

Abstract
Purpose of review
To summarize the current knowledge about the biology and clinical management of adult desmoid tumors.
Recent findings
In the past decade, we have learned that desmoid tumors are driven by alterations of the Wnt/APC/β-catenin pathway, sporadic desmoid tumors are associated with somatic mutations of CTNNB1, and germline mutations of APC and somatic mutations of CTNNB1 are probably mutually exclusive. One-third of desmoid tumors are misdiagnosed; a second pathological opinion is therefore of major importance for desmoid tumor. Surgery is no longer regarded as the cornerstone of desmoid tumors; several retrospective studies have demonstrated the safety of a ‘wait and see’ policy in sporadic abdominal wall desmoid tumor. Desmoid tumors is no longer regarded as an absolute contraindication for pregnancy. At least two new investigational drugs targeting the Wnt/APC/β-catenin pathway are currently being developed.
Summary
The management of desmoid tumors requires multidisciplinary expertise by an experienced team. We must fully understand the physiopathology of the disease (factors influencing the natural history of the disease) and learn how to avoid desmoid tumors occurrence in patients with APC germline mutations, identify reliable prognostic/predictive factors and better assess the efficacy of systemic treatment.
Keywords: APC, CTNNB1, desmoid tumor, wait and see
INTRODUCTION
Desmoid tumor, also known as aggressive fibromatosis, is a rare, locally invasive, nonmetastasizing but potentially multifocal proliferation of mesenchymal stem cell progenitors [1]. Desmoid tumors constitute a soft tissue mass arising at any part of the body in different types of connective tissues, including muscle, fascia, and aponeurosis. The most common primary sites are the abdominal wall, limbs, girdles, and mesenteric area. Desmoid tumors infiltrate the surrounding structures and spread along planes and muscle. Desmoid tumors can lead to severe pain, functional impairment and, more rarely, a life-threatening condition. Desmoid tumors are typically diagnosed in young adults (peak incidence at 35–40 years), mainly in women at reproductive age [2▪▪]. The course of desmoid tumor is unpredictable, as spontaneous regression, long-lasting stable disease and disease progression can occur, and reliable and validated predictive factors are lacking. Desmoid tumors comprise at least two different clinico-pathological entities: sporadic desmoid tumor and desmoid tumor associated with germline mutation of APC. Here, we aim to summarize the most recent data about the biology and management of adult desmoid tumor as well as to describe the current ongoing clinical trials.
Box 1.
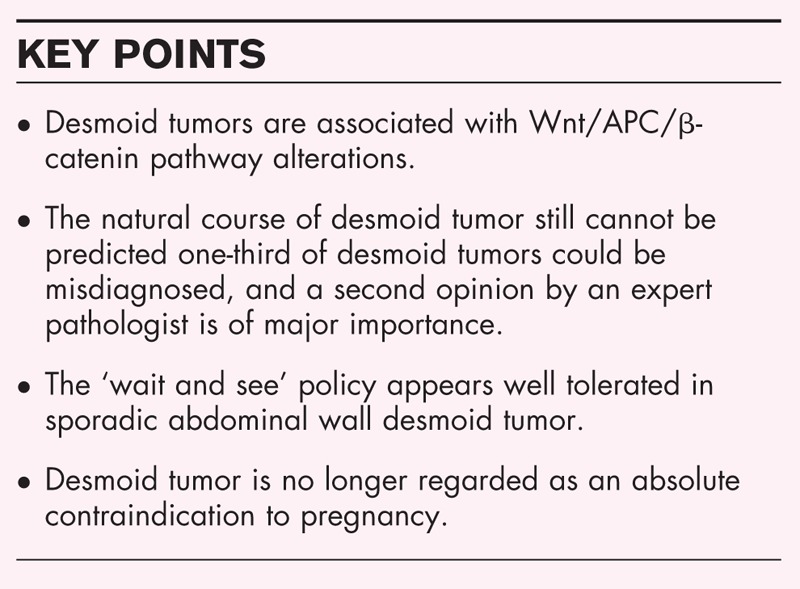
no caption available
CLINICO-PATHOLOGICAL ENTITIES
The two entities (sporadic desmoid tumor vs. desmoid tumor associated with APC mutation) are associated with specific mutually exclusive molecular alterations (CTNNB1 mutation in sporadic desmoid tumor vs APC mutation in desmoid tumor with germline mutation of APC) [3–5,6▪▪]. Because no other alteration has been identified as recurrent by whole-exome sequencing and genomic analysis, these Wnt/APC/β-catenin pathway alterations are considered to be the driver of tumor cell proliferation.
Sporadic desmoid tumor
The CTNNB1 gene encodes for β-catenin, a proto-oncogene. β-Catenin regulates cell adhesion and cell transcription. In desmoid tumor, there is an abnormal stabilization and nuclear accumulation of β-catenin. The accumulated β-catenin binds transducin beta-like protein 1 (TBL1/TBLR1), and this complex stimulates the expression of several Wnt/APC/β-catenin pathway target genes, including some proliferative factors, such as S100A4 or CTHRC1[4]. Three major mutation hotspots have been described in CTNNB1 exon 3, T41A, S45F, and S45P [5,6▪▪]. These hotspots affect the site of APC/β-catenin interaction and alter the ability of APC to drive β-catenin degradation by the proteasome [4].
Most desmoid tumors (85–90%) are sporadic and associated with somatic CTNNB1 mutations [7–9]. In sporadic desmoid tumor, a female predominance is obvious (sex ratio of approximately 0.5), and the median age at diagnosis is approximately 40 [2▪▪]. In the largest series of sporadic desmoid tumor (n = 254), 88% had CTNNB1 mutations identified by direct sequencing, whereas no mutations were detected in 175 potential morphologic mimics [8]. The most common CTNNB1 mutations are T41A (approximately 55%), S45F (approximately 35%), and S45P (approximately 10%) [6▪▪,10]. Rarer mutations or deletions have been described [6▪▪,10]. Interestingly, Doyen et al.[11] recently reported 2 cases of patients with multiple metachronous sporadic desmoid tumor associated with distinct CTNNB1 mutations.
Desmoid tumor associated with germline APC mutations
In cases of FAP, a truncated APC protein with low affinity to β-catenin is created. This process results in the nuclear accumulation of β-catenin and, consequently, the deleterious overexpression of its target genes [4].
Approximately 10–15% of desmoid tumors are associated with germline mutations of APC, providing different syndromic associations: desmoid tumor associated with FAP, desmoid tumor as part of Gardner syndrome (FAP, desmoid tumor, and osteomatosis of the skull and the mandible, sebaceous cysts, and cutaneous and subcutaneous fibromas) or desmoid tumor associated with Turcot syndrome (FAP and brain tumor) [12]. Here, the sex ratio is close to 1 [13–15].
A meta-analysis of five European FAP registries (2260 patients and 912 families) showed that the occurrence of desmoid tumor is approximately 10% (220/2260), the median age at diagnosis is 31 years, and the first desmoid tumors are often in the intra-abdominal space (52.9%) or abdominal wall (24.6%) [15]. Desmoid tumor is diagnosed after prophylactic surgery in 72.0% of cases and before surgery in 28.0%. The median time between prophylactic surgery and the diagnosis of desmoid tumor was 36 months (range, ref). The APC mutation is located beyond codon 1444 in 65.0% of cases associated with DT. Risk factors for desmoid tumor in univariate analysis are APC mutation beyond codon 1444 (P < 0.0001), age at first surgery less than 31 years (P = 0.003), and prior abdominal surgery (P = 0.003). In multivariate analysis, the APC mutation site (OR = 3.0, P < 0.0001) and prior surgery (OR = 2.58, P = 0.0004) were the two independent risk factors for desmoid tumor [15].
Other studies confirm that compared to other mutations, APC mutation beyond codon 1444 is a significant risk factor for desmoid tumor [16–18]. Prior familial history of desmoid tumor is a risk factor for desmoid tumor, but this phenotype probably reflects the underlying germline mutation [19].
Desmoid tumors are a major concern in FAP patients and cause significant morbidity and mortality, as most cases are intra-abdominal (in the small bowel mesentery) and can cause bowel obstruction or ulceration and ureter stenosis. Desmoid tumor was the main cause of death in some series of patients with FAP with desmoid tumor [13].
FAP requires prophylactic colectomy to reduce recto-colic cancer mortality. However, this necessary surgical procedure plays a key role in the initiation and promotion of desmoid tumor. Regarding these facts, the appropriate surgical procedure (ileorectal anastomosis vs. proctectomy) is debated [14]. On one hand, some authors suggest that proctectomy is a simple procedure that avoids the risk of re-operation, which could increase the occurrence of desmoid tumor. On the other hand, some authors claim that FAP associated with desmoid tumor is usually an attenuated form that does not require rectum ablation and is accessible for careful follow-up of the remaining rectum [20]. In the international study mentioned above, the type of prophylactic surgery was not a risk factor for desmoid tumor occurrence (P = 0.53) [15]. Ultimately, the issue of the ideal prophylactic surgical procedure is still an open question.
‘Wild-type’ desmoid tumor
CTNNB1 or APC mutations are mutually exclusive and nearly universal in desmoid tumor; in a series of 117 cases, Crago et al. found mutation of CTNNB1 in 101 cases and mutation of APC in 10 cases, as well as other gene alterations affecting the Wnt/APC/β-catenin pathway, including chromosome 6 loss (two cases) and BMI1 mutation (one case) [6▪▪]. Ultimately, the so-called ‘wild-type’ desmoid tumor (without CTNNB1 or APC mutations) does not exist, or is misdiagnosed (other spindle cell proliferation mimicking desmoid tumor) [2▪▪,8] or desmoid tumor with CTNNB1 or APC mutations unrecognized by nonsensitive standard diagnostic methods [6▪▪], and exceptional desmoid tumor with other molecular alterations of the Wnt/APC/β-catenin pathway [6▪▪].
MANAGEMENT OF DESMOID TUMOR
Diagnosis
Magnetic resonance imaging (MRI) is regarded as the most appropriate imaging method to better characterize the initial extension of desmoid tumor and to monitor the outcome [21]. Imaging-guided core-needle biopsy is required to formally diagnose desmoid tumor. However, mesenteric masses could be difficult or hazardous to biopsy. Histologically, DTs are composed of monoclonal spindle-shaped cells separated by an abundant collagenous matrix. A French nationwide survey demonstrated that one-third of desmoid tumors are misdiagnosed (the most challenging differential diagnoses are nodular fasciitis and low grade fibromyxoid sarcoma) [2▪▪]. A second opinion by an expert pathologist is of major importance in such diseases [2▪▪]. The nuclear overexpression of β-catenin is a useful diagnostic tool, but its sensitivity depends on the immunohistochemistry method used. We strongly believe that a second opinion by an expert pathologist is necessary.
There is no consensus on initial rectocolonoscopy in the initial work-up of desmoid tumor. Desmoid tumor could be the first manifestation of FAP (initial germline mutation or unrecognized inherited mutation in cases of attenuated FAP). Thus, initial rectocolonoscopy could be considered. Nevertheless, because APC and CTNNB1 mutations appear to be mutually exclusive, rectocolonoscopy could be avoided in cases with a desmoid tumor harboring somatic mutations of CTNNB1.
Treatment
The multidisciplinary management of desmoid tumor requires cautious assessment of the expected benefits and risks associated with the proposed treatment; ideally, it is a decision shared with the patient.
Large en-bloc surgery is no longer regarded as the cornerstone treatment for desmoid tumor because the rate of relapse after surgery exceeds 60% in larger series [22]. The risk of relapse is similar in the context of APC germline mutations (approximately 22/42) [13]. The local relapse could be larger than the primary tumor. Some clinical parameters are associated with a high risk of relapse after surgery in different studies; these parameters include young age [22,23], large tumor size [22–24], and positive margins [22,24]. Crago et al.[22] developed and then validated a predictive nomogram based on age, tumor size and primary location. Several studies suggest that the S45F CTNNB1 mutation is an important risk factor for local recurrence after curative-intent surgery for primary desmoid tumor (Table 1) [25–29]. Salas et al.[30] developed a 36-gene molecular signature that predicted relapse after surgery with an accuracy of 78%. In this model, the top two genes that were upregulated in the recurrence group were FECH (ferrochelatase, which is involved in protoheme biosynthesis) and stomatin-like protein 2, which regulates mitochondrial functions. The top gene upregulated in the no recurrence group was thyroid hormone receptor-interacting protein 6, which is involved in cell adhesion.
Table 1.
The S45F mutation as a risk factor for local relapse after surgery for primary desmoid tumor
| n | Median follow-up (months) | Relapse, n (%) | S45F mutation, n (%) | S45F as a risk factor | Ref. | |
| Crude hazard ratio, HR (95% CI) | Adjusted hazard ratio (1), HR (95% CI) | |||||
| 89 | 62 | Not done | 19 (21.3) | 4.28 (1.75–10.48) | 4.28 (1.75–10.48) | [26] |
| 179 | 50 | 48 (26.8) | 39 (21.7) | Not done | 2.59 (1.19–5.65) | [27] |
| 101 | Not done | 50 (49.5) | 37 (36.6) | Not done | Not done | [28] |
| 95 | 31 | Not done | 23 (24.2) | Not done | Not done | [29] |
| 101 | 41 | 17 (16.8) | 18 (17.8) | 8.50 (1.85–39.00) | 6.20 (2.24–17.15) | [30] |
Therefore, there has been a shift to a more conservative approach, the ‘wait-and-see’ policy [31]. Currently available data suggest that only a small percentage of desmoid tumors are progressive and that most progressions are seen within 36 months following diagnosis [23,32,33]. French and Italian studies suggest that the ‘wait and see’ policy is well tolerated, particularly in cases of sporadic abdominal wall desmoid tumor (Table 2) [23,32,34]. This approach has been assessed by different prospective trials. It has been recommended by several authors and has been proposed as the standard of care, at least in Europe [35]. The ‘wait and see’ policy avoids inadequate treatment for desmoid tumor that could spontaneously regress and discourages treatment for stable and pauci-symptomatic desmoid tumor. Reasons for treatment (including surgery) are volumetric progression and symptom worsening. However, the precise definition of the population of patients which should be proposed for watch and wait is missing.
Table 2.
Sporadic desmoid tumor: ‘wait and see’ experiences
Radiotherapy is rarely used in desmoid tumor management. Adjuvant radiotherapy is proposed in cases of R1/R2 resection and in cases located at critical sites, without demonstration of its utility. Nevertheless, regarding the young age of these patients, close follow-up is preferred because of the potential long-term toxicity of radiotherapy, especially when tumors develop in irradiated fields. Radiotherapy at a dose of 56 Gy in 28 daily fractions of 2 Gy has been shown to provide adequate local control in the vast majority of progressive patients, with a local control rate of 77% and a complete response rate of 17% [36]. Long-term follow-up is required to better analyze the risk--benefit ratio of this option [36]. Other local procedures could be discussed in locally advanced and progressive desmoid tumor: cryoablation and isolated limb perfusion [37].
Different systemic treatments could be used in cases of desmoid tumor progression (Table 3) [21,37–45]. Today, none of these treatments could be considered as a standard of care, as available data have only come from retrospective studies with a limited number of cases or nonrandomized phase II studies. A large retrospective case series (n = 134) showed that 85% of patients with desmoid tumor treated with antiestrogen achieve progression arrest. This rate is similar in sporadic desmoid tumor and FAP-associated desmoid tumor [47]. Some authors regard anthracycline-based regimens as those providing a higher objective response rate (approximately 50%); however, the available data are scarce (Table 3). A phase II trial assessing the activity of the methotrexate-vinblastine is very interesting because it showed that approximately 60% of patients experience symptom palliation, and the long-term efficacy was impressive (10-year progression-free survival of approximately 60%) [40]. Tyrosine kinase inhibitors have shown some signs of activity (Table 3). However, overall, in the absence of an internal control or randomization, it is difficult to attribute the observed slowing of tumor progression to the natural history of the disease or to the real activity of systemic treatment. Currently, there is no reliable tool for choosing the best systemic treatment in progressive desmoid tumor. However, some studies suggest that desmoid tumors harboring CTTNB1 mutations are more sensitive to imatinib than desmoid tumors without CTTNB1 mutations [44], and the CTTNB1 S45F mutation is associated with resistance to NSAIDs (meloxicam) compared to other mutations or the absence of mutations (4/4 vs. 9/29) [48]. Further clinical trials are urgently needed to identify predictive factors and properly assess the activity of the treatment using randomized trials and trials using quality of life data.
Table 3.
Systemic therapy options in adults with desmoid tumors: ‘best available’ evidence
| Treatment | Nature of the study | n | Objective response rate | Other activity endpoints | Ref. |
| Sulindac | Retrospective | 14 | 57% | [38] | |
| Toremifene | Retrospective | 27 | 22% | 6-month PFS: 76% | [39] |
| Methotrexate-Vinblastine | Phase II | 27 | 15% | 10-year PFS: 67% | [40] |
| Pegylated doxorubicin | Retrospective | 14 | 33% | [41] | |
| Doxorubicin + dacarbazine | Retrospective | 12 | 50% | [42] | |
| Imatinib (800 mg/day) | Phase II | 51 | 6% | 1-year PFS: 66% | [43] |
| Imatinib (800 mg/day) | Phase II | 37 | 3% | 6-month PFS: 65% | [44] |
| Imatinib (400 mg/day) | Phase II | 50 | 12% | 1-year PFS: 67% | [45] |
| Sunitinib | Phase II | 19 | 26% | 1-year PFS: 80% | [46] |
| Sorafenib | Retrospective | 26 | 26% | [21] |
PFS, progression-free survival.
Supportive care
Pain management is a major issue for patients with DT. In a limited case series (n = 16), Emori et al.[49] showed that desmoid tumor-related pain is associated with the overexpression of cyclooxygenase-2. Patients affected by desmoid tumor are young and will face long-lasting morbidity; therefore, functional impairments must be managed by physiotherapists and social workers to avoid work and social exclusion. Supportive care is of major importance in this condition.
Pregnancy is another complex issue for women with sporadic desmoid tumor. We know that a large number of cases occur within the 24 months following pregnancy; these cases typically arise in the abdominal wall. This observation suggests that hormone signaling, mechanical constraints, inherent trauma, or postpartum healing play a role in the development of desmoid tumor [50]. A large multicenter retrospective study of cases diagnosed during pregnancy provided the following evidence: the probability of spontaneous regression after pregnancy was approximately 10%, the documented progression after the ‘wait and see’ policy was 60%, the failure of medical treatment occurred in 10%, and the risk of relapse after surgery was 13% [50]. In patients with preexisting desmoid tumor, the occurrence of pregnancy was associated with a risk of relapse or progression of approximately 40%; this relapse/progression spontaneously regressed in approximately 10% of cases, and medical therapy was effective in approximately 90% of cases [50]. Furthermore, no major obstetric complications (spontaneous vaginal delivery in most cases) were documented in this large retrospective series of cases managed in the reference centers. Ultimately, desmoid tumor cannot be regarded as a definitive contraindication to pregnancy; pregnancy and desmoid tumor can be safely managed in reference centers, when the patient is fully informed of the state of the art regarding the impact of pregnancy on her condition. No recommendations could be made for cases of mesenteric desmoid tumor associated with FAP. Furthermore, in the absence of reliable data, there was no recommendation for hormonal contraceptive treatments or the hormonal treatment of infertility.
FUTURE DIRECTIONS
Ongoing trials are summarized in SDC1 (online appendix). Furthermore, 2 investigational drugs targeting β-catenin are of major interest: tegatrabetan (BC-2059) and the gamma-secretase inhibitor PF-03084014. In vitro, tegatrabetan directly stimulates β-catenin degradation. In culture, tegatrabetan induces the apoptosis of desmoid tumor cells. A phase I/II trial is planned. The gamma-secretase inhibitor PF-03084014 stimulates the Notch pathway, which interacts with and indirectly regulates the Wnt/APC/β-catenin pathway. A phase I trial showed a high rate of objective responses among patients with desmoid tumor (five of seven) [51]. A phase II trial is ongoing (SDC1, online appendix).
CONCLUSION
Factors influencing the outcome of desmoid tumor remain unknown. Reliable and validated predictive and prognostic factors have not yet been identified. Desmoid tumors are misdiagnosed in a large number of outside reference centers. Surgery is no longer regarded as the standard of care, and the ‘wait and see’ policy seems well tolerated, at least in sporadic abdominal wall desmoid tumor. There is no consensus on treatment in cases of progressive desmoid tumor. At least 2 new investigational drugs targeting the Wnt/APC/β-catenin pathway are currently being developed.
Acknowledgements
We would like to thank Séverine Marchant for her assistance with manuscript editing.
Financial support and sponsorship
None.
Conflicts of interest
There are no conflicts of interest.
REFERENCES AND RECOMMENDED READING
Papers of particular interest, published within the annual period of review, have been highlighted as:
▪ of special interest
▪▪ of outstanding interest
REFERENCES
- 1.Wu C, Nik-Amini S, Nadesan P, et al. Aggressive fibromatosis (desmoid tumor) is derived from mesenchymal progenitor cells. Cancer Res 2010; 70:7690–7698. [DOI] [PubMed] [Google Scholar]
- 2▪▪.Penel N, Coindre JM, Bonvalot S, et al. Management of desmoid tumours: a nationwide survey of labelled reference centre networks in France. Eur J Cancer 2016; 58:90–96. [DOI] [PubMed] [Google Scholar]; This nationwide 4-year survey demonstrates that one third of desmoid tumors are misdiagnosed outside reference center and stresses the importance of second opinion by expert pathologist. Moreover, the set-up of reference network was associated with dramatic decrease in delay between initial diagnosis and first consultation experienced physician in management of desmoid tumor (from 400 to 60 days).
- 3.Cheon SS, Cheah AY, Turley S, et al. Beta-catenin stabilization dysregulates mesenchymal cell proliferation; motility, and invasiveness and causes aggressive fibromatosis and hyperplasic cutaneous 189 wounds. Proc Natl Acad Sci USA 2002; 99:6973–6978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li J, Wang CY. TBL1-TBLR1 and beta-catenin recruit each other to Wnt target-gene promoter for transcription activation and oncogenesis. Nat Cell Biol 2008; 10:160–169. [DOI] [PubMed] [Google Scholar]
- 5.Salas S, Chibon F, Noguchi T, et al. Molecular 194 characterization by array comparative genomic hybridization and DNA sequencing of 194 desmoid tumors. Genes Chromosomes Cancer 2010; 49:560–568. [DOI] [PubMed] [Google Scholar]
- 6▪▪.Crago AM, Chmielecki J, Rosenberg M, et al. Near universal detection of alterations in CTNNB1 and wnt pathway regulators in desmoid-type fibromatosis by whole-exome sequencing and genomic analysis. Genes Chromosomes Cancer 2015; 54:606–615. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study establishes that CTNNB1 and APC mutations are probably mutually exclusive in desmoid tumor and are nearly universal in desmoid tumor.
- 7.Huss S, Nehles J, Binot E, et al. Beta-catenin (CTNNB1) mutations and clinicopathological features of mesenteric desmoid-type fibromatosis. Histopathology 2013; 62:294–304. [DOI] [PubMed] [Google Scholar]
- 8.Le Guellec S, Soubeyran I, Rochaix P, et al. CTNNB1 mutation analysis is a useful tool for the diagnosis of desmoid tumors: a study of 260 desmoid tumors and 191 potential morphologic mimics. Mod Pathol 2012; 25:1551–1558. [DOI] [PubMed] [Google Scholar]
- 9.Tejpar S, Nollet F, Li C, et al. Predominance of beta-catenin mutations and beta-catenin dysregulation in sporadic aggressive fibromatosis (desmoid tumor). Oncogene 1999; 18:6615–6620. [DOI] [PubMed] [Google Scholar]
- 10.Aitken SJ, Presneau N, Kalimuthu S, et al. Next-generation sequencing is highly sensitive for the detection of beta-catenin mutations in desmoid-type fibromatoses. Virchow Arch 2015; 467:203–211. [DOI] [PubMed] [Google Scholar]
- 11.Doyen J, Duranton-Tanneur V, Hostein I, et al. Spatio-temporal genetic heterogeneity of CTNNB1 mutations in sporadic desmoid type fibromatosis lesions. Virchows Arch 2016; 468:369–374. [DOI] [PubMed] [Google Scholar]
- 12.Fritch LSA, Yi JA, Hall BAC, Moertel CL. A novel APC gene mutation associated with a severe phenotype in a patient with Turcot Syndrome. J Pediatr Hematol Oncol 2014; 36:e177–e179. [DOI] [PubMed] [Google Scholar]
- 13.Desurmont T, Lefèvre JH, Shields C, et al. Desmoid tumour in familial adenomatous polyposis patients: responses to treatments. Fam Cancer 2015; 14:31–39. [DOI] [PubMed] [Google Scholar]
- 14.Burgess A, Xhaja X, Church j. Does intra-abdominal desmoid disease affect patients with an ileal pouch differently than those with an ileorectal anastomosis? Dis Colon Rectum 2011; 54:1388–1391. [DOI] [PubMed] [Google Scholar]
- 15.Nieuwenhuis MH, Lefevre JH, Bülow S, et al. Family history surgery and APC mutation are risk factors for desmoid tumors in familial adenomatous polyposis: an international cohort study. Dis Colon Rectum 2011; 54:1229–1234. [DOI] [PubMed] [Google Scholar]
- 16.Caspari R, Olschwang S, Friedl W, et al. Familial adenomatous polyposis: desmoid tumours and lack of ophthalmic lesions (CHRPE) associated with APC mutations beyond codon 1444. Hum Mol Genet 1995; 4:337–340. [DOI] [PubMed] [Google Scholar]
- 17.Sturt NJ, Gallagher MC, Bassett P, et al. Evidence for genetic predisposition to desmoid tumours in familial adenomatous polyposis independent of the germline APC mutation. Gut 2004; 53:1832–1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schiessling S, Kihm M, Ganschow P, et al. Desmoid tumour biology in patients with familial adenomatous coli. Br J Surg 2013; 100:694–703. [DOI] [PubMed] [Google Scholar]
- 19.Nieuwenhuis MH, De Vos Tot Nederveen Cappel W, Botma A, et al. Desmoid tumors is a Dutch cohort of patents with familial adenomatous polyposis. Clin Gastroenterol Hepatol 2008; 6:215–219. [DOI] [PubMed] [Google Scholar]
- 20.Church JM, Xhaja X, Warrier SK, et al. Desmoid tumors do not prevent proctectomy following abdominal colectomy and ileorectal anastomosis in patients with familial adenomatous polyposis. Dis Colon Rectum 2014; 57:343–347. [DOI] [PubMed] [Google Scholar]
- 21.Gounder MM, Lefkowitz RA, Keohan ML, et al. Activity of Sorafenib against desmoid tumor/deep fibromatosis. Clin Cancer Res 2011; 17:4082–4090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Crago AM, Denton B, Salas S, et al. A prognostic nomogram for prediction of recurrence in desmoid fibromatosis. Ann Surg 2013; 258:347–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Salas S, Dufresne A, Bui B, et al. Prognostic factors influencing progression-free survival determined from a series of sporadic desmoid tumors: a wait-and-see policy according to tumor presentation. J Clin Oncol 2011; 29:3553–3558. [DOI] [PubMed] [Google Scholar]
- 24.Huang K, Wang CM, Chen JG, et al. Prognostic factors influencing event-free survival and treatments in desmoid-type fibromatosis: an analysis from a large institution. Am J Surg 2014; 207:847–854. [DOI] [PubMed] [Google Scholar]
- 25.Lazar AJ, Tuvin D, Hajibashi S, et al. Specific mutations in the betacatenin gene (CTNNB1) correlate with local recurrence in sporadic desmoid tumors. Am J Pathol 2008; 173:1518–1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Colombo C, Miceli R, Lazar AJ, et al. CTNNB1 45F mutation is a molecular prognosticator of increased postoperative primary desmoid tumor recurrence. Cancer 2013; 119:3696–3702. [DOI] [PubMed] [Google Scholar]
- 27.Domont J, Salas S, Lacroix L, et al. High frequency of beta-catenin heterozygous mutations in extra-abdominal fibromatosis: a potential molecular tool for disease management. Br J Cancer 2010; 102:1032–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mullen JT, DeLaney TF, Rosenberg AE, et al. β-Catenin mutation status and outcomes in sporadic desmoid tumors. Oncologist 2013; 18:1043–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Van Broekhoven DLM, Verhoef C, Grünhaven DJ, et al. Prognostic value of CTNNB1 gene mutation in primary sporadic aggressive fibromatosis. Ann Surg Oncol 2015; 22:1464–1470. [DOI] [PubMed] [Google Scholar]
- 30.Salas S, Brulard C, Terrier P, et al. Gene expression profiling of desmoid tumors by cDNA microarrays and correlation with progression-free survival. Clin Cancer Res 2015; 21:4194–4200. [DOI] [PubMed] [Google Scholar]
- 31.Bonvalot S, Eldweny H, Haddad V, et al. Extra-abdominal primary fibromatosis: aggressive management could be avoided in a subgroup of patients. Eur J Surg Oncol 2008; 34:462–468. [DOI] [PubMed] [Google Scholar]
- 32.Fiore M, Rimareix F, Mariani L, et al. Desmoid-type fibromatosis: a front-line conservative approach to select patients for surgical treatment. Ann Surg Oncol 2009; 16:2587–2593. [DOI] [PubMed] [Google Scholar]
- 33.Bonvalot S, Ternès N, Fiore M, et al. Spontaneous regression of primary abdominal wall desmoid tumors: more common than previously thought. Ann Surg Oncol 2013; 20:4096–4102. [DOI] [PubMed] [Google Scholar]
- 34.Colombo C, Miceli R, Le Pechoux C, et al. Sporadic extra abdominal wall desmoid-type fibromatosis: surgical resection can be safely limited to a minority of patients. Eur J Cancer 2015; 51:186–192. [DOI] [PubMed] [Google Scholar]
- 35.Kasper B, Baumgarten C, Bonvalot S, et al. Management of sporadic desmoid-type fibromatosis: a European consensus approach based on patients’ and professionals’ expertise - a sarcoma patients EuroNet and European Organisation for Research and Treatment of Cancer/Soft Tissue and Bone Sarcoma Group initiative. Eur J Cancer 2015; 51:127–136. [DOI] [PubMed] [Google Scholar]
- 36.Keus RB, Nout RA, Blay JY, et al. Results of a phase II pilot study of moderate dose radiotherapy for inoperable desmoid-type fibromatosis - an EORTC STBSG and ROG study (EORTC 62991-22998). Ann Oncol 2013; 24:2672–2676. [DOI] [PubMed] [Google Scholar]
- 37.Bonvalot S, Rimareix F, Causeret S, et al. Hyperthermic isolated limb perfusion in locally advanced soft tissue sarcoma and progressive desmoid-type fibromatosis with TNF 1 mg and melphalan (T1-M HILP) is safe and efficient. Ann Surg Oncol 2009; 16:3350–3357. [DOI] [PubMed] [Google Scholar]
- 38.Tsukada K, Church JM, Jagelman DG, et al. Noncytotoxic drug therapy for intraabdominal desmoid tumor in patients with familial adenomatous polyposis. Dis Colon Rectum 1992; 35:29–33. [DOI] [PubMed] [Google Scholar]
- 39.Fiore M, Colombo C, Radaelli S, et al. Activity of toremifene in sporadic desmoid-type fibromatosis. J Clin Oncol 2011; 29: (abstract 10033). [Google Scholar]
- 40.Azzarelli A, Gronchi A, Bertulli R, et al. Low-dose chemotherapy with methotrexate and vinblastine for patients with advanced aggressive fibromatosis. Cancer 2001; 92:1259–1264. [DOI] [PubMed] [Google Scholar]
- 41.Constantinidou A, Jones RL, Scurr M, et al. Pegylated liposomal doxorubicin, an effective, well tolerated treatment for refractory aggressive fibromatosis. Eur J Cancer 2009; 45:2930–2934. [DOI] [PubMed] [Google Scholar]
- 42.Patel S, Evans H, Benjamin R. Combination chemotherapy in adult desmoid tumors. Cancer 1993; 72:3244–3247. [DOI] [PubMed] [Google Scholar]
- 43.Chugh R, Wathen JK, Patel SR, et al. Efficacy of imatinib in aggressive fibromatosis: results of a phase II multicenter Sarcoma Alliance for Research through Collaboration (SARC) trial. Clin Cancer Res 2010; 16:4884–4891. [DOI] [PubMed] [Google Scholar]
- 44.Kasper B, Gruenwald V, Reichardt P, et al. Correlation of CTNNB1 mutation status with progression arrest rate in RECIST progressive desmoid-type fibromatosis treated with imatinib: translational research results from a phase 2 study of the German Interdisciplinary Sarcoma Group (GISG-01). Ann Surg Oncol 2016; 23:1924–1927. [DOI] [PubMed] [Google Scholar]
- 45.Penel N, Le Cesne A, Bui BN, et al. Imatinib for progressive and recurrent aggressive fibromatosis (desmoid tumors): an FNCLCC/French Sarcoma Group phase II trial with a long-term follow-up. Ann Oncol 2011; 22:452–457. [DOI] [PubMed] [Google Scholar]
- 46.Jo JC, Hong YS, Kim KP, et al. A prospective multicenter phase II study of sunitinib in patients with advanced aggressive fibromatosis. Invest New Drugs 2014; 32:369–372. [DOI] [PubMed] [Google Scholar]
- 47.Quast DR, Schneider R, Burdzik E, et al. Long-term outcome of sporadic and FAP-associated desmoid tumors treated with high-dose selective estrogen receptor modulators and sulindac: a single-center long-term observational study in 134 patients. Fam Cancer 2016; 15:31–40. [DOI] [PubMed] [Google Scholar]
- 48.Hamada S, Futamura N, Ikuta K, et al. CTNNB1 S45F mutation predicts poor efficacy of meloxicam treatment for desmoid tumors: a pilot study. PLOS One 2014; 9:e96391.1-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Emori M, Kaya M, Mitsuhashi T, et al. Desmoid tumor-associated pain is dependent on mast cell expression of cyclooyxygenase. Diagnostic Pathology 2014; 9:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fiore M, Coppola S, Cannell AJ. Desmoid-type fibromatosis and pregnancy. A multiinstitutional analysis of recurrence and obstetric. Ann Surg 2014; 259:973–978. [DOI] [PubMed] [Google Scholar]
- 51.Messersmith WA, Shapiro GI, Cleary JM, et al. A phase I, dose-finding study in patients with advanced solid malignancies of the oral gamma-secretase inhibitor PF-03084014. Clin Cancer Res 2015; 21:60–67. [DOI] [PubMed] [Google Scholar]