

Abstract
Purpose of review
The goal of this review is to summarize recent advances in our understanding of the regulation of redox homeostasis and the subtype-specific role of antioxidant enzymes in B-cell-derived malignancies. Furthermore, it presents selected prooxidative therapeutic strategies against B-cell neoplasms.
Recent findings
Recent reports have shown that the disturbed redox homeostasis in B-cell malignancies is regulated by cancer-specific signaling pathways and therefore varies between the individual subtypes. For instance, in a subtype of diffuse large B-cell lymphoma with increased oxidative phosphorylation, elevated reactive oxygen species are accompanied by higher levels of thioredoxin and glutathione and inhibition of either of these systems is selectively toxic to this subtype. In addition, growing number of small molecule inhibitors targeting antioxidant enzymes, such as auranofin, SK053, adenanthin, or decreasing glutathione level, such as imexon, buthionine sulfoximine, and L-cysteinase, trigger specific cytotoxic effects against B-cell malignancies. Lastly, attention is drawn to recent reports of effective treatment modalities involving prooxidative agents and interfering with redox homeostasis provided by stromal cells.
Summary
Recent findings reveal important differences in redox homeostasis within the distinct subsets of B-cell-derived malignancies that can be therapeutically exploited to improve existing treatment and to overcome drug resistance.
Keywords: antioxidant enzymes, B cell, leukemia, lymphoma, reactive oxygen species
INTRODUCTION
Constant generation of reactive oxygen species (ROS) is a natural biochemical event in the life of all aerobic organisms. Data accumulated in recent years suggest that cancer cells, including certain subtypes of B-cell malignancies, generate higher levels of ROS mainly because of metabolic dysregulation. As ROS are potentially mutagenic, a tentative strategy was to use antioxidant compounds in hope to prevent carcinogenesis and delay cancer progression. However, this approach has been largely disappointing [1]. The interplay between ROS and cancer was proved far more complex and multifaceted than expected. ROS appeared to be not solely an end waste product of cellular metabolism, but an important oncogenic signaling second messenger and mediator of drug resistance. In line with these findings, tumors in patients with advanced disease and poor prognosis have dysregulated expression of certain ROS-metabolizing enzymes and exhibit prooxidant phenotype [2,3]. Thus, a simple antioxidant treatment of advanced tumors may actually help cancer cells to cope with oxidative stress conditions and progress more rapidly [4,5]. In recent years, the new hope has been placed into using targeted and synergism-based prooxidant therapies to push cancer cells toward death. An increasing evidence suggest that this kind of approach seems especially promising in B-cell malignancies [6,7▪▪].
Malignant B cells have higher ROS levels compared with normal counterparts because of altered metabolism [8,9▪▪] and oncogenic signaling, such as MYC proto-oncogene [10] or BCR-ABL1 fusion protein. [11]. Moderate ROS levels, via oxidative damage to DNA, lead to genomic instability and facilitate tumor progression [12]. In addition, elevated ROS can react with sulfur-containing amino acids side chains, leading to oxidation of sulfhydryl groups and inactivation of cellular phosphatases. This favors B-cell receptor (BCR) and oncogenic kinase signaling, and thereby promotes malignant B-cell survival [13]. However, in contrast to tumor-supporting role of moderate ROS levels, the excessive production of ROS induces damage to cellular biomolecules and organelles and leads to cell death. Thus, eliminating or inhibiting cellular mechanisms that neutralize and maintain ‘safe’ ROS levels might actually shift the balance of protumorigenic ROS activity toward cancer cell death. Experimental therapeutic approaches, following this concept, have been investigated for at least a decade. However, these studies are only beginning to elucidate specific redox targets, or their combinations, in distinct subtypes of B-cell neoplasms. Malignant cells utilize several, to some extent redundant, antioxidant systems, including low-molecular-weight compounds, for example, glutathione (GSH), and antioxidant enzymes, such as superoxide dismutases (SODs), catalases (CAT), peroxiredoxins (PRDXs), and thioredoxin (TRX) system (Table 1, Fig. 1, reviewed extensively in [22–24]). It is now being recognized that exaggerated antioxidant response, although quite commonly occurring in many types of B-cell cancers, is regulated by cancer-specific signaling pathways and, therefore, is distinct in individual subtypes of the disease. Herein, we summarize the current knowledge on redox homeostasis in malignant B cells and present selected prooxidative strategies to treat B-cell cancers.
Table 1.
Major families of antioxidant enzymes in cells
| Function | Enzyme | Isoforms and localization | References |
| Dismutation of superoxide | Superoxide dismutase | SOD1 (c)SOD2 (m)SOD3 (e) | [14] |
| Reduction of H2O2 and/or peroxides | Catalase | CAT (p, c, m) | [15] |
| Glutathione peroxidase | GPX1 (c)GPX2 (c)GPX3 (e)GPX4 (c, m)GPX5 (e)GPX6 (e)GPX7 (er)GPX8 (er) | [16] | |
| Peroxiredoxins | PRDX1 (c)PRDX2 (c)PRDX3 (m)PRDX4 (er)PRDX5 (m, p, c)PRDX6 (c) | [17] | |
| Reduction of disulfide bonds | Thioredoxin | TRX1 (c)TRX2 (m) | [18] |
| Glutaredoxin | GRX1 (c)GRX2 (c, m)GRX3 (n)GRX5 (m) | [19] | |
| Re-cycling of oxidized enzymes/substrates | Thioredoxin reductase | TR1 (c)TR2 (m) | [18] |
| Glutathione reductase | GR (c, m) | [20] | |
| Sulphiredoxin (SRX) | SRX (c, m) | [21] |
c, cytosol; e, extracellular; er, endoplasmic reticulum; m, mitochondria; n, nucleus; p, peroxisomes.
FIGURE 1.
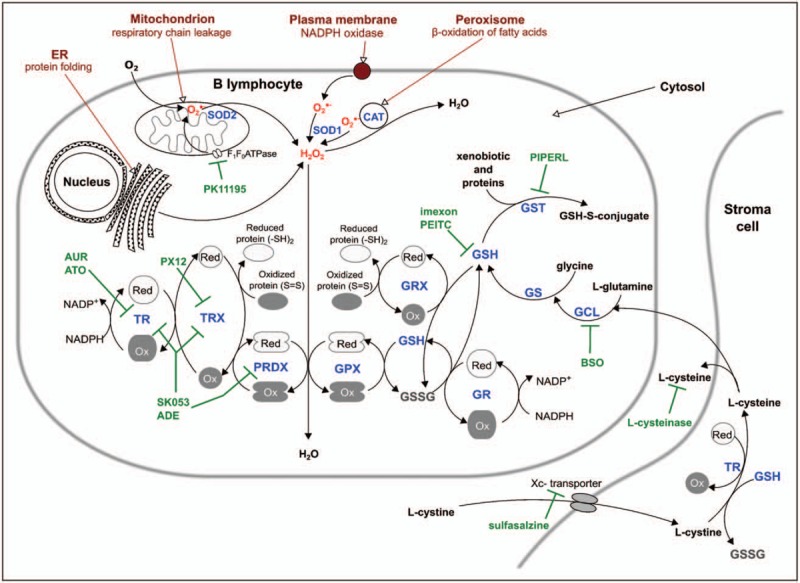
Schematic representation of cellular antioxidant pathways and their inhibitors. Major sources of cellular ROS (in red): mitochondrion, peroxisome, ER, NOX. Enzymes of the thioredoxin system: TRX, TR, PRDX. Enzymes and substrates of the glutathione system: GSH, GSSG, GPX, GRX, GR, GST. Enzymes involved in GSH synthesis: GCL, GS. Inhibitors targeting antioxidant pathways (in green). Abbreviations: ADE, adenanthin; ATO, arsenic trioxide; AUR, auranofin; BSO, buthionine sulfoximine, ER, endoplasmic reticulum; GCL, glutamate cysteine ligase; GPX, glutathione peroxidase; GR, glutathione reductase; GRX, glutaredoxins; GS, glutathione synthetase; GSH, reduced glutathione; GSSG, oxidized glutathione; GST, glutathione-S-transferase; NOX, NADPH oxidase; Ox, oxidized enzyme; PEITC, phenethyl isothiocyanate; PIPERL, piperlongumine; PK11195, inhibitor of F1F0 ATP synthase; PRDX, peroxiredoxin; TR, thioredoxin reductase; TRX, thioredoxin; Xc-transporter, L-cystine-L-glutamate antiporter system; Red, reduced enzyme.
Box 1.

no caption available
REDOX SIGNATURES IN B-CELL MALIGNANCIES
Genes controlling the redox homeostasis are important players in the pathogenesis of multiple types of cancer. Consistent with the hypothesis suggesting the causative role of ROS in cancer initiation, single-nucleotide polymorphisms of certain genes involved in the production and detoxification of ROS significantly increase the risk of diffuse large B-cell lymphoma (DLBCL) development [25,26]. In addition, in patients with established disease, single-nucleotide polymorphisms within the myloperoxidase gene are associated with shorter overall survival and progression-free survival [27]. In accordance with this, DLBCL patients with the worst prognosis exhibit decreased expression of SOD2 and vitamin D-upregulated protein (VDUP1, a protein that inhibits TRX activity), but increased TRX expression [3].
Although these earlier results clearly highlight the pathogenic relevance of redox homeostasis in the biological and clinical characteristics of the disease, these studies do not take into account the inherent molecular heterogeneity of the disease [28,29]. For example, a large subset of DLBCLs is reliant on BCR activity (’BCR’-DLBCLs). Additional subset of tumors that do not exhibit addiction to BCR signaling features transcriptional program associated with increased oxidative phosphorylation (‘OxPhos’–DLBCLs) [29]. Energetic metabolism of these DLBCLs is fueled predominantly by palmitate β-oxidation and oxidative phosphorylation of palmitate-derived carbons [30]. Accordingly, ‘OxPhos’-DLBCLs overexpress multiple components of mitochondrial electron transport chain (subunits of complex I, II, and V) that increase mitochondrial superoxide production. The mechanism to counteract this burden potentially relies on overexpression of multiple enzymes involved in ROS detoxification, for example, SOD2 and TRX [9▪▪,30]. Efficient clearance of ROS would also ensure that oxidative phosphorylation could remain elevated in ‘OxPhos’-DLBCLs. Consistent with this, inhibition of either antioxidant system was selectively toxic to this lymphoma subtype. In addition, cells lacking TRX were uniformly more sensitive to ROS and doxorubicin-induced apoptosis than control cells. Toxicity of TRX inhibition in ‘OxPhos-DLBCL’ cells was at least partially dependent on reduced proapoptotic activity of Forkhead box protein O1 (FOXO1) [9▪▪]. TRX reduces disulfide bonds between FOXO1 and p300, formed in response to oxidative stress. As acetylation of FOXO1 increases its apoptotic potential, depletion of TRX facilitated p300-mediated acetylation of FOXO1, and favored FOXO1-dependent cell death. Knockdown of FOXO1 in ‘OxPhos-DLBCL’ cells with silenced TRX expression markedly reduced cell apoptosis in response to ROS, demonstrating that FOXO1 is a redox-sensitive mediator of DLBCL cells’ responses to oxidative stress [9▪▪]. These findings underscore the role of TRX in the pathogenesis of DLBCL and are a substantial step toward therapeutic exploitation of the redox environment modulation in DLBCLs.
Recently, a very interesting drug-resistance mechanism involving antioxidant enzymes was also reported for a subset of DLBCL with chronic active BCR signaling, termed ‘activated B-cell (ABC)-like DLBCL’ [31▪]. In ABC–DLBCLs, doxorubicin, a major component of lymphoma treatment protocols, localizes predominantly to the cytoplasm and triggers cytoplasmic oxidative stress instead of nuclear DNA damage response. Activity of signal transducer and activator of transcription 3 (STAT3) in these DLBCLs led to upregulation of antioxidant enzyme SOD2 and mediated doxorubicin resistance [31▪]. Small molecule STAT3 inhibitor overrode this SOD2-dependent resistance mechanism and restored doxorubicin sensitivity in preclinical models.
The elevated levels of oxidative stress biomarkers were also reported in serum of B-cell chronic lymphocytic leukemia (CLL) patients when compared with healthy study participants. It was accompanied by elevated expression of selected antioxidant enzymes, free thiols, and higher overall antioxidant capacity, presumably as an adaptation to elevated ROS [8]. In addition, mitochondrial biogenesis and increased oxidative phosphorylation were observed in a subset of patients with the highest ROS levels. Interestingly, this subset was sensitive to prooxidative treatment with a benzodiazepine derivative, an inhibitor of ATP synthase composed of subunits F1 and F0[8].
INVOLVEMENT OF REDOX PATHWAYS IN STROMA-MALIGNANT CELL COMMUNICATION
During their differentiation in bone marrow niches and maturation in secondary lymphoid organs, normal B cells rely to a large extent on the support from the surrounding microenvironment. In recent years, a considerable amount of evidence has been gathered regarding the communication between B-cell-derived cancer cells and the microenvironment via BCR-mediated signals, direct cell–cell interactions, and by soluble factors (reviewed in [32,33]). Some studies also highlighted biochemical and metabolic pathways that are crucial for stromal support of malignant cell growth, including the management of cancer-related oxidative stress. The study by Zhang et al.[34] reported a potent reduction of oxidative stress in CLL cells by bone marrow-derived mesenchymal stromal cells (BMSC) via the highly specific cystine–glutamate antiporter system, Xc-. Stromal cells utilize this system to import L-cystine and convert it to L-Cys, which is then released into the tumor microenvironment; subsequently, CLL cells uptake the reduced amino acid to facilitate synthesis of GSH (Fig. 1). This observation is in accordance with the previous report that B-cell-derived malignancies can intrinsically display limited capacity for L-Cys uptake and, therefore, are more prone to ROS-induced cell death [35].
The role of microenvironment in alleviation of cellular oxidative stress has been also documented in DLBCL. In patient-derived DLBCL xenografts (a system that retains microenvironmental components) GSH supply from stromal cells to lymphoma cells has been identified as a major mechanism responsible for adaptation of DLBCL cells to oxidative stress. Importantly, this mechanism was targetable with a small molecule compound, pyrvinium pamoate, which exhibited potent antitumor in vitro and in vivo activity [36]. The dependence on stromal support has also been reported in certain precursor B-cell acute lymphoblastic leukemia (B-ALL) [37]. Moreover, bone marrow-derived stromal cells (BMSC) induced a multidrug resistant phenotype of B-ALL cells, at least in part by secretion of soluble factors that facilitate adaptation of tumor cells to oxidative stress. In a coculture model, BMSC-conditioned medium triggered upregulation of antioxidant enzymes (SOD2, glutathione peroxidase 1/2) and antiapoptotic protein Myeloid cell leukemia 1 (Mcl-1) in B-ALL cells. Blocking such metabolic remodeling by inhibiting antioxidant production restored drug sensitivity, indicating that metabolic plasticity in leukemic cells is a targetable mechanism of chemoresistance, and potentially disease recurrence [38]. Similarly, a recent study has demonstrated that adipocytes, a prominent component of the bone marrow microenvironment, support survival of daunorubicin-treated B-ALL cells via oxidative stress response [39].
Taken together, these data highlight the crucial role of stromal support in the management of oxidative stress and induction of drug resistance phenotype in precursor and mature B-cell malignancies alike. Nonetheless, it has to be noted that most of these studies involved in vitro coculture models; even though 3-D extracellular scaffold systems are quite advanced in recapitulating tumor – microenvironment interactions, the translational value of these findings need to be further confirmed in additional in vivo studies.
INHIBITION OF ANTIOXIDANT DEFENSE AND OTHER REACTIVE OXYGEN SPECIES-INDUCING THERAPEUTIC STRATEGIES: PRECLINICAL AND CLINICAL STUDIES
Increased production of ROS in certain tumor cells because of metabolic dysregulation and specific reliance on antioxidant systems opens a way to specific targeting these pathways in tumor cells. In addition, as multiple classical chemotherapeutics (such as doxorubicin or melphalan) have been shown to produce ROS, these strategies might be synergistic with them. In a number of studies, the researchers have tried to utilize pharmacological inhibition of antioxidant enzymes or other ROS-inducing compounds. In the current work, we mainly describe the use of compounds specifically targeting antioxidant defenses or inducing ROS production. Table 2 and Fig. 1 present several examples of such compounds.
Table 2.
Examples of oxidative stress-inducing agents utilized against B-cell malignancies
| Compound | Antioxidant defense target | Malignancy | Research phase | Reference/clinical trial identifier |
| Auranofin | TR | CLLcHL | in vitro, murine modelsClinical Trial Phase 2in vitro, murine models | [6]NCT01419691[40] |
| SK053 | TRX, TR, PRDX1–4 | Burkitt lymphoma | in vitro | [7▪▪,41] |
| Adenanthin | TRX, TR, PRDX1, PRDX2 | Burkitt lymphoma | in vitro | [42] |
| ATO | TR | CLLBurkitt lymphomaB-cell lymphoma | in vitroin vitroin vitro | [43][44][45] |
| BSO | GCL, inhibits GSH biosynthesis | DLBCL, mantle-cell lymphoma | in vitro | [46] |
| Imexon | GSH-depletion | B-cell NHL | Clinical Trial Phase 2 | [47]NCT01314014 |
| Piperlongumine | GST-π | Burkitt lymphoma | in vitro | [48] |
| Sulfasalazine | Xc- cystine transporter | DLBCL | in vitro | [49] |
| Ellagic acid | Pro-oxidant | CLL | in vitro | [50] |
| Ascorbic acid (vitamin C) | Pro-oxidant | Burkitt lymphomaNHL | in vitroPilot Clinical Trial | [51]NCT00626444 |
ATO, As2O3, arsenic trioxide; BSO, buthionine-sulfoximine; cHL, classical Hodgkin lymphoma; CLL, chronic lymphoblastic leukemia; DLBCL, Diffuse Large B-Cell Lymphoma; GCL, glutamate-cysteine ligase; NHL, non-Hodgkin lymphoma.
TRX and GSH are two major antioxidant systems in cells. Thus, several small molecule inhibitors of various components of these systems have been investigated, so far mostly in preclinical models of B-cell cancers. SK053, which directly binds and inhibits both TRX [52] and dimeric PRDXs, the H2O2-scavenging enzymes, triggers ROS-induced extracellular signal–regulated kinases 1/2 (ERK1/2) activation, cell cycle arrest, and apoptosis in Burkitt lymphoma cell line models [7▪▪]. Similar antilymphoma effects were also reported for another inhibitor with similar specificity – adenanthin [42]. Accordingly, auranofin, the inhibitor of thioredoxin reductase, an upstream enzyme of TRX system, has recently demonstrated antitumor activity in preclinical models of CLL [6] and classical Hodgkin lymphoma [40]. Auranofin, a gold complex approved decades ago for the treatment of rheumatoid arthritis, has recently been evaluated in the phase I/II clinical trial in CLL and small lymphocytic lymphoma patients. The final results of the study have not been published yet, however, preliminary conference reports reveal only transient response and limited clinical efficacy of auranofin used as monotherapy [53]. Similarly, inhibitors of GSH biosynthesis, like buthionine sulfoximine, are effective only in combinations with other therapeutic agents (see the paragraph below) [6,46].
Another long known, though still not fully understood phenomenon, is the particular susceptibility of B-cell lymphoma cells to ROS-inducing agents. As published by Chen et al.[51], lymphoma cells are much more sensitive to direct exposure to exogenous H2O2 as compared to normal B cells. Accordingly, lymphoma cells are among the most sensitive to L-ascorbate (LD50 ∼ 0.5 mmol/l), which generates exogenous H2O2[51]. Despite the high sensitivity in vitro, the antilymphoma activity of high dose parenteral L-ascorbate in vivo is limited [54]. However, another clinically applicable ROS-inducing compounds are being investigated in clinics. A prominent example is imexon, a cyanoaziridine antineoplastic agent binding intracellular thiols and thus depleting stores of cysteine and GSH, which in consequence increases ROS levels [55]. The results of Phase II study published in 2014 revealed 30% overall response rate and a good correlation of the clinical response with redox parameters, supporting the use of imexon against lymphoma [47].
SYNERGISTIC COMBINATIONS INVOLVING INHIBITION OF ANTIOXIDANT DEFENSES
As highlighted above, numerous small molecule inhibitors targeting redox homeostasis are investigated for the treatment of B-cell cancers, yet, in majority of cases their efficacy in monotherapy is limited. However, increasing amount of research reports on synergistic effects of inhibitors of antioxidant enzymes combined with other drugs. Importantly, in many cases these therapeutic modalities present high degree of selectivity toward malignant cells. In Table 3 we present several examples of such combination therapies.
Table 3.
Redox-based combination regimens used for the treatment of B-cell malignancies
| Mechanism | Combined compounds | Malignancy, model | References |
| Enhancement of apoptosis-inducing agents | BSO and SMAC mimetics (BV6, LCL161) | B-ALL cell lines:RehNalmTanouePrimary B-ALL blasts | [56,57] |
| Auranofin and SMAC mimetic (LCL161) | B-ALL cell lines:Reh,Tanoue | [57] | |
| Auranofin and doxorubicine, cisplatin, gemcitabine | cHL cell lines:L-1236HDLM-2 | [40] | |
| PEITC and fludarabine | Primary CLL cells | [34] | |
| PX-12 and doxorubicin | DLBCL cells lines | [58] | |
| TRX-downregulation and doxorubicin | DLBCL cell lines | [9▪▪] | |
| Induction of nonapoptotic cell death | BSO and etoposide BSO and SN-38 | Bcl-2 overexpressing 697 human B-ALL cell line | [59] |
| Dual targeting of antioxidant defense pathways | BSO and auranofin | DLBCL cell lines:SUD-HL6,OCI-LY10MCL cell lines: Rec-1, Granta | [46] |
| BSO and ATO | Burkitt lymphoma cell lines:U937, Namalwa | [60] | |
| Ethacrynic acid and ATO | Burkitt lymphoma cell lines: Raji, Namalwa, Daudi | [61] | |
| PRDX1 and PRDX2 downregulation | Burkitt lymphoma cell lines: Raji, Namalwa | [7▪▪] | |
| Inhibition of stromal support | PEITC and fludarabine/oxaliplatine | Primary CCL cocultured with MSC - HS5 | [34] |
| PEITC and SAHA | Primary CCL cocultured with MSC - HS5 | [62] | |
| L-cysteinase and fludarabine | TCL1-Tg:p53−/− mouse model Primary CLL co-cultured with stromal NKTert cells | [63▪▪] | |
| PEITC and L-asparaginasepiperlongumine and L-asparaginase | Primary B-ALL cells and B-ALL cell lines (REH, SUP-B15) cultured in MSC-derived conditioned medium | [38] |
As2O3, arsenic trioxide; B-ALL, B-cell acute lymphoblastic leukemia; BSO, buthionine-sulfoximine; cHL, classical Hodgkins lymphoma; CLL, chronic lymphocytic leukemia; DLBCL, diffuse large B-cell lymphoma; GCL, glutamate-cysteine ligase; MCL, mantle cell lymphoma; MSC, mesenchymal stromal cells; NHL, non-Hodgkin lymphoma; PEITC, phenethyl isothiocyanate; SAHA, Suberoylanilide Hydroxamic Acid.
It is well established that redox status affects vulnerability to apoptosis and that several antioxidant enzymes including TRX1, PRDX1, play antiapoptotic functions [64,65]. Overcoming apoptosis resistance with inhibitors of antioxidant defense system was also reported for B-cell neoplasms. In B-ALL cell lines and primary cells, buthionine sulfoximine, an inhibitor of GSH synthesis, potentiated the efficacy of SMAC mimetics, molecules that antagonize inhibitors of apoptosis. Similar effect was also reported for a combination of SMAC mimetics with auranofin. Interestingly, the therapies were not toxic to nonmalignant leukocytes nor to mesenchymal stromal cells, underscoring tumor selectivity [56,57].
A likely reason for limited efficacy of antioxidant enzymes’ inhibitors used as single agents is the occurrence of several antioxidant systems, which, at least to some extent, play redundant functions. Indeed, it was shown that thioredoxin reductase inhibition leads to a compensatory increase in the activity of GSH system [66]. Accordingly, concomitant targeting of both TRX and GSH redox systems resulted in synergistic cytotoxicity in cell line models of CLL [6], DLBCL [46], and Burkitt lymphoma [60]. Similar synergistic effect was also observed in primary CLL [6] and mantle cell lymphoma cells but not in normal B cells [46]. Hence, the use of inhibitors with broader specificity, such as SK053 or adenanthin, which simultaneously inhibit several antioxidant enzymes, may contribute to better efficacy. In support of this hypothesis, the concomitant downregulation of both PRDX1 and PRDX2 contributed to significant attenuation of Burkitt lymphoma cell growth [7▪▪].
Finally, the restriction of GSH biosynthesis, either by thiol depletion or inhibition of stromal support, emerges as a novel strategy to overcome chemotherapy resistance. As mentioned above, stromal cells provide malignant B cells with L-Cys, a substrate for GSH biosynthesis, which may contribute to drug resistance. In accordance, GSH depleting agent β-phenylethyl isothiocyanate enhances cytotoxic effects of fludarabine and oxaliplatine in primary CLL-stroma coculture models, including cells derived from patients with drug-resistant 17p-deletion [34]. Similarly, phenylethyl isothiocyanate enhances sensitivity to L-asparaginase of B-ALL cells cultured in a medium containing soluble factors secreted by stromal cells [38]. Another, recently presented strategy to hamper GSH biosynthesis, is to deplete L-Cys via administration of L-cysteinase, an enzyme which degrades L-Cys. L-cysteinase significantly reduces tumor growth as well as increases the efficacy of fludarabine in TCL1-Tg:p53−/−mice, a transgenic model of CLL. Importantly, systemic administration of this enzyme is well tolerated, without any signs of toxicity. Moreover, L-cysteinase enhances fludarabine cytotoxicity to primary, patient-derived CLL cells cultured in vitro with stroma. The advantage of L-cysteinase over other GSH-depleting agents is that it concomitantly reduces GSH and L-Cys supply, what is manifested with a remarkable in vivo efficacy [63▪▪]. It would be interesting to see the efficacy of the combinations of the enzyme with other treatment modalities.
CONCLUSION
Despite a significant progress in our understanding of the role of redox homeostasis in B-cell malignancies in recent years, one must conclude that the purely prooxidant therapeutic approaches against B-cell cancers are still in their early development. Further studies are needed to identify subtype-specific targets and selective inhibitors for such. Importantly, the synergistic effects of prooxidant therapeutics with chemotherapy agents are encouraging and should stimulate further research for testing their combinations with modern drugs used in clinics.
Acknowledgements
We would like to thank Professor Jakub Golab for his continued support.
Financial support and sponsorship
The work was supported by National Science Center 2015/18/E/NZ5/00723 (MF), 2014/13/N/NZ6/02081 (MS), European Commission Horizon 2020 Programme 692180-STREAM-H2020-TWINN-2015 (RZ), the Medical University of Warsaw 1M19/PM1/16 (AGJ) grants.
Conflicts of interest
There are conflicts of interest.
REFERENCES AND RECOMMENDED READING
Papers of particular interest, published within the annual period of review, have been highlighted as:
▪ of special interest
▪▪ of outstanding interest
Footnotes
Malgorzata Firczuk and Przemyslaw Juszczynski equally contribute to this article.
REFERENCES
- 1.Omenn GS, Goodman GE, Thornquist MD, et al. Effects of a combination of beta carotene and vitamin A on lung cancer and cardiovascular disease. N Engl J Med 1996; 334:1150–1155. [DOI] [PubMed] [Google Scholar]
- 2.Honda M, Yamada Y, Tomonaga M, et al. Correlation of urinary 8-hydroxy-2′-deoxyguanosine (8-OHdG), a biomarker of oxidative DNA damage, and clinical features of hematological disorders: a pilot study. Leuk Res 2000; 24:461–468. [DOI] [PubMed] [Google Scholar]
- 3.Tome ME, Johnson DB, Rimsza LM, et al. A redox signature score identifies diffuse large B-cell lymphoma patients with a poor prognosis. Blood 2005; 106:3594–3601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Le Gal K, Ibrahim MX, Wiel C, et al. Antioxidants can increase melanoma metastasis in mice. Sci Transl Med 2015; 7:308re8. [DOI] [PubMed] [Google Scholar]
- 5.Klein EA, Thompson IM, Jr, Tangen CM, et al. Vitamin E and the risk of prostate cancer: the Selenium and Vitamin E Cancer Prevention Trial (SELECT). JAMA 2011; 306:1549–1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fiskus W, Saba N, Shen M, et al. Auranofin induces lethal oxidative and endoplasmic reticulum stress and exerts potent preclinical activity against chronic lymphocytic leukemia. Cancer Res 2014; 74:2520–2532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7▪▪.Trzeciecka A, Klossowski S, Bajor M, et al. Dimeric peroxiredoxins are druggable targets in human burkitt lymphoma. Oncotarget 2016; 7:1717–1731. [DOI] [PMC free article] [PubMed] [Google Scholar]; The study shows that BL cells and cell lines have elevated levels of PRDX 1 and 2 and that targeting of these enzymes either by knockdown or peptidomimetic inhibitor SK053 causes increased accumulation of ROS, cell cycle arrest, and apoptosis.
- 8.Jitschin R, Hofmann AD, Bruns H, et al. Mitochondrial metabolism contributes to oxidative stress and reveals therapeutic targets in chronic lymphocytic leukemia. Blood 2014; 123:2663–2672. [DOI] [PubMed] [Google Scholar]
- 9▪▪.Sewastianik T, Szydlowski M, Jablonska E, et al. FOXO1 is a TXN- and p300-dependent sensor and effector of oxidative stress in diffuse large B-cell lymphomas characterized by increased oxidative metabolism. Oncogene 2016; 35:5989–6000. [DOI] [PubMed] [Google Scholar]; The study shows that elevated TRX level in OxPhos–DLBCL blocks ROS dependent p300-mediated FOXO1 acetylation, its nuclear translocation and in this way attenuate FOXO1 transcriptional activity toward genes involved in apoptosis and cell cycle inhibition.
- 10.Vafa O, Wade M, Kern S, et al. c-Myc can induce DNA damage, increase reactive oxygen species, and mitigate p53 function: a mechanism for oncogene-induced genetic instability. Mol Cell 2002; 9:1031–1044. [DOI] [PubMed] [Google Scholar]
- 11.Naughton R, Quiney C, Turner SD, Cotter TG. Bcr-Abl-mediated redox regulation of the PI3K/AKT pathway. Leukemia 2009; 23:1432–1440. [DOI] [PubMed] [Google Scholar]
- 12.Kantner HP, Warsch W, Delogu A, et al. ETV6/RUNX1 induces reactive oxygen species and drives the accumulation of DNA damage in B cells. Neoplasia 2013; 15:1292–1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Reth M. Hydrogen peroxide as second messenger in lymphocyte activation. Nat Immunol 2002; 3:1129–1134. [DOI] [PubMed] [Google Scholar]
- 14.Fukai T, Ushio-Fukai M. Superoxide dismutases: role in redox signaling, vascular function, and diseases. Antioxid Redox Signal 2011; 15:1583–1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Glorieux C, Zamocky M, Sandoval JM, et al. Regulation of catalase expression in healthy and cancerous cells. Free Radic Biol Med 2015; 87:84–97. [DOI] [PubMed] [Google Scholar]
- 16.Brigelius-Flohe R, Maiorino M. Glutathione peroxidases. Biochim Biophys Acta 2013; 1830:3289–3303. [DOI] [PubMed] [Google Scholar]
- 17.Wood ZA, Schroder E, Robin Harris J, Poole LB. Structure, mechanism and regulation of peroxiredoxins. Trends Biochem Sci 2003; 28:32–40. [DOI] [PubMed] [Google Scholar]
- 18.Arner ES, Holmgren A. Physiological functions of thioredoxin and thioredoxin reductase. Eur J Biochem 2000; 267:6102–6109. [DOI] [PubMed] [Google Scholar]
- 19.Mesecke N, Spang A, Deponte M, Herrmann JM. A novel group of glutaredoxins in the cis-golgi critical for oxidative stress resistance. Mol Biol Cell 2008; 19:2673–2680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Couto N, Wood J, Barber J. The role of glutathione reductase and related enzymes on cellular redox homoeostasis network. Free Radic Biol Med 2016; 95:27–42. [DOI] [PubMed] [Google Scholar]
- 21.Biteau B, Labarre J, Toledano MB. ATP-dependent reduction of cysteine-sulphinic acid by S. cerevisiae sulphiredoxin. Nature 2003; 425:980–984. [DOI] [PubMed] [Google Scholar]
- 22.Glasauer A, Chandel NS. Targeting antioxidants for cancer therapy. Biochem Pharmacol 2014; 92:90–101. [DOI] [PubMed] [Google Scholar]
- 23.Gorrini C, Harris IS, Mak TW. Modulation of oxidative stress as an anticancer strategy. Nat Rev Drug Discov 2013; 12:931–947. [DOI] [PubMed] [Google Scholar]
- 24.Hanschmann EM, Godoy JR, Berndt C, et al. Thioredoxins, glutaredoxins, and peroxiredoxins: molecular mechanisms and health significance: from cofactors to antioxidants to redox signaling. Antioxid Redox Signal 2013; 19:1539–1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lan Q, Zheng T, Shen M, et al. Genetic polymorphisms in the oxidative stress pathway and susceptibility to non-Hodgkin lymphoma. Hum Genet 2007; 121:161–168. [DOI] [PubMed] [Google Scholar]
- 26.Wang SS, Davis S, Cerhan JR, et al. Polymorphisms in oxidative stress genes and risk for non-Hodgkin lymphoma. Carcinogenesis 2006; 27:1828–1834. [DOI] [PubMed] [Google Scholar]
- 27.Gustafson HL, Yao S, Goldman BH, et al. Genetic polymorphisms in oxidative stress-related genes are associated with outcomes following treatment for aggressive B-cell non-Hodgkin lymphoma. Am J Hematol 2014; 89:639–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Alizadeh AA, Eisen MB, Davis RE, et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature 2000; 403:503–511. [DOI] [PubMed] [Google Scholar]
- 29.Monti S, Savage KJ, Kutok JL, et al. Molecular profiling of diffuse large B-cell lymphoma identifies robust subtypes including one characterized by host inflammatory response. Blood 2005; 105:1851–1861. [DOI] [PubMed] [Google Scholar]
- 30.Caro P, Kishan AU, Norberg E, et al. Metabolic signatures uncover distinct targets in molecular subsets of diffuse large B cell lymphoma. Cancer Cell 2012; 22:547–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31▪.Mai Y, Yu JJ, Bartholdy B, et al. An oxidative stress-based mechanism of doxorubicin cytotoxicity suggests new therapeutic strategies in ABC-DLBCL. Blood 2016; 128:2797–2807. [DOI] [PMC free article] [PubMed] [Google Scholar]; The study shows the mechanism of ABC–DLBCL resistance to CHOP and rituximab combined with CHOP regimen in which doxorubicin present mainly in cytosolic compartment cause oxidative stress that trigger STAT3-mediated upregulation of SOD2 which in turn confers doxorubicin resistance in ABC–DLBCLs.
- 32.Krause DS, Scadden DT. A hostel for the hostile: the bone marrow niche in hematologic neoplasms. Haematologica 2015; 100:1376–1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Scott DW, Gascoyne RD. The tumour microenvironment in B cell lymphomas. Nat Rev Cancer 2014; 14:517–534. [DOI] [PubMed] [Google Scholar]
- 34.Zhang W, Trachootham D, Liu J, et al. Stromal control of cystine metabolism promotes cancer cell survival in chronic lymphocytic leukaemia. Nat Cell Biol 2012; 14:276–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Banjac A, Perisic T, Sato H, et al. The cystine/cysteine cycle: a redox cycle regulating susceptibility versus resistance to cell death. Oncogene 2008; 27:1618–1628. [DOI] [PubMed] [Google Scholar]
- 36.Sugimoto K, Hayakawa F, Shimada S, et al. Discovery of a drug targeting microenvironmental support for lymphoma cells by screening using patient-derived xenograft cells. Sci Rep 2015; 5:13054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Boutter J, Huang Y, Marovca B, et al. Image-based RNA interference screening reveals an individual dependence of acute lymphoblastic leukemia on stromal cysteine support. Oncotarget 2014; 5:11501–11512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu J, Masurekar A, Johnson S, et al. Stromal cell-mediated mitochondrial redox adaptation regulates drug resistance in childhood acute lymphoblastic leukemia. Oncotarget 2015; 6:43048–43064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sheng X, Tucci J, Parmentier JH, et al. Adipocytes cause leukemia cell resistance to daunorubicin via oxidative stress response. Oncotarget 2016; 7:73147–73159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Celegato M, Borghese C, Casagrande N, et al. Preclinical activity of the repurposed drug auranofin in classical Hodgkin lymphoma. Blood 2015; 126:1394–1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Muchowicz A, Firczuk M, Wachowska M, et al. SK053 triggers tumor cells apoptosis by oxidative stress-mediated endoplasmic reticulum stress. Biochem Pharmacol 2015; 93:418–427. [DOI] [PubMed] [Google Scholar]
- 42.Muchowicz A, Firczuk M, Chlebowska J, et al. Adenanthin targets proteins involved in the regulation of disulphide bonds. Biochem Pharmacol 2014; 89:210–216. [DOI] [PubMed] [Google Scholar]
- 43.Lozano-Santos C, Amigo-Jimenez I, Nova-Gurumeta S, et al. Arsenic trioxide synergistically potentiates the cytotoxic effect of fludarabine in chronic lymphocytic leukemia cells by further inactivating the Akt and ERK signaling pathways. Biochem Biophys Res Commun 2015; 461:243–248. [DOI] [PubMed] [Google Scholar]
- 44.Lu D, Bai XC, Gui L, et al. Arsenic trioxide-induced apoptosis of human malignant lymphoma cell lines and its mechanisms. Di Yi Jun Yi Da Xue Xue Bao 2003; 23:997–1001. [PubMed] [Google Scholar]
- 45.Shen L, Chen TX, Wang YP, et al. As2O3 induces apoptosis of the human B lymphoma cell line MBC-1. J Biol Regul Homeost Agents 2000; 14:116–119. [PubMed] [Google Scholar]
- 46.Kiebala M, Skalska J, Casulo C, et al. Dual targeting of the thioredoxin and glutathione antioxidant systems in malignant B cells: a novel synergistic therapeutic approach. Exp Hematol 2015; 43:89–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Barr PM, Miller TP, Friedberg JW, et al. Phase 2 study of imexon, a prooxidant molecule, in relapsed and refractory B-cell non-Hodgkin lymphoma. Blood 2014; 124:1259–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Han SS, Son DJ, Yun H, et al. Piperlongumine inhibits proliferation and survival of Burkitt lymphoma in vitro. Leuk Res 2013; 37:146–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gout PW, Buckley AR, Simms CR, Bruchovsky N. Sulfasalazine, a potent suppressor of lymphoma growth by inhibition of the x(c)-cystine transporter: a new action for an old drug. Leukemia 2001; 15:1633–1640. [DOI] [PubMed] [Google Scholar]
- 50.Salimi A, Roudkenar MH, Sadeghi L, et al. Ellagic acid, a polyphenolic compound, selectively induces ROS-mediated apoptosis in cancerous B-lymphocytes of CLL patients by directly targeting mitochondria. Redox Biol 2015; 6:461–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chen Q, Espey MG, Krishna MC, et al. Pharmacologic ascorbic acid concentrations selectively kill cancer cells: action as a pro-drug to deliver hydrogen peroxide to tissues. Proc Natl Acad Sci U S A 2005; 102:13604–13609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Klossowski S, Muchowicz A, Firczuk M, et al. Studies toward novel peptidomimetic inhibitors of thioredoxin-thioredoxin reductase system. J Med Chem 2012; 55:55–67. [DOI] [PubMed] [Google Scholar]
- 53.Saba NS, Ghias M, Manepalli R, et al. Auranofin Induces a Reversible In-Viva Stress Response That Correlates With a Transient Clinical Effect In Patients With Chronic Lymphocyte Leukemia. In American Society of Hematology Meeting. Edited by: Blood; 2013:3819. Vol 122. [Google Scholar]
- 54.Shatzer AN, Espey MG, Chavez M, et al. Ascorbic acid kills Epstein-Barr virus positive Burkitt lymphoma cells and Epstein-Barr virus transformed B-cells in vitro, but not in vivo. Leuk Lymphoma 2013; 54:1069–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Iyengar BS, Dorr RT, Remers WA. Chemical basis for the biological activity of imexon and related cyanoaziridines. J Med Chem 2004; 47:218–223. [DOI] [PubMed] [Google Scholar]
- 56.Schoeneberger H, Belz K, Schenk B, Fulda S. Impairment of antioxidant defense via glutathione depletion sensitizes acute lymphoblastic leukemia cells for SMAC mimetic-induced cell death. Oncogene 2015; 34:4032–4043. [DOI] [PubMed] [Google Scholar]
- 57.Hass C, Belz K, Schoeneberger H, Fulda S. Sensitization of acute lymphoblastic leukemia cells for LCL161-induced cell death by targeting redox homeostasis. Biochem Pharmacol 2016; 105:14–22. [DOI] [PubMed] [Google Scholar]
- 58.Li C, Thompson MA, Tamayo AT, et al. Over-expression of Thioredoxin-1 mediates growth, survival, and chemoresistance and is a druggable target in diffuse large B-cell lymphoma. Oncotarget 2012; 3:314–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yoshida A, Takemura H, Inoue H, et al. Inhibition of glutathione synthesis overcomes Bcl-2-mediated topoisomerase inhibitor resistance and induces nonapoptotic cell death via mitochondrial-independent pathway. Cancer Res 2006; 66:5772–5780. [DOI] [PubMed] [Google Scholar]
- 60.Chen D, Chan R, Waxman S, Jing Y. Buthionine sulfoximine enhancement of arsenic trioxide-induced apoptosis in leukemia and lymphoma cells is mediated via activation of c-Jun NH2-terminal kinase and up-regulation of death receptors. Cancer Res 2006; 66:11416–11423. [DOI] [PubMed] [Google Scholar]
- 61.Wang R, Liu C, Xia L, et al. Ethacrynic acid and a derivative enhance apoptosis in arsenic trioxide-treated myeloid leukemia and lymphoma cells: the role of glutathione S-transferase p1-1. Clin Cancer Res 2012; 18:6690–6701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhang W, Pelicano H, Yin R, et al. Effective elimination of chronic lymphocytic leukemia cells in the stromal microenvironment by a novel drug combination strategy using redox-mediated mechanisms. Mol Med Rep 2015; 12:7374–7388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63▪▪.Cramer SL, Saha A, Liu J, et al. Systemic depletion of L-cyst(e)ine with cyst(e)inase increases reactive oxygen species and suppresses tumor growth. Nat Med 2017; 23:120–127. [DOI] [PMC free article] [PubMed] [Google Scholar]; The study shows that administration of an engineered human L-cysteinase depletes extracellular L-cysteine and L-cystine pool in mice and nonhuman primates and causes cell cycle arrest and death selectively in cancer cells. This study shows promising and nontoxic approach in overcoming stromal support.
- 64.Kim SY, Kim TJ, Lee KY. A novel function of peroxiredoxin 1 (Prx-1) in apoptosis signal-regulating kinase 1 (ASK1)-mediated signaling pathway. FEBS Lett 2008; 582:1913–1918. [DOI] [PubMed] [Google Scholar]
- 65.Saitoh M, Nishitoh H, Fujii M, et al. Mammalian thioredoxin is a direct inhibitor of apoptosis signal-regulating kinase (ASK) 1. EMBO J 1998; 17:2596–2606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mandal PK, Schneider M, Kolle P, et al. Loss of thioredoxin reductase 1 renders tumors highly susceptible to pharmacologic glutathione deprivation. Cancer Res 2010; 70:9505–9514. [DOI] [PubMed] [Google Scholar]