. 1, http://tanlab.ucdenver.edu/Mucosal_Melanoma/MucosalData/).
SF3B1 is a component of the spliceosome and plays a key role in early stages of pre-mRNA splicing (Fig. 3c). Mutations in SF3B1 are rare in cutaneous melanomas 14, and none of our in-house 135 cutaneous melanomas had an SF3B1 mutation. This suggests that SF3B1 might be exploited as a novel prognostic and/or therapeutic target, specifically in mucosal melanomas for anorectal and vulvovaginal sites. Figure 4a illustrates the known SF3B1 mutations found in this study and other published cancer studies from cBioPortal 7,18. Both uveal melanoma and breast cancer harbor mutations in the HEAT domain of SF3B1, with a majority of samples having mutations at positions R625 and K700, respectively (Fig. 4a) 6,8,17. Mutations in the HEAT (Huntingtin, Elongation factor 3, protein phosphatase 2A, Targets of rapamycin 1) domain of SF3B1, such as R625 and K700, have been associated with alternative splicing events, primarily through the induction of aberrant 3′ splice site selection 6,8,19.
Fig. 4.
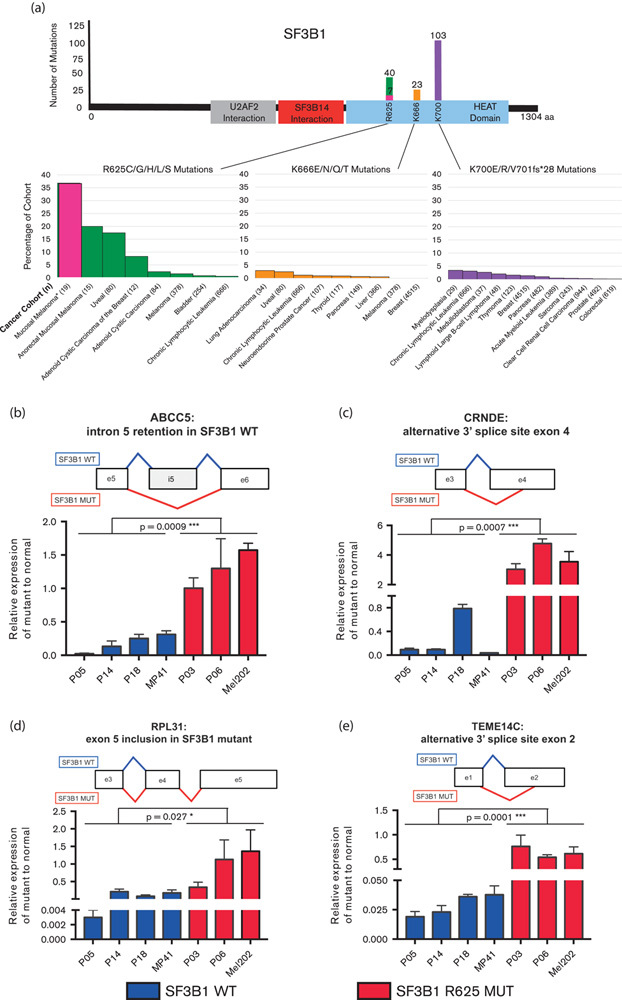
(a) Number and location of SF3B1 mutations found in the literature and cBioPortal, followed by the proportion of each cancer’s subtype. Highlighted in pink is the current study representing the largest percentage SF3B1 mutations of any cohort to date. (b–e) Analysis of differentially spliced genes identified in SF3B1-mutant uveal melanoma and breast cancer. (b) CRNDE, (c) RPL31, (d) ABCC5, and (e) TMEM14C were validated using quantitative reverse-transcription PCR in three SF3B1 wild-type (blue) and two SF3B1 R625 (red) mucosal melanoma PDX samples. Uveal melanoma cell lines were used as controls, MP41 (SF3B1 wild-type) and Mel202 (SF3B1 R625G). Unpaired two-tail parametric t-tests were used to determine significance between mutant and wild-type. *P value less than 0.05 was considered statistically significant. Schematic representations of the splicing events is illustrated above each graph. Exons are represented as boxes, with lines representing splicing events occurring in SF3B1 wild-type (blue) and SF3B1 mutant (red). ***P value of less than 0.01.
Functional validation of differential splicing genes in SF3B1-mutant mucosal melanomas
Recent functional studies in uveal melanoma, breast cancer, and hematological malignancies have identified genes that are differentially spliced in SF3B1-mutant tumors compared with wild type 6,8,20. Considering the functional role of mutant SF3B1 in inducing differential splicing, we hypothesized that SF3B1 R625H/S/C mutations in mucosal melanoma would also lead to differential splicing of genes as compared with SF3B1 wild-type tumors.
To test this hypothesis, we performed qRT-PCR using specific primer design to target alternatively spliced genes previously found in SF3B1-mutant samples. We selected seven genes (ABCC5, ANKHD1, CRNDE, GUSPB11, RPL31, TMEM14C, and UQCC) that have been previously identified by means of RNA sequencing and validated using qRT-PCR to be differentially spliced in SF3B1 R625 and K700E mutants compared with wild-type uveal melanomas and breast cancer 6,8. To validate these alternatively spliced genes in mucosal melanomas, we tested SF3B1 R625H-mutant (n=2) and wild-type (n=3) patient-derived xenografts established from our mucosal melanoma primary samples. Uveal melanoma cell lines Mel202 (SF3B1 R625G) and MP41 (SF3B1 wild-type) were utilized as positive and negative controls, respectively, for differential splicing analysis. This analysis identified all seven genes to be differentially spliced in mucosal melanomas with SF3B1 mutant as compared with wild-type samples (Fig. 4b–e and Supplementary Fig. 2, http://tanlab.ucdenver.edu/Mucosal_Melanoma/MucosalData/). This demonstrates that SF3B1 R625-mutant mucosal melanomas are functionally involved in alternative splicing of genes, similar to SF3B1 R625 and K700E mutants found in uveal melanoma and breast cancer.
Correlation of SF3B1-mutant mucosal melanomas with clinical outcome
The clinical data for patients harboring all SF3B1 mutations is summarized in Table 1. Of the eight patients with SF3B1 mutations, five had developed metastatic disease and had relatively longer survival times compared with those patients with wild-type SF3B1. However, the number of patients is too small to make any definitive statements.
Discussion
Mucosal melanomas represent a small fraction of the total melanoma patient population, and understanding their mutational landscape is important in developing more effective treatment(s) for this unique group of patients. This has been, and remains, a formidable challenge 4,5. They often present late, are difficult to diagnose, and have a poor prognosis compared with cutaneous melanoma. Until recently, there was little understanding about the molecular mechanisms underlying the development of these diseases.
In accordance with prior studies, we found a low frequency of the known driver mutations commonly found in cutaneous melanomas (Fig. 1). Mutations in BRAF were seen but these were not the driver V600 mutations described in cutaneous melanoma (Table 1). Similar to other studies 7, frequent mutations in KIT were found, especially in the vulvovaginal and anorectal subtypes. In these two groups of patients, the KIT mutations were comutated with NF1 in 32% of the samples. We also noted that a large percentage (80%) of the nasopharyngeal group was pan negative, meaning they lacked the common driver mutation classifications of BRAF, NRAS, NF1, or KIT. The frequency of the pan-negative genotype in nasopharyngeal melanomas was also higher than that seen in our cutaneous melanoma database (19%) (Fig. 2).
Activating mutations in KIT have been previously described in mucosal melanomas, and occasionally patients respond to the tyrosine kinase inhibitor imatinib 21. In the present study, KIT mutations, particularly in the vulvovaginal and anorectal subtypes, were often comutated with NF1, 22 and 60%, respectively. NF1 is a tumor suppressor that normally suppresses RAS signaling. Mutations in NF1 also activate RAS signaling, similar to some of the KIT mutations. Interestingly, mutations in either NF1 or KIT have been implicated as pivotal for the development of gastrointestinal stromal tumors 21. Comutations of KIT and NF1 have also been reported in gastrointestinal stromal tumors, although they are rare 21. We reason that mutations in either gene may only weakly activate the downstream signaling, and not be sufficient for tumorigenesis, whereas comutations in both genes lead to stronger and sufficient activation for tumor development. We contemplate whether combinations of inhibitors of MAPK and PI3K signaling may be explored for these patients.
NF1 mutations have also been found concurrent with BRAF mutations in melanoma, and loss of NF1 can contribute to melanoma resistance to BRAF inhibitors (ref). In addition, loss of NF1 can cooperate with mutated BRAF to promote melanocyte proliferation and tumorigenesis 22. We conjecture that mutations in NF1 may also contribute to the resistance to imatinib in some KIT-mutated mucosal melanoma. Further investigation is needed to determine whether there is cooperation between KIT and NF1 mutations in mucosal melanoma to uncover new areas for therapy.
Another important and intriguing finding was the high frequency of mutations in SF3B1, subunit 1 of splicing factor 3b. SF3B1 is a part of the U2 small nuclear ribonucleoprotein (U2 snRNP) complex along with p14 and U2AF65, which plays a key role in the early stages of pre-mRNA splicing by facilitating branch point recognition and selection of the 3′ splice site (Fig. 3c) 23. SF3B1 mutations have previously been described in myelodysplastic syndrome, chronic lymphocytic leukemia (CLL), breast cancer, and uveal melanomas, within exons 12–15, which correspond to the C-terminal HEAT domain of SF3B1 (Fig. 4a) 6,17,24. SF3B1 is the most frequently mutated spliceosome gene in cancer and is mutated in 20% of hematological malignancies (myelodysplastic syndrome and CLL), 18% of uveal melanomas, 5% of pancreatic cancers, 1.8% of breast cancers, and less than 1% in cutaneous melanoma 8,17,25–27. The frequency of mutations in SF3B1 in this study (8/19, 42%) is higher than that reported in any other cancer type (Fig. 4a). Furthermore, we identified that mutations in other spliceosome pathway genes occurred at a higher frequency in mucosal melanoma as compared with our 135 sun-exposed cutaneous melanomas (Fig. 3b). We observed that patient 1 harbored the most mutations in our cohort; this may explain the higher comutation rate of SF3B1 with other spliceosomal component genes. For other patients, the mutations could be detected and predicted to be nonsynonymous and deleterious by the algorithms. Future work will be needed to determine their functional roles in the splicing process in mucosal melanoma.
In our study, seven (37%) of the mucosal melanoma patients carried the recurrent SF3B1 mutation at codon 625 (Fig. 4a). Five of the mucosal melanoma patients harbored the mutation R625H, which is also a common mutation found in uveal melanoma 17. The other two patients carried mutation R625C in SF3B1, and one variant R625S. A recent targeted sequencing of 15 anorectal mucosal melanoma patients described five patients with the identical SF3B1 R625 mutation, confirming our findings 7. WES in cutaneous melanoma found only a very low frequency of SF3B1 R625H/C mutations, 2/231 cutaneous patient samples 14. A review of our WES database of 135 cutaneous melanoma patients revealed no other mutations in SF3B1. A previous study found four patients with SF3B1 mutation in 20 desmoplastic melanoma patient; none of these mutations was at codon 625 28.
Recent studies have elucidated a functional role for recurrent hotspot mutations found in the C-terminal HEAT domains in SF3B1, indicating that these mutations result in alternative splicing, primarily by inducing aberrant 3′ splice site selection 19,27. Thus, it was hypothesized that the SF3B1 R625 mutation results in differential interaction with the pre-mRNA or other splicing factors of the U2 snRNP complex, such as p14 or U2AF65, resulting in binding to a noncanonical branch point, thus facilitating alternative 3′ splice site selection 19,29. Furthermore, approximately half of the aberrantly spliced mRNAs are subject to non-sense-mediated decay resulting in the downregulation of gene and protein expression 19. However, the functional role of alternatively spliced genes that do not undergo non-sense-mediated decay remains to be determined, as it is possible that these aberrant transcripts are processed into aberrant proteins with altered functions.
In our current study, we show for the first time that SF3B1 mutations in mucosal melanoma are associated with alternative splicing (Fig. 4b–e and Supplementary Fig. 2, http://tanlab.ucdenver.edu/Mucosal_Melanoma/MucosalData/). Utilizing qRT-PCR, we analyzed seven previously reported genes to be alternatively spliced in SF3B1-mutant uveal melanoma and breast cancer. We validated all these genes to be differentially spliced in mucosal melanoma, highlighting a functional role for mutations in SF3B1. Specifically, we found ABCC5, CRNDE, RPL31, and TMEM14C, in which the alternatively spliced variants were highly expressed in SF3B1 mutant as compared with wild-type mucosal melanoma samples. ABCC5 encodes for an ABC transport protein that is involved in multidrug resistance, and was observed to retain introns in SF3B1 wild-type uveal melanoma samples, but not in SF3B1-mutant samples perhaps due to increased splicing efficiency 6. We validated this finding in our SF3B1-mutant mucosal melanoma samples (Fig. 4b). CRNDE is a long noncoding RNA and has been shown to be associated with development and found to be upregulated in colorectal cancer and leukemias 30. We found that the ratio of the alternative spliced form of CRNDE compared with the canonical spliced form is higher in SF3B1-mutant samples (Fig. 4c). In common with SF3B1-mutant breast cancer tumors, we also observed differential splicing of RPL31, a ribosomal protein found to be overexpressed in colorectal tumors in familial adenomatous polyposis patients (Fig. 4d) 31. We observed a higher expression of RPL31 in SF3B1-mutant samples (Fig. 4d). Finally, we also observed a higher ratio of the differentially spliced form of TMEM14C, a transmembrane protein important for heme biosynthesis, in SF3B1-mutant samples (Fig. 4e). Further experiments are needed to elucidate the role of these differentially spliced genes expressed in SF3B1-mutant mucosal melanomas.
Interestingly, SF3B1 wild-type overexpression in cell lines does not recapitulate the alternative splicing events induced by SF3B1 mutant, indicating that mutations in SF3B1 are neomorphic 19,23. Using an inducible genetic inhibition model, Zhou et al. 32 identified that selective inhibition of SF3B1 mutant, but not wild-type alleles, resulted in differential splicing without effecting cell growth. However, inhibition of SF3B1 wild-type resulted in growth inhibition in vitro, indicating that cells are reliant on SF3B1 wild-type for growth 32. Alternatively, SF3B1-mutant pancreatic cells were found to be more sensitive to pharmacological inhibition of SF3B1 as compared with wild-type 8. Thus, it remains to be determined whether the effects of inhibition of SF3B1 are dependent on SF3B1 mutational status and tumor type. Recently, a phase I study of E7101, a novel spliceosome inhibitor, whose main target is SF3B1, has begun in solid tumors 33. Other preclinical leads of SF3B1 inhibitors are currently being developed by the pharmaceutical industry 34–36. Identifying the effects of SF3B1 inhibition in mucosal melanoma may open new avenues for the treatment of mucosal melanomas with an SF3B1 mutation.
There were too few patients in our study to make significant survival correlations between patients with SF3B1 mutations and those with wild-type SF3B1. The presence of SF3B1 mutations in CLL correlates to a worse prognosis, whereas SF3B1 hotspot mutations appear to correlate with better prognosis in uveal melanomas 17,24. More patient samples are needed to establish the correlation between SF3B1 mutation and overall survival in mucosal melanomas.
For future work, we plan to perform RNA sequencing in the mucosal melanoma samples to compare the alternatively spliced transcripts in SF3B1 mutant as compared with wild-type as previously described 19,23. In addition, as a part of our future research, we plan to test and validate the splicing transcripts in fresh tumors (when we received new patients) or formalin-fixed paraffin-embedded tissues (archival samples).
In summary, this study is the largest cohort of mucosal melanomas reported with WES. This provides new insight into the mutational landscape of these rare subtypes of melanoma. We identified comutations of KIT and NF1 (32%), as well as recurrent SF3B1 mutations (42%). SF3B1 mutations have not been previously reported in vulvovaginal or nasopharyngeal mucosal melanoma, and only once previous study found mutations in anorectal mucosal melanoma using targeted sequencing 7. In addition, our report is the first study in demonstrating the functional role of SF3B1 R625 mutations inducing alternative splicing in mucosal melanoma. Future studies will focus on the wide spread effects of SF3B1 mutations on aberrant mRNA isoforms, and the effect of alternate splicing on tumor progression in mucosal melanoma. These findings advance our understanding of mucosal melanoma and offer potential targetable mutations that could improve treatment for these rare forms of melanoma.
Supplementary Material
All supplementary digital content is available directly from the corresponding author.
Acknowledgements
The authors thank members of the International Melanoma Biorepository and Research Laboratory (IMBRL) for comments and assistance in this research. They also thank Dr Rita Gonzalez for isolating and extracting DNA from the blood samples, Drs Audrey Hendricks and Matthew Rioth for critical comments of this manuscript, and Dr Bifeng Gao, Katrina Diener, Todd Woessner, and Ted Shade from the Genomics and Microarray Shared Resource for providing technical assistance in performing the whole-exome sequencing of this research.
This work is partly supported by the National Institutes of Health P30CA046934, Cancer League of Colorado, the David F. and Margaret T. Grohne Family Foundation, the Rifkin Endowed Chair (WAR), the Amy Davis Foundation and the Moore Family Foundation.
Conflicts of interest
There are no conflicts of interest.
References
- 1.Furney SJ, Turajlic S, Stamp G, Nohadani M, Carlisle A, Thomas JM, et al. Genome sequencing of mucosal melanomas reveals that they are driven by distinct mechanisms from cutaneous melanoma. J Pathol 2013; 230:261–269. [DOI] [PubMed] [Google Scholar]
- 2.Curtin JA, Fridlyand J, Kageshita T, Patel HN, Busam KJ, Kutzner H, et al. Distinct sets of genetic alterations in melanoma. N Engl J Med 2005; 353:2135–2147. [DOI] [PubMed] [Google Scholar]
- 3.Del Vecchio M, Di Guardo L, Ascierto PA, Grimaldi AM, Sileni VC, Pigozzo J, et al. Efficacy and safety of ipilimumab 3 mg/kg in patients with pretreated, metastatic, mucosal melanoma. Eur J Cancer 2014; 50:121–127. [DOI] [PubMed] [Google Scholar]
- 4.Spencer KR, Mehnert JM. Mucosal melanoma: epidemiology, biology and treatment. Cancer Treat Res 2016; 167:295–320. [DOI] [PubMed] [Google Scholar]
- 5.Postow MA, Hamid O, Carvajal RD. Mucosal melanoma: pathogenesis, clinical behavior, and management. Curr Oncol Rep 2012; 14:441–448. [DOI] [PubMed] [Google Scholar]
- 6.Furney SJ, Pedersen M, Gentien D, Dumont AG, Rapinat A, Desjardins L, et al. SF3B1 mutations are associated with alternative splicing in uveal melanoma. Cancer Discov 2013; 3:1122–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yang HM, Hsiao SJ, Schaeffer DF, Lai C, Remotti HE, Horst D, et al. Identification of recurrent mutational events in anorectal melanoma. Mod Pathol 2017; 30:286–296. [DOI] [PubMed] [Google Scholar]
- 8.Maguire SL, Leonidou A, Wai P, Marchiò C, Ng CK, Sapino A, et al. SF3B1 mutations constitute a novel therapeutic target in breast cancer. J Pathol 2015; 235:571–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hintzsche J, Kim J, Yadav V, Amato C, Robinson SE, Seelenfreund E, et al. IMPACT: a whole-exome sequencing analysis pipeline for integrating molecular profiles with actionable therapeutics in clinical samples. J Am Med Inform Assoc 2016; 23:721–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc 2009; 4:1073–1081. [DOI] [PubMed] [Google Scholar]
- 11.Adzhubei I, Jordan DM, Sunyaev SR. Predicting functional effect of human missense mutations using PolyPhen-2. Curr Protoc Hum Genet 2013; Chapter 7:Unit 7.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Forbes SA, Beare D, Gunasekaran P, Leung K, Bindal N, Boutselakis H, et al. COSMIC: exploring the world’s knowledge of somatic mutations in human cancer. Nucleic Acids Res 2015; 43:D805–D811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kanehisa M, Sato Y, Kawashima M, Furumichi M, Tanabe M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res 2016; 44:D457–D462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kong Y, Krauthammer M, Halaban R. Rare SF3B1 R625 mutations in cutaneous melanoma. Melanoma Res 2014; 24:332–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Micel LN, Tan AC, Selby HM, Brunkow KL, Robertson KM, Davis SL, et al. Antitumor activity of the MEK inhibitor TAK-733 against melanoma cell lines and patient-derived tumor explants. Mol Cancer Ther 2015; 14:317–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wong KM, Micel LN, Selby HM, Tan AC, Bagby SM, Spreafico A, et al. Targeting the protein ubiquitination machinery in melanoma by the NEDD8-activating enzyme inhibitor pevonedistat (MLN4924). Invest New Drugs 2016; 35:11–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Harbour JW, Roberson EDO, Anbunathan H, Onken MD, Worley LA, Bowcock AM. Recurrent mutations at codon 625 of the splicing factor SF3B1 in uveal melanoma. Nat Genet 2013; 45:133–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 2013; 6:pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Darman RB, Seiler M, Agrawal AA, Lim KH, Peng S, Aird D, et al. Cancer-associated SF3B1 hotspot mutations induce cryptic 3′ splice site selection through use of a different branch point. Cell Rep 2015; 13:1033–1045. [DOI] [PubMed] [Google Scholar]
- 20.Dolatshad H, Pellagatti A, Fernandez-Mercado M, Yip BH, Malcovati L, Attwood M, et al. Disruption of SF3B1 results in deregulated expression and splicing of key genes and pathways in myelodysplastic syndrome hematopoietic stem and progenitor cells. Leukemia 2015; 29:1092–1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bello DM, Dematteo RP, Ariyan CE. The GIST of targeted therapy for malignant melanoma. Ann Surg Oncol 2014; 21:2059–2067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Maertens O, Johnson B, Hollstein P, Frederick DT, Cooper ZA, Messiaen L, et al. Elucidating distinct roles for NF1 in melanomagenesis. Cancer Discov 2013; 3:338–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Alsafadi S, Houy A, Battistella A, Popova T, Wassef M, Henry E, et al. Cancer-associated SF3B1 mutations affect alternative splicing by promoting alternative branchpoint usage. Nat Commun 2016; 7:10615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hahn CN, Venugopal P, Scott HS, Hiwase DK. Splice factor mutations and alternative splicing as drivers of hematopoietic malignancy. Immunol Rev 2015; 263:257–278. [DOI] [PubMed] [Google Scholar]
- 25.Biankin AV, Waddell N, Kassahn KS, Gingras M-C, Muthuswamy LB, Johns AL, et al. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature 2012; 491:399–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Papaemmanuil E, Cazzola M, Boultwood J, Malcovati L, Vyas P, Bowen D, et al. Somatic SF3B1 mutation in myelodysplasia with ring sideroblasts. N Engl J Med 2011; 365:1384–1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang L, Lawrence MS, Wan Y, Stojanov P, Sougnez C, Stevenson K, et al. SF3B1and other novel cancer genes in chronic lymphocytic leukemia. N Engl J Med 2011; 365:2497–2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Krauthammer M, Kong Y, Ha BH, Evans P, Bacchiocchi A, McCusker JP, et al. Exome sequencing identifies recurrent somatic RAC1 mutations in melanoma. Nat Genet 2012; 44:1006–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cretu C, Schmitzová J, Ponce-Salvatierra A, Dybkov O, De Laurentiis EI, Sharma K, et al. Molecular architecture of SF3b and structural consequences of its cancer-related mutations. Mol Cell 2016; 64:307–319. [DOI] [PubMed] [Google Scholar]
- 30.Ellis BC, Molloy PL, Graham LD. CRNDE: a long non-coding RNA involved in CanceR, Neurobiology, and DEvelopment. Front Genet 2012; 3:270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chester KA, Robson L, Begent RH, Talbot IC, Pringle JH, Primrose L, et al. Identification of a human ribosomal protein mRNA with increased expression in colorectal tumours. Biochim Biophys Acta 1989; 1009:297–300. [DOI] [PubMed] [Google Scholar]
- 32.Zhou Q, Derti A, Ruddy D, Rakiec D, Kao I, Lira M, et al. A chemical genetics approach for the functional assessment of novel cancer genes. Cancer Res 2015; 75:1949–1958. [DOI] [PubMed] [Google Scholar]
- 33.Eskens FALM, Ramos FJ, Burger H, O’Brien JP, Piera A, de Jonge MJA, et al. Phase I pharmacokinetic and pharmacodynamic study of the first-in-class spliceosome inhibitor E7107 in patients with advanced solid tumors. Clin Cancer Res 2013; 19:6296–6304. [DOI] [PubMed] [Google Scholar]
- 34.Effenberger KA, Urabe VK, Prichard BE, Ghosh AK, Jurica MS. Interchangeable SF3B1 inhibitors interfere with pre-mRNA splicing at multiple stages. RNA 2016; 22:350–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Larrayoz M, Blakemore SJ, Dobson RC, Blunt MD, Rose-Zerilli MJJ, Walewska R, et al. The SF3B1 inhibitor spliceostatin A (SSA) elicits apoptosis in chronic lymphocytic leukaemia cells through downregulation of Mcl-1. Leukemia 2016; 30:351–360. [DOI] [PubMed] [Google Scholar]
- 36.Yoshida K, Ogawa S. Splicing factor mutations and cancer. Wiley Interdiscip Rev RNA 2014; 5:445–459. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
All supplementary digital content is available directly from the corresponding author.