

Abstract
Maintenance of genomic stability is a critical determinant of cell survival and is necessary for growth and progression of malignant cells. Interstrand crosslinking (ICL) agents, including platinum-based agents, are first-line chemotherapy treatment for many solid human cancers. In malignant cells, ICL triggers the DNA damage response (DDR). When the damage burden is high and lesions cannot be repaired, malignant cells are unable to divide and ultimately undergo cell death either through mitotic catastrophe or apoptosis. The activities of ICL agents, in particular platinum-based therapies, establish a “molecular landscape,” i.e., a pattern of DNA damage that can potentially be further exploited therapeutically with DDR-targeting agents. If the molecular landscape created by platinum-based agents could be better defined at the molecular level, a systematic, mechanistic rationale(s) could be developed for the use of DDR-targeting therapies in combination/maintenance protocols for specific, clinically advanced malignancies. New therapeutic drugs such as poly(ADP-ribose) polymerase (PARP) inhibitors are examples of DDR-targeting therapies that could potentially increase the DNA damage and replication stress imposed by platinum-based agents in tumor cells and provide therapeutic benefit for patients with advanced malignancies. Recent studies have shown that the use of PARP inhibitors together with platinum-based agents is a promising therapy strategy for ovarian cancer patients with ”BRCAness”, i.e., a phenotypic characteristic of tumors that not only can involve loss-of-function mutations in either BRCA1 or BRCA2, but also encompasses the molecular features of BRCA-mutant tumors. On the basis of these promising results, additional mechanism-based studies focused on the use of various DDR-targeting therapies in combination with platinum-based agents should be considered. This review discusses, in general, (1) ICL agents, primarily platinum-based agents, that establish a molecular landscape that can be further exploited therapeutically; (2) multiple points of potential intervention after ICL agent–induced crosslinking that further predispose to cell death and can be incorporated into a systematic, therapeutic rationale for combination/maintenance therapy using DDR-targeting agents; and (3) available agents that can be considered for use in combination/maintenance clinical protocols with platinum-based agents for patients with advanced malignancies.
Keywords: Platinum-based agents, DDR targeting agents, PARP, WEE-1, ATR, Chk-1/Chk-2
INTRODUCTION
Human cellular components, including cellular DNA, are subject to a variety of endogenous and environmental damages [1]. Much of this damage can interfere with fundamental cellular processes and lead to cell death. Damaged cells have developed multiple repair mechanisms to restore genomic integrity and to maintain viability. These repair processes are used not only by normal cells but also by cancer cells, to achieve continuous growth and progression. Malignant cells possess an extensive network of repair mechanisms that allows resolution of DNA damage and maintenance of genomic integrity [2]. Knowledge of these pathways has allowed for the development and application of specific chemotherapeutic drugs that target DNA, through DNA crosslinking, and exploitation of the limitations of DNA damage response (DDR) and DNA repair in malignant cells.
DNA crosslinking produces toxic lesions and can be divided into two major categories: intrastrand crosslinking, which occurs when two nucleotides of the same DNA strand are covalently linked, and interstrand crosslinking (ICL), which occurs when nucleotides belonging to different DNA strands are covalently linked. The linkage of nucleotides on different DNA strands through an intermediate alkylating agent requires the agent to have two separate reactive groups. In these “bifunctional” alkylating agents, each reactive group of the agent covalently binds to a different nucleotide and the ICL is formed. Crosslinking agents have been described as more clastogenic than mutagenic, meaning that they perform their action at the chromosomal level rather than the DNA sequence level [3].
ICL agents have been used in clinical practice for more than 70 years, following the introduction of a nitrogen mustard for the treatment of leukemia and Hodgkin disease [4]. Platinum-based agents, one of the most widely used categories of ICL agents, were introduced later when cisplatin, the first such agent, was first shown to have anti-tumor properties by Rosenberg in 1969 [5]. The number of antitumor drugs with ICL properties used for clinical purposes has increased in recent years. Cisplatin-based combinations remain first-line therapy for a number of solid tumors including lung, bladder, ovarian, and testicular cancers [6]. ICLs block the separation of DNA strands, thus interfering with the fundamental processes of DNA transcription and replication. If ICLs are not repaired before mitosis, they can lead to cell death via the apoptosis pathway. Multiple pathways participate in the recognition of DNA crosslinks, including MAPK, ATR, and p53, which can result in activation of Caspase 9 or FasL to initiate apoptosis [7, 8].
ICL drugs can be categorized into two broad groups: alkylating agents and psoralens-parent compounds of the larger family of plant-derived furocoumarins (Figure 1). The larger family of alkylating agents consists mainly of nitrogen mustards, mitomycin C, and platinum-based agents. Psoralens, which are mostly used in dermatological diseases such as psoriasis and vitiligo, first require activation by UV light before they can intercalate into DNA and form ICLs with thymines [9].
Fig 1.
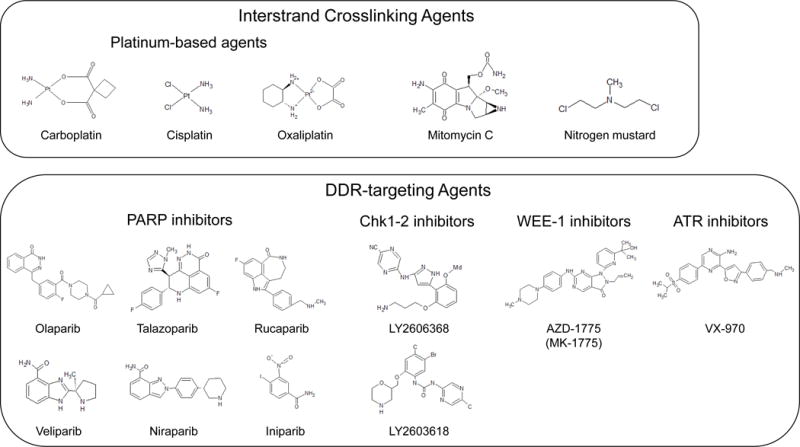
Stereotypical chemical types of interstrand crosslinking agents, including platinum-based agents, mitomycin C, and nitrogen mustards; platinum-based agents; PARP inhibitors; Chk1-2 inhibitors; WEE-1 inhibitor; and ATR inhibitors currently used in clinical trials.
Note: DDR, DNA damage response; PARP, poly(ADP-ribose) polymerase; ATR, Ataxia telangiectasia and Rad3 related
In this review, we focus on combination platinum-based and DDR-targeting cancer therapy. Platinum-based agents are widely used for solid tumors and have been studied extensively in terms of efficacy, mechanism of action, and side effects. The most commonly used DDR-targeting agents are poly(ADP-ribose) polymerase (PARP) inhibitors. In recent years, a relatively large number of these agents have been developed, and many are being tested in clinical trials. The broader field of DDR-targeting agents includes ATR inhibitors, WEE-1 inhibitors, and Chk1/2 inhibitors. These agents have only recently been used clinically, and information about their antitumor activity is limited. In general, the use of PARP inhibitors and other DDR-targeting agents has been predicated on the induction of synthetic lethality, i.e., “a situation in which a defect in one gene or protein is compatible with cell viability, but results in cell death when combined (synthesized) with another gene or protein defect” [10]. Although the concept of synthetic lethality originated with early geneticists using the fruit fly [11], its recently appreciated relevance to oncology science has been dramatically accelerated by technical advances in genomic medicine [10]. Closely associated with the concept of synthetic lethality is “BRCAness.” BRCAness has generally been defined as a phenotypic characteristic of tumors that not only can involve loss-of-function mutations in either BRCA1 or BRCA2, but also encompasses the molecular features of BRCA-mutant tumors [10, 12]. The assumption in the search to further define BRCAness within the context of experimental and clinical oncology is that tumors with BRCAness will respond to specific DDR-targeting therapies similarly to tumors with either BRCA1 or BRCA2 loss of function mutations [10, 12].
A principle of modern oncological science and clinical practice is to achieve better responses while avoiding side effects and the development of resistance. While a therapeutic response of cancers to PARP inhibition due to BRCA1 and BRCA2 mutations has been demonstrated, a wider use of PARP inhibitors in cancers with other “native” or drug-induced deficiencies in other homologous recombination genes and/or DDR genes has not been widely investigated. This approach would incorporate the concept of using platinum-based drugs, in part, to create a favorable “molecular landscape” that could potentiate the activities of DDR-targeting agents to maintain the platinum responses or possibly induce synthetic lethality.
MOLECULAR LANDSCAPE OF PLATINUM-BASED AGENTS AND RESISTANCE MECHANISMS
Platinum-based agents establish a “molecular landscape” that can be further exploited therapeutically
An understanding of the “molecular landscape” that platinum-based agents establish in cancer cells requires a comprehensive understanding of their mechanism of action. If the platinum-based agent is not biologically neutralized by the cancer cell’s transmembrane transporter system, then it can react with the cell’s DNA, RNA, and proteins [13]. It is widely accepted that platinum-based agents exert their cytotoxic effects predominantly via nuclear DNA damage, yet their effects on other compartments of the cell should not be ignored [14]. Platinum-based drugs exert pleiotropic effects that include widespread interactions with cellular molecules that can lead to clinical side effects, limiting their usage. The broad range of intracellular molecular targets of platinum-based agents was clearly demonstrated by Wisnovsky et al., who showed that platinum-induced mitochondrial DNA (mtDNA) damage is sufficient to mediate the activity of a platinum-based chemotherapeutic drug, even without damaging nuclear DNA [15].
Platinum-based agents generate DNA lesions on the purine bases (adenine [A] and guanine [G]) of DNA; the N7 atoms of the imidazole rings are the most accessible site for platinum-based drugs to bind DNA [16]. The first reactive event that takes place is the production of a monofunctional DNA adduct, which following subsequent reactions, can lead to a variety of DNA lesions [17]. These lesions could be summarized as (1) DNA monoadducts, (2) intrastrand crosslinks, (3) DNA-protein crosslinks, and (4) ICL [14, 18] (Figure 2).
Fig 2.
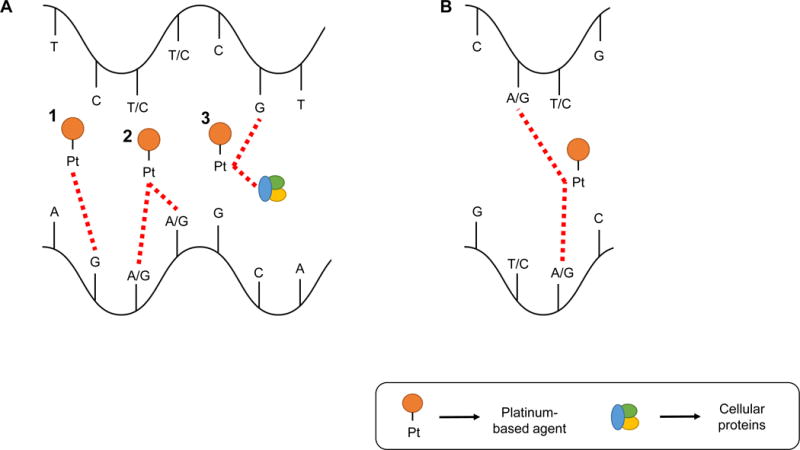
Mechanisms of action of platinum-based agents. A. Single DNA strand actions of platinum-based agents; 1. DNA monoadducts; 2. Intrastrand crosslinks; 3. DNA-protein crosslinks. B. Double DNA strand actions of platinum-based agents; Interstrand crosslinks.
Note: Pt, Platinum
Monoadducts are generated when a platinum-based agent loses a molecule of water and then reacts with a purine (Figure 2.A.1). Monoadduct creation is also the first step in the development of intrastrand crosslinks. The majority of intrastrand crosslinks are 1,2-d(GpG) crosslinks, followed by a small number of 1,2-d(ApG) [14] (Figure 2.A.2). ICLs occur when the platinum-based agent links a guanine/adenine from one strand with a guanine/adenine of the other strand. The most common position of interaction between the agent and the DNA is the N7 position of guanine [3, 19].
The nature of ICL lesions, i.e., both DNA strands targeted, leads to inhibition of DNA replication fork separation, thus affecting critical cellular processes such as DNA replication and gene transcription. Although platinum-based agents are considered to lack cell cycle specificity, cytotoxicity is increased with exposure to the drug during S phase. Cisplatin causes cell cycle arrest in G2 phase and accumulation of cancer cells in G2/M phase, ultimately leading to apoptosis [17, 19–21].
As discussed above, the effects of platinum-based agents are not limited to DNA but extend to RNA and proteins. Osborn et al. reported that platinum-based drugs can interact with tRNA and rRNA in vitro, demonstrating that these drugs can potentially interact with RNA-based regulatory pathways [22]. Karasawa et al. identified a variety of proteins that interact with platinum-based drugs after they traverse the cellular membrane. These interacting proteins largely included heat shock and ribosomal proteins, and the authors suggested that these types of interactions are the major cause of cisplatin’s cytotoxic side effects in specific organ systems (nephrotoxicity and ototoxicity) [23].
Extensive research has revealed multiple mechanisms that underlie recognition and repair of DNA damage induced by platinum-based agents. A better understanding of these mechanisms has become important because virtually all cancers become highly resistant to this type of chemotherapy. The cellular repair response to platinum-based agent–induced DNA damage cannot be predicted due to the complexity of these lesions, and the alkylation damage can elicit a variety of repair mechanisms. The most important DNA repair mechanisms implicated within the context of platinum-based agents are the three excision repair pathways—the nucleotide excision repair (NER), base excision repair (BER), and mismatch repair (MMR) pathways, which are involved in repairing DNA (SSBs)—and the homologous recombination (HR) and non-homologous end-joining (NHEJ) pathways, that repair DSBs [24]. Importantly, these pathways are also related to clinical resistance to platinum-based agents.
The NER pathway involves more than 30 protein factors and is the major pathway for repairing DNA lesions induced by platinum-based agents. Platinum-based agent-induced DNA damage is initially recognized by the heterodimer XPC/hHR23B, which binds with higher affinity to helix-distorting DNA lesions than to non-damaged, double-stranded DNA [25, 26]. The binding of XPC/hHR23B to the DNA lesions provides a substrate for the TFIIH complex to attach and function as a repair factor [27]. The enzymes of this complex detect and verify the DNA damage. After this step, 24–30 base pairs of the lesion-containing area are excised and then repaired [28].
Another DNA repair pathway involved in repair of SSBs generated by platinum-based agents that is functionally related to the NER pathway is BER. A major difference between NER and BER is the size of the lesion that can be recognized. In BER, a DNA glycosylase enzyme recognizes and then removes the nitrogenous base part of the affected nucleotide. After this step, an AP endonuclease removes the abasic site; a DNA polymerase fills the gap, and a DNA ligase seals the DNA area [29]. Studies have shown that patients who have specific single nucleotide polymorphisms (SNPs) in major BER genes show a better response to platinum-based treatment, supporting the significance of the BER pathway in the repair platinum-induced DNA lesions [30].
MMR is also required for the recognition and resolution of specific DNA lesions generated by platinum-based agents [31]. The lesions that are detected by MMR are mispaired or unpaired bases. After detection, assembly of multiple factors takes place to excise the error, and DNA polymerases also gather to repair the base sequence [32]. The human MutS protein homolog 2 protein has been shown to detect 1,2-d(GpG) intrastrand DNA crosslinks induced by cisplatin [33]. Tumor cells that have a deficient MMR system are not able to repair DNA areas with unpaired bases, leading to accumulation of these lesions, a process called “microsatellite instability,” that can ultimately lead to cell death [34].
HR is another major DNA repair pathway that plays an important role in repairing the DNA lesions induced by platinum-based agents [19]. After NER-mediated identification and resolution of a DNA lesion, the process of HR “fills in” the resulting DNA gaps. HR-dependent ICL repair occurs during late S or G2 phase [19]. It is well-documented in the literature, that critical components of the HR mechanism which mediate the repair of DSBs are BRCA1 and BRCA2 [35, 36]. Although BRCA1 and BRCA2 have many pivotal roles in DNA repair, one of the most crucial roles seems to be direct binding of BRCA2 to the RAD51 protein to form a complex on the DNA. This complex leads to the recruitment of proteins to the DNA lesions that initiates the HR repair process [12, 37]. HR repair of DNA lesions requires a second, homologous DNA that can act as a template for the repair reaction [38–42]. Information from the homologous ‘donor’ sequence is copied into the damaged site, allowing for the precise restoration of the original DNA sequence [42]. Ovarian cancer patients with germline mutations in BRCA1 and/or BRCA2 exhibit impaired ability to repair DNA lesions via HR. This event could potentially explain the increased sensitivity that these tumors, and some prostate cancers, have to platinum-based drugs [43, 44].
Finally, NHEJ is also involved in the repair of ICL DNA lesions, but to a lesser extent compared with the repair pathways discussed above [45]. In contrast to HR, which repairs DSBs maintaining the genetic information contained in the homologous DNA strand, NHEJ often introduces errors while repairing the DNA lesion. NHEJ functions throughout the cell cycle to directly ligate broken DNA ends. First, the end-binding KU70/80 heterodimer complex binds to the DNA ends and activates DNA-dependent protein kinase catalytic subunits. Then the DNA lesion is removed, and finally, a DNA ligase IV-XRCC4 rejoins the two DNA ends. In contrast to HR, NHEJ can generate translocations and/or deletions and is generally considered to be error-prone [45, 46].
Mechanisms of resistance to platinum-based agents
The mechanisms associated with resistance to platinum-based agents involve three related activities: altered cellular accumulation of the agent, cytosolic inactivation/metabolism of the agent, and altered DNA repair. These activities can overcome the therapy effects of the platinum-based agent [13]. Amable et al. [13] reviewed these resistance mechanisms within the context of cisplatin treatment, but these resistance pathways are relevant to all platinum-based agents. Each of these mechanisms can singly or in combination contribute to cancer cell resistance.
A reduction in intracellular accumulation of platinum-based agents can be achieved by cancer cells either by increased drug efflux or by decreased drug influx. Thus far, it has been reported that a significant number of membrane transporters facilitate the influx of platinum-based agents into cancer cells. One of the most studied and understood is the copper transport protein 1 (CTR1). CTR1 mRNA levels were found to be high in ovarian cancer patients who had a good response and an increased disease-free survival period after treatment with cisplatin [47]. High CTR1 levels have the capacity to transfer more drug into the cell and achieve better therapeutic results. Cells resistant to cisplatin demonstrate low levels of CTR1 mRNA and also decreased levels of cisplatin [48]. Kim et al. reported that reduced drug accumulation in cancer tissues is an important mechanism of platinum resistance in patients with non-small cell lung cancer [49]. Additionally, the increased efflux of platinum-based agents has also been suggested as a resistance mechanism; however, it is thought to be of relatively minor significance compared with the decreased influx mechanisms [13]. Thus far, two efflux mechanisms have been reported: P-type ATPase transporters and ATP-binding cassette transporters.
Cytosolic inactivation/metabolism of platinum-based agents is an alternative cellular mechanism of platinum resistance. The major pathway of this mechanism is conjugation of the platinum-based agent with glutathione (GSH), and neutralization through exportation of the GSH-conjugated molecules [50].
Finally, alterations in DNA repair mechanisms, mainly those involved in the NER pathway, can induce resistance to platinum-based agents. Increased NER-related gene expressions have been correlated with resistance to platinum-based drugs [51–53]. The excision repair cross-complementation group 1 factor (ERCC1) is one of the best-studied examples, being highly expressed in platinum-resistant ovarian and gastric cancers. ERCC1 forms a heterodimer complex with ERCC4, also called XPF endonuclease, forming the ERCC1-XPF enzyme complex [54]. Recently, Amable et al. reviewed a comprehensive list of studies that found correlations between the high levels of ERCC1 and cisplatin resistance [13]. These studies included data from ovarian [55], cervical [56], lung [57], liver [58], and gastric cancers [59], and indicated that high ERCC1 expression may be a promising potential biomarker for cisplatin resistance [13]. On the other hand, low ERCC1-XPF expression was found in testicular cancers, which are very responsive to platinum-based agents [14]. Additionally, ERCC1-XPF expression has been suggested as a determinant of cisplatin therapy efficacy in non-small cell lung cancer [60].
GENE MUTATIONS AND SIGNALING PATHWAYS THAT REGULATE RESPONSE TO PLATINUM-BASED AGENTS
The development of resistance to platinum-based agents and the consequential limitation of treatment choices within this type of chemotherapy necessitate the development of new therapeutic strategies and approaches. Identification of specific gene mutations, and gene expression profiles that can predict response to platinum-based agents is of paramount importance. This will require a greater understanding of specific cell signaling pathways that mediate the response to these agents. Pharmaceutical regulation of such genes and pathways could dramatically improve the effects of platinum-based agents.
Zhao et al. reported that SNPs in the BER pathway affected the prognosis of platinum-based chemotherapy in patients who had advanced non-small cell lung cancer. Patients who had specific SNPs in three major BER genes were found to have a better response to platinum-based treatment [30]. This finding can be explained by the fact that these SNPs might cause defects in BER pathway efficacy and thus decreased levels of DNA repair after DNA damaging platinum-based chemotherapy.
Reles et al. studied nearly 200 ovarian carcinomas to analyze the effect of p53 genetic alterations on the response to platinum-based chemotherapy. They found that p53 alterations were significantly correlated with resistance to platinum-based chemotherapy, early relapse, and shortened overall survival [61].
Cass et al. [43], and later Tan et al. [62] reported that ovarian cancers with germline BRCA gene mutations have higher response rates to platinum-based chemotherapy. The latter authors supported the existence of a clinical syndrome of BRCAness [62]. In addition, Konstantinopoulos et al. reported that the BRCAness profile correlated with responsiveness to platinum-based agents and PARP inhibitors in patients with ovarian cancer and also identified a subset of sporadic patients with improved outcomes [63]. This profile could potentially be expanded to many other types of cancer with similar genetic background.
Thus, specific gene mutations that occur in molecular pathways involved in the repair of DNA damage induced by platinum-based agents may provide a foundation for testing and application of specific combination therapy approaches that exploit this potential vulnerability.
COMBINATION THERAPY USING PLATINUM-BASED AGENTS AND DDR-TARGETING AGENTS
Platinum-based agents and PARP inhibitors: suppression of PARP catalytic activities and PARP trapping
DDR-targeting agents may be uniquely able to potentiate or maintain therapy responses to platinum-based agents. Multiple DDR-targeting agents are in clinical trials, with others in active development. Among the most promising agents are PARP inhibitors. These agents can not only directly lead to synthetic lethality in specific malignancies with genetic deficiencies in the HR DNA repair pathway, but may also potentiate the therapeutic effects of platinum-based agents through alterations in the transcriptional regulation of various genes and signaling activities that may interact with DNA damage induced by these chemotherapy agents.
PARP inhibitors are prototypic representatives of DDR-targeting therapy with regard to their use in combination with platinum-based agents. The important role of PARP in DDR and repair was first reported in 1979 when it was found that PARP activity is increased after administration of chemotherapy and radiation treatment [64]. PARPs compose a family of more than 18 enzymes that play fundamental roles in multiple cellular actions such as DNA replication, DNA transcription, and DNA damage repair [65]. Many of these enzymes play a catalytic role in repairing damaged DNA. PARP-1, the most studied member of the PARP family, is an important factor for repairing SSBs. SSBs are intermediates in both the BER and NER pathways, and any alteration in PARP activity could have an influence on these pathways. Flohr et al. reported that PARP activation in some cell models is one of the earliest cellular mechanisms involved in DNA repair, independently of the type of DNA lesion. Activation of PARP leads to increased activities of the BER and NER pathways in cells with a fully functional DDR/DNA repair capacity, but not in cells in which PARP is inhibited [66]. The BER complex consists mainly of DNA ligase III, DNA polymerase β, and the XRCC1 [67]. PARP binds to SSBs through its N-terminal zinc finger motifs, which activate its C-terminal domain to hydrolyze NAD+. This event produces long chains of ADP-ribose units, i.e., PARylation [68, 69] and recruits factors of the BER complex [70, 71]. It has also been reported that PARP activation after exposure to ultraviolet (UV) radiation promotes interaction between PARP1 and XPA, which is an important component of the NER pathway [72]. D’Amours et al. showed that PARP can undergo automodification in cases of DSBs and can be one of the main acceptors of ADP-riboses [73].
In experimental models in which the PARP-1 gene is deleted, it has been shown that SSBs collapse at the points of replication forks and result in DSBs [74]. DSBs trigger HR repair pathways to restore the DNA sequences that were disrupted. Farmer et al. showed that when BRCA1 or BRCA2 are defective, cells become sensitized PARP inhibition, and this event results in chromosomal instability, cell cycle arrest, and apoptosis [75]. Additionally, Bryant et al. reported that BRCA2-deficient cells are acutely sensitive to PARP inhibitors, supporting the idea that deficiency in HR, such as in BRCA2-deficient cells, results in arrest of the repair process, and collapse of the replication forks [76].
The overarching therapy rationale behind targeting PARP is that, because of high levels of DNA damage, cancer cells are highly dependent on effective DNA repair and replication, but are very often deficient in DDR pathways, such as HR (i.e., BRCAness); and this condition can be exploited by PARP inhibition, i.e., a synthetic lethality strategy. A phase I clinical trial conducted in 2005 provided preliminary clinical evidence that a synthetic lethality strategy could have efficacy in patients with BRCA-mutant tumors [77]. Importantly, the results of this study validated the original concepts and preclinical studies that had shown the potential of synthetic lethality [10]. Recent studies have also demonstrated that approximately 25% of metastatic castration-resistant prostate cancers (mCRPC) harbor genomic alterations in DDR genes [78, 79]. In heavily pre-treated patients with mCRPC, 14 of 16 (88%) with alterations in DNA-repair genes (including BRCA1/2, ATM, Fanconi’s anemia genes, and CHEK2) responded to olaparib [80]. Based on these encouraging results, the US Food and Drug Administration (FDA) has granted breakthrough therapy designation for olaparib treatment of BRCA1/2 or ATM gene mutated mCRPC in patients who have received a prior taxane-based chemotherapy and at least one newer hormonal agent (abiraterone or enzalutamide). These new developments and other positive information has led to clinical trials with PARP inhibitors as combination therapy with platinum-based chemotherapy, leading to the approval of olaparib (AZD2281, brand name Lynparza) by the FDA for the treatment of heavily pretreated advanced ovarian cancer associated with defective BRCA genes [81, 82]. Although the initiation of new clinical trials represent important developments in the field, additional experimental and clinical data are needed to determine whether combined or sequential administration of platinum-based agents and PARP inhibitors would offer better therapeutic outcomes.
Olaparib (AZD2281), veliparib (ABT-888), niraparib (MK-4827), and talazoparib (MDV3800) are currently the most widely used PARP inhibitors. All of these agents are effective in inhibiting PARP catalytic activity [83]. Until recently, it was assumed that PARP inhibitors perform their anticancer effects only via their catalytic inhibition of PARPs in a landscape of BRCA deficiency. This rationale, however, could not explain the different results in experiments using PARP inhibition or gene deletion. To explain these results, Helleday et al. and Kedar et al. proposed a model of PARP complex trapping on DNA by the PARP inhibitors [84, 85]. The trapped PARP complex on the DNA strand constitutes another potential mechanism of synthetic lethality for these therapeutic agents in patients with the BRCAness phenotype.
Talazoparib is a novel PARP inhibitor that may have important advantages over other clinically available agents of this class [86, 87]. In addition to relatively higher activity regarding PARP catalytic inhibition, talazoparib had highly superior PARP trapping activity compared with other inhibitors including olaparib [86–89]. Murai et al. showed the relevance of the trapping mechanism of PARP inhibitors to their antiproliferative/anticancer activity [89]. Furthermore, they showed that trapping potency of the existing PARP inhibitors is relatively low compared with their potency to inhibit PARP catalytic activity [89].
DNA damage that is introduced by platinum-based agents induces DNA repair pathways that require PARP-1 activity, thus establishing a basis for convergence of specific types of DNA lesions and PARP inhibition. One example is the induction of the BER pathway by platinum-based agents. Lavrik et al. showed that PARP-1 can bind intermediates of the BER pathway [90]. When PARP-1 auto-PARylates, a variety of BER pathway proteins such as XRCC1, DNA polymerase, and DNA ligase are activated and recruited to the sites of DNA damage [91]. Under conditions of PARP inhibition, PARP-1 binds at the DNA-damaged area, but its catalytic activity is limited. Therefore, BER enzyme recruitment is decreased while PARP is stabilized on the DNA strands, impeding DNA repair [89, 91].
Another potential DDR pathway induced by platinum-based agents and also regulated by PARP-1 is the NER pathway. Pines et al. reported that DDB2, an essential protein for recognition and removal of DNA lesions by NER, is regulated by PARP-1. DDB2 PARylation leads to the protein’s stabilization and further recruitment [92]. Furthermore, Maltseva et al. demonstrated that XPC-RAD23B, an essential complex of the NER pathway, is regulated by PARP-1 [93]. Induction of the NER pathway by platinum-based agents and simultaneous inhibition of PARP-1 could potentially suppress DNA repair.
Michels et al. have demonstrated synergistic interaction between cisplatin and PARP inhibitors in non-small cell lung cancer. They showed that the combined treatment of cisplatin and PARP inhibitor elicits apoptosis. [94]. Cooperative interactions between chemotherapy-mediated ssDNA breaks and PARP catalytic and trapping activities are critical determinants of cytotoxicity and tumor growth suppression.
Platinum-based agents and PARP inhibitors: alterations of PARP regulation of gene transcription
Although the role of PARP-1 in DNA damage repair and the consequent therapeutic interference with PARP in cancer therapy have been studied in detail with regard to PARP catalytic activities and PARP trapping, the role of PARP-1 in gene transcriptional regulation and exploitation of these activities for cancer therapy have not been extensively explored (Figure 1 and 3).
Fig 3.
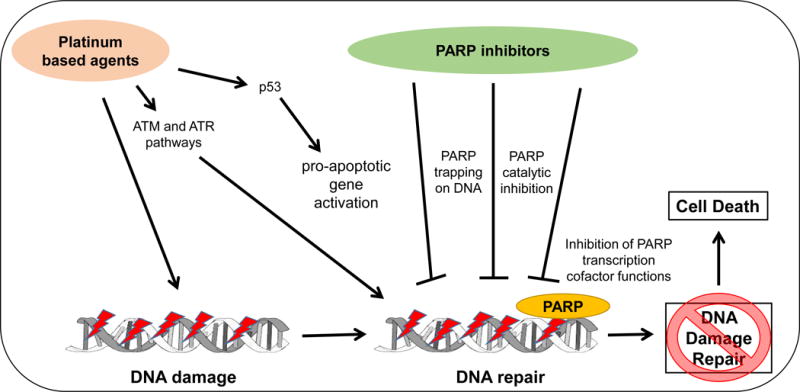
Convergence and interactions of platinum – based agents and DNA damage response – targeting agents.
Note: PARP, poly(ADP-ribose) polymerase; ATM, Ataxia telangiectasia; ATR: Ataxia telangiectasia and Rad3 related
Schreiber et al. reviewed the important role of PARP-1 in the epigenetic regulation of chromatin and gene expression. They categorized these effects of PARP-1 into two major groups: PARP-1’s modulation of chromatin structure and regulation of the transcriptional machinery. The second regulatory activity links PARP as a direct modulator of gene transcription via its interference with transcription factors and cofactors [95]. Thus far, a large number of transcription factors, such as PAX6, Sox2, Oct-1AP-2, b-Myb, NF-κB and others, have been shown to be associated with PARP activity, some with stimulatory effects and others with inhibitory effects [95, 96]. Interestingly, PARP-1 also binds to E2F-1, a transcription factor implicated in the activation of S-phase genes as well as induction of apoptosis [97]. Binding of PARP-1 to E2F-1 enhances binding to the E2F-1 promoter, regulating E2F-1–mediated transcription.
A recent study showed that PARP inhibition was associated with downregulation of specific DDR genes including Rad51, Chk1, and BRCA1 and with specific E2F-1 gene targets including EZH2 [98]. Interestingly, the same study found that combination treatment with PARP inhibition, cisplatin, and etoposide was more effective than PARP inhibition alone in vitro. These results warrant additional investigations of PARP-mediated gene expression and molecular pathways that lead to DDR.
Further, Schiewer et al. described the pro-tumorigenic effects of PARP-1 in androgen receptor (AR)-positive cancer cells in vitro and in vivo. They found that PARP-1 is a potent modulator of AR function and that PARP-1 ablation could increase the sensitivity of prostate cancer cells to androgen depletion. PARP-1 enzymatic activity was found to be required for AR transcriptional function [99].
Given that PARP has been shown to modify AR function, a critical question is the set of AR target genes that are regulated by PARP-mediated modifications of AR function. Recent publications have demonstrated that AR signaling upregulates expression of specific DDR genes [100–102] and that these genes are associated with prostate cancer metastasis [102]. These studies have established a novel mechanism of DDR regulation and potentially a novel platform for the development of biomarkers and therapeutic approaches in prostate cancer, expanding the synthetic lethality concept. Upregulation of DDR expression by AR sets the stage for consideration of specific combinations of AR signaling inhibitors and DDR-targeting therapies. In addition, these studies also suggest that PARP inhibition may be able to act in combination with platinum-based agents through alteration of DDR gene expression. Additional focus on PARP target gene activities may reveal specific pathways that, when suppressed by PARP inhibition or specific inhibitors of PARP target genes, synergize with specific mutations induced by platinum-based agents.
DDR-targeting Agents: New Weapons in the Oncological Armamentarium
Although less studied within the context of combination treatment with platinum-based agents, DDR-targeting agents other than PARP inhibitors are worth consideration. WEE-1 inhibitors are a relatively new group of agents targeting the DNA damage response pathway. WEE-1 kinase is a G2-M checkpoint kinase that catalyzes an inhibitory phosphorylation of CDK1 and CDK2 [103, 104]. When WEE-1 is inhibited, cell cycle arrest and proper DNA repair are abrogated [105]. The WEE-1 inhibitor most widely used in clinical practice is AZD1775 (MK-1775), and to date, only the results of a phase I clinical study of patients with refractory solid tumors have been reported [106]. Additional experimental and clinical studies testing AZD1775 in combination with radiation or chemotherapy will be critical for determining the clinical potential of WEE-1 inhibitors.
Ataxia Telangiectasia and Rad3-realted protein (ATR) serine/threonine kinase inhibitors, with VE-821 being the most commonly used agent, also interact with central kinases involved in the DDR pathway [107]. ATR is a large kinase activated by many different types of DNA damage, including DSBs, base adducts, and crosslinks. The DNA lesion becomes coated by the replication protein A (RPA) and the complex of RPA-DNA activates ATR-interacting protein. This event leads to ATR activation and further phosphorylation of substrates to regulate DNA repair [108]. ATR inhibitors could potentially be used in combination therapy with platinum-based agents to contribute to DNA damage generated by platinum-based agents and to impede DNA repair.
CHK1 is a critical kinase involved in halting the cell cycle when the DNA damage could potentially lead to cell death [109]. A mechanistic understanding behind combining platinum-based agents and CHK1 inhibitors is still not clear. The main concept is that cancer treatment with DNA-damaging agents, such as platinum-based agents, ultimately results in CHK1 activation, CDC25 inhibition, and cell cycle arrest. By inhibiting CHK1, CDC25 is activated, leading to an uncontrolled cell cycle and finally cell death [109]. Currently, two CHK1 inhibitors, LY2606368 and LY2603618, are being tested in phase I clinical trials [110, 111]. On the basis of these results, LY2606368 has been proposed for use in phase II trials for patients with squamous cell carcinoma [111]. Similarly, LY2603618 has been proposed for phase II studies [110].
Recently Li et al. demonstrated synergistic therapeutic approaches by pharmacological targeting of PARP and the c-Myb-TOPBP1-ATR-Chk1 pathway. Silencing of c-Myb or its downstream targets, BRCA1 or TOPBP1, showed synergistic effects with olaparib in experimental models of AR-negative prostate cancer. It has been shown that c-Myb and AR share a common set of DDR target genes [102]. Reaper et al. also reported that inhibition of ATR further sensitizes cancer cells to cisplatin [112]. Induction therapies with platinum-based agents lead to extensive DNA damage and activation of DDR pathways. Blocking or silencing of the downstream factors of these pathways with PARP and/or ATR inhibitors may potentially sensitize cancer cells to platinum-based agents and increase their effects.
CLINICAL TRIALS OF PLATINUM-BASED AGENTS AND DDR-TARGETING AGENTS
As discussed above, a relatively large number of ongoing clinical trials are assessing the clinical efficacy of DDR-targeting agents as combination therapy with platinum-based agents. To ascertain the total number of trials and their details, we searched the clinical trial database (www.clinicaltrials.gov, as of April 10, 2015), using the search terms ‘PARP,’ ‘ATR,’ ‘WEE-1,’ and ‘CHK-1.’ The results are presented in Tables 1 and 2. Table 1 shows that the most widely tested DDR-targeting agents are PARP inhibitors with 45 clinical trials. In contrast, other DDR-targeting agents are used as combined treatment with platinum-based agents in fewer clinical trials (Table 2). The only platinum-based agents that were used in all the above-mentioned clinical trials were carboplatin and cisplatin. Clinical trials with other types of alkylating agents were excluded.
Table 1.
Clinical trials of platinum-based agents and PARP inhibitors
| Clinical Trial ID | Study Name | Conditions (cancer or disease) | PARP inhibitor used | Platinum-based agent used | Other agent used | |
|---|---|---|---|---|---|---|
| 1. | NCT00516724 | Study to Assess the Safety and Tolerability of a PARP Inhibitor in Combination with Carboplatin and/or Paclitaxel | Triple negative metastatic breast cancer and advanced ovarian cancer | Olaparib | Carboplatin | Paclitaxel |
| 2. | NCT01237067 | Olaparib in Combination with Carboplatin for Refractory or Recurrent Women’s Cancers | Cervical, ovarian, fallopian tube, breast, endometrial and primary peritoneal carcinoma | Olaparib | Carboplatin | |
| 3. | NCT01445418 | AZD2281 Plus Carboplatin to Treat Breast and Ovarian Cancer | Breast and ovarian cancer | Olaparib | Carboplatin | |
| 4. | NCT01081951 | Study to Compare the Efficacy and Safety of Olaparib When Given in Combination with Carboplatin and Paclitaxel, Compared with Carboplatin and Paclitaxel in Patients with Advanced Ovarian Cancer | Ovarian cancer | Olaparib | Carboplatin | Paclitaxel |
| 5. | NCT02033551 | A Study Evaluating Veliparib as a Single Agent or in Combination with Chemotherapy in Subjects with Solid Tumors | Breast, ovarian, colon, lung and gastric cancer; solid tumors | Veliparib | Carboplatin | Paclitaxel, FOLFIRI |
| 6. | NCT01063816 | A Study of ABT-888 in Combination with Carboplatin and Gemcitabine in Subjects with Advanced Solid Tumors | Advanced solid tumors | Veliparib | Carboplatin | Gemcitabine |
| 7. | NCT01459380 | Veliparib, Pegylated Liposomal Doxorubicin Hydrochloride, Carboplatin, and Bevacizumab in Treating Patients with Recurrent Ovarian Cancer, Primary Peritoneal Cancer, or Fallopian Tube Cancer | Ovarian tumors: malignant, mixed epithelial tumor, clear cell cystadenocarcinoma, endometrioid adenocarcinoma, serous cystadenocarcinoma, recurrent carcinoma, undifferentiated carcinoma; recurrent fallopian tube and recurrent primary peritoneal carcinoma | Veliparib | Carboplatin | Bevacizumab, pegylated liposomal doxorubicin hydrochloride |
| 8. | NCT01251874 | Veliparib and Carboplatin in Treating Patients with HER2-Negative Metastatic Breast Cancer | Breast cancer: BRCA1 or BRCA2 mutation carrier, estrogen receptor negative/positive, HER2/Neu negative, male breast carcinoma, progesterone receptor negative/positive, stage IIIB/IIIC/IV breast cancer, triple-negative carcinoma | Veliparib | Carboplatin | |
| 9. | NCT01149083 | Veliparib with or without Carboplatin in Treating Patients with Stage III or Stage IV Breast Cancer | Breast cancer: BRCA1/BRCA2 mutation carrier, recurrent breast carcinoma, stage IIIB/IIIC/IV breast cancer | Veliparib | Carboplatin | |
| 10. | NCT00588991 | Veliparib and Topotecan with or without Carboplatin in Treating Patients with Relapsed or Refractory Acute Leukemia, High-Risk Myelodysplasia, or Aggressive Myeloproliferative Disorders | Adult acute leukemia – subtypes: megakaryoblastic, monoblastic, monocytic, myeloid with Inv(16)(p13.1q22), CBFB-MYH11, myeloid with maturation, myeloid with minimal differentiation, myeloid with t(16;16)(p13.1;q22), CBFB-MYH11, myeloid with t(8;21)(q22;q22), RUNX1-RUNX1T1, myeloid with t(9;11)(p22;q23), MLLT3-MLL, myeloid leukemia without maturation, myelomonocytic leukemia; adult erythroleukemia; adult pure erythroid leukemia; chronic myelomonocytic leukemia; de novo myelodysplastic syndrome; essential thrombocythemia; hematopoietic and lymphoid cell neoplasm; Philadelphia chromosome negative; BCR-ABL1 positive chronic myelogenous leukemia; polycythemia vera; previously treated myelodysplastic syndrome; recurrent adult acute lymphoblastic leukemia; recurrent adult acute myeloid leukemia; recurrent disease; secondary myelodysplastic syndrome | Veliparib | Carboplatin | Topotecan |
| 11. | NCT02483104 | Veliparib in Combination with Carboplatin and Weekly Paclitaxel in Japanese Subjects with Ovarian Cancer | Ovarian cancer | Veliparib | Carboplatin | Paclitaxel |
| 12. | NCT02106546 | Randomized, Double-Blind, Multicenter, Study Comparing Veliparib Plus Carboplatin and Paclitaxel Versus Placebo Plus Carboplatin and Paclitaxel in Previously Untreated Advanced or Metastatic Squamous Non-Small Cell Lung Cancer | Lung cancer – squamous non-small cell | Veliparib | Carboplatin | Paclitaxel |
| 13. | NCT01009190 | A Study of Poly (ADP-Ribose) Polymerase Inhibitor PF-01367338 In Combination with Several Chemotherapeutic Regimens | Advanced solid tumors | PF-1367338 | Carboplatin | |
| 14. | NCT01560104 | A Clinical Study Conducted in Multiple Centers Comparing Veliparib in Combination with Carboplatin and Paclitaxel Versus a Placebo in Combination with Carboplatin and Paclitaxel in Patients with Advanced Non-small Cell Lung Cancer | Lung cancer – non-small cell | Veliparib | Carboplatin | Paclitaxel |
| 15. | NCT01281150 | Veliparib in Combination with Carboplatin and Paclitaxel in Treating Patients with Locally Advanced or Metastatic Solid Tumors | Breast cancer: adult solid neoplasm, estrogen receptor negative/positive, HER2/Neu negative, male breast carcinoma, progesterone receptor negative, recurrent breast carcinoma, triple-negative, stage IIIB/IIIC/IV breast cancer | Veliparib | Carboplatin | Paclitaxel |
| 16. | NCT00535119 | Veliparib, Carboplatin, and Paclitaxel in Treating Patients with Advanced Solid Cancer | Adult solid neoplasm, BRCA1/BRCA2mutation carrier, hereditary breast and ovarian cancer syndrome | Veliparib | Carboplatin | Paclitaxel |
| 17. | NCT01045304 | Study of SAR240550 (BSI-201) in Combination with Gemcitabine/Carboplatin, in Patients with Metastatic Triple Negative Breast Cancer | Breast cancer – metastatic | Iniparib | Carboplatin | Gemcitabine |
| 18. | NCT00687687 | Evaluation of Paclitaxel (Taxol, NSC #673089), Carboplatin (Paraplatin, NSC #241240), and BSI-201 (NSC #746045, IND #71,677) in the Treatment of Advanced, Persistent, or Recurrent Uterine Carcinosarcoma | Uterine carcinosarcoma | BSI-201 | Carboplatin | Paclitaxel |
| 19. | NCT00540358 | A Phase 2 Trial of Standard Chemotherapy, with or without BSI-201, in Patients with Triple Negative Metastatic Breast Cancer | Breast cancer | Iniparib | Carboplatin | Gemcitabine |
| 20. | NCT00938652 | A Phase 3, Multi-Center Study of Gemcitabine/Carboplatin, with or without BSI-201, in Patients with ER-, PR-, and Her2-Negative Metastatic Breast Cancer | Breast cancer | Iniparib | Carboplatin | Gemcitabine |
| 21. | NCT01455532 | A Dose Escalation Study of Iniparib as a Single Agent and in Combination in Solid Tumors | Neoplasm malignant | Iniparib | Carboplatin | Gemcitabine |
| 22. | NCT01213381 | Safety and Pharmacokinetics of SAR240550 (BSI-201) Twice Weekly in Patients with Advanced Solid Tumors | Advance solid tumors | Iniparib | Carboplatin | Gemcitabine |
| 23. | NCT02163694 | A Phase 3 Randomized, Placebo-controlled Trial of Carboplatin and Paclitaxel with or without Veliparib (ABT-888) in HER2-negative Metastatic or Locally Advanced Unresectable BRCA-associated Breast Cancer | Breast cancer – metastatic | Veliparib | Carboplatin | Paclitaxel, placebo |
| 24. | NCT02470585 | Veliparib with Carboplatin and Paclitaxel and as Continuation Maintenance Therapy in Subjects with Newly Diagnosed Stage III or IV, High-grade Serous, Epithelial Ovarian, Fallopian Tube, or Primary Peritoneal Cancer | Ovarian cancer/neoplasm | Veliparib | Carboplatin | Paclitaxel |
| 25. | NCT02412371 | A Study Evaluating the Efficacy and Tolerability of Veliparib in Combination with Paclitaxel/Carboplatin-Based Chemoradiotherapy Followed by Veliparib and Paclitaxel/Carboplatin Consolidation in Subjects with Stage III Non-Small Cell Lung Cancer | Lung cancer – non-small cell stage III | Veliparib | Carboplatin | Paclitaxel |
| 26. | NCT01366144 | Veliparib, Paclitaxel, and Carboplatin in Treating Patients with Solid Tumors That Are Metastatic or Cannot Be Removed by Surgery and Liver or Kidney Dysfunction | Bladder, breast, unknown primary origin, endometrial, esophageal, lung, renal pelvis urothelial, ureter urothelial and urethral carcinoma; malignant head and neck neoplasm; melanoma; ovarian neoplasm; testicular lymphoma | Veliparib | Carboplatin | Paclitaxel |
| 27. | NCT02289690 | Dose Escalation and Double-blind Study of Veliparib in Combination with Carboplatin and Etoposide in Treatment-Naive Extensive Stage Disease Small Cell Lung Cancer | Lung cancer – small cell | Veliparib | Carboplatin | Etoposide |
| 28. | NCT01082549 | Trial of Gemcitabine/Carboplatin with or without Iniparib (SAR240550) (a PARP1 Inhibitor) in Subjects with Previously Untreated Stage IV Squamous Non-Small Cell Lung Cancer (NSCLC) | Lung cancer – squamous cell | Iniparib | Carboplatin | Gemcitabine |
| 29. | NCT00813956 | A Phase 2 Study of Standard Chemotherapy Plus BSI-201 (a PARP Inhibitor) in the Neoadjuvant Treatment of Triple Negative Breast Cancer | Breast cancer – triple negative | BSI-201 | Carboplatin | Gemcitabine |
| 30. | NCT01711541 | Combination Chemotherapy with or without Veliparib in Treating Patients with Stage IV Head and Neck Cancer | Human papillomavirus infection; salivary gland squamous cell carcinoma stage IVA/IVB laryngeal/hypopharyngeal/lip and oral cavity/nasal cavity and paranasal sinus squamous cell carcinoma; stage IVA/IVB major salivary gland carcinoma, tongue carcinoma | Veliparib | Carboplatin, Cisplatin | Fluorouracil, hydroxyurea, paclitaxel |
| 31. | NCT00989651 | Carboplatin, Paclitaxel, Bevacizumab, and Veliparib in Treating Patients with Newly Diagnosed Stage II-IV Ovarian Epithelial, Fallopian Tube, or Primary Peritoneal Cancer | Fallopian tube: clear cell, endometrioid, mucinous and undifferentiated adenocarcinoma; carcinosarcoma; serous neoplasm; transitional cell carcinoma; Cancer stage IIA/IIB/IIC,IIIA/IIIB/IV Ovarian: Brenner tumor; carcinosarcoma; clear cell, endometrioid, mucinous, serous and undifferentiated adenocarcinoma; mixed epithelial tumor; transitional cell carcinoma; cancer stage IIA/IIB/IIC,IIIA/IIIB/IV; primary peritoneal serous adenocarcinoma stage IIIA/IIIB/IIIC/IV primary peritoneal cancer |
Veliparib | Carboplatin, Cisplatin | Bevacizumab |
| 32. | NCT02264990 | Study Comparing Veliparib Plus Carboplatin and Paclitaxel Versus Investigator’s Choice of Standard Chemotherapy in Subjects Receiving First Cytotoxic Chemotherapy for Metastatic or Advanced Non-Squamous Non-Small Cell Lung Cancer Who Are Current or Former Smokers | Lung cancer – non-squamous non-small cell | Veliparib | Carboplatin, Cisplatin | Paclitaxel, pemetrexed |
| 33. | NCT00422682 | A Study Evaluating BSI-201 in Combination with Chemotherapeutic Regimens in Subjects with Advanced Solid Tumors | Tumors | BSI-201 | Carboplatin, Temozolomide | Topotecan, gemcitabine, paclitaxel |
| 34. | NCT01506609 | The Study Evaluating Efficacy and Tolerability Of Veliparib in Combination with Temozolomide or In Combination with Carboplatin and Paclitaxel Versus Placebo in Subjects with BRCA1 and BRCA2 Mutation and Metastatic Breast Cancer | Breast cancer – metastatic | Veliparib | Carboplatin, Temozolomide | Paclitaxel |
| 35. | NCT01074970 | PARP Inhibition for Triple Negative Breast Cancer (ER-/PR-/HER2-) with BRCA1/2 Mutations | Breast cancer | Rucaparib | Cisplatin | |
| 36. | NCT01642251 | Cisplatin and Etoposide with or without Veliparib in Treating Patients with Extensive Stage Small Cell Lung Cancer or Metastatic Large Cell Neuroendocrine Non-small Cell Lung Cancer | Carcinoma: unknown primary origin, extensive stage small cell lung, large cell lung, neuroendocrine, newly diagnosed of unknown primary origin, stage IV non-small cell lung | Veliparib | Cisplatin | Etoposide |
| 37. | NCT01281852 | Paclitaxel, Cisplatin, and Veliparib in Treating Patients with Advanced, Persistent, or Recurrent Cervical Cancer | Cervical cancer: adenocarcinoma, adenosquamous, squamous cell, recurrent, stage IVB | Veliparib | Cisplatin | Paclitaxel |
| 38. | NCT01104259 | Veliparib, Cisplatin, and Vinorelbine Ditartrate in Treating Patients with Recurrent and/or Metastatic Breast Cancer | Breast cancer: estrogen receptor-negative, HER2-negative, hereditary breast/ovarian cancer – BRCA1/BRCA2, male breast cancer, progesterone receptor-negative, recurrent, stage IV, triple-negative | Veliparib | Cisplatin | Vinorelbine tartrate |
| 39. | NCT00678132 | AZD2281 and Cisplatin Plus Gemcitabine to Treat Solid Tumor Cancers | Neoplasms | Olaparib | Cisplatin | Gemcitabine |
| 40. | NCT01086254 | SAR240550 in Combination with Gemcitabine/Cisplatin in Non-small Cell Lung Cancer | Lung cancer – non-small cell stage IV | Iniparib | Cisplatin | Gemcitabine |
| 41. | NCT01345357 | Study of CEP-9722 in Combination with Gemcitabine and Cisplatin in Patients with Advanced Solid Tumors or Mantle Cell Lymphoma | Solid tumors or mantle cell lymphoma | CEP-9722 | Cisplatin | Gemcitabine |
| 42. | NCT02723864 | Veliparib (ABT-888), an Oral PARP Inhibitor, and VX-970, an ATR Inhibitor, in Combination with Cisplatin in People with Refractory Solid Tumors | Neoplasms | Veliparib | Cisplatin | VX-970 |
| 43. | NCT02595905 | Cisplatin with or without Veliparib in Treating Patients with Stage IV Triple-Negative and/or BRCA Mutation-Associated Breast Cancer | Breast cancer: BRCA1/BRCA2 mutation carrier, estrogen receptor negative, HER2/Neu negative, progesterone receptor negative, stage IV, triple-negative | Veliparib | Cisplatin | |
| 44. | NCT02308072 | Phase I Study of Olaparib Combined with Cisplatin-based Chemoradiotherapy to Treat Locally Advanced Head and Neck Cancer | Head and neck cancer | Olaparib | Cisplatin | |
| 45. | NCT01585805 | Gemcitabine Hydrochloride and Cisplatin with or without Veliparib or Veliparib Alone in Treating Patients with Locally Advanced or Metastatic Pancreatic Cancer | Breast cancer: BRCA1/BRCA2 mutation carrier; pancreatic cancer: non-metastatic, metastatic, recurrent, stage III, stage IV | Veliparib | Cisplatin | Gemcitabine |
Source: www.clinicaltrials.gov
Table 2.
Clinical trials of platinum-based agents and ATR kinase inhibitors, WEE-1 inhibitors, and Chk1-2 inhibitors
| Clinical Trial ID | Study Name | Conditions (cancer or disease) | DDR targeting agent used | Platinum-based agent used | Other agent used | |
|---|---|---|---|---|---|---|
| Clinical Trials for ATR kinase inhibitors | ||||||
| 1. | NCT02567422 | VX-970, Cisplatin, and Radiation Therapy in Treating Patients with Locally Advanced HPV-Negative Head and Neck Squamous Cell Carcinoma | Head and neck squamous cell carcinoma; stage III/IVA/IVB/IVC nasal cavity and paranasal sinus squamous cell carcinoma; stage III/IVA/IVB/IVC oropharyngeal squamous cell carcinoma, | ATR kinase inhibitor VX-970 | Cisplatin | |
| 2. | NCT02567409 | Cisplatin and Gemcitabine Hydrochloride with or without ATR Kinase Inhibitor VX-970 in Treating Patients with Metastatic Urothelial Cancer | Urothelial carcinoma: metastatic to the renal pelvis and ureter; Stage IV bladder cancer | ATR kinase inhibitor VX-970 | Cisplatin | Gemcitabine |
| 3. | NCT02627443 | Carboplatin and Gemcitabine Hydrochloride with or without ATR Kinase Inhibitor VX-970 in Treating Patients with Recurrent and Metastatic Ovarian, Primary Peritoneal, or Fallopian Tube Cancer | Ovarian: high-grade serous adenocarcinoma, endometrioid tumor, recurrent carcinoma, stage IV cancer Fallopian tube: recurrent carcinoma, stage IV cancer Primary peritoneal carcinoma: recurrent, stage IV |
ATR kinase inhibitor VX-970 | Carboplatin | Gemcitabine |
| Clinical Trials for WEE-1 inhibitors | ||||||
| 1. | NCT01849146 | WEE1 Inhibitor AZD1775, Radiation Therapy, and Temozolomide in Treating Patients with Newly Diagnosed or Recurrent Glioblastoma Multiforme | Adult glioblastoma and recurrent adult brain neoplasm | AZD1775 | Temozolomide | |
| 2. | NCT02196168 | Cisplatin with or without WEE1 Inhibitor MK-1775 in Treating Patients with Recurrent or Metastatic Head and Neck Cancer | Recurrent oropharyngeal/hypopharyngeal/laryngeal/lip and oral cavity/nasal cavity and paranasal sinus squamous cell carcinoma; recurrent laryngeal/oral cavity verrucous carcinoma; recurrent metastatic squamous cell carcinoma in the neck with occult primary; squamous cell carcinoma metastatic in the neck with occult primary; stage IV hypopharyngeal squamous cell carcinoma; stage IVA/IVB/IVC laryngeal squamous cell carcinoma; stage IVA/IVB/IVC laryngeal verrucous carcinoma; stage IVA/IVB/IVC lip and oral cavity squamous cell carcinoma; stage IVA/IVB/IVC nasal cavity and paranasal sinus squamous cell carcinoma; stage IVA/IVB/IVC oral cavity verrucous carcinoma; stage IVA/IVB/IVC oropharyngeal squamous cell carcinoma, tongue carcinoma | AZD1775 | Cisplatin | |
| 3. | NCT01958658 | AZD1775, Cisplatin, and Radiation Therapy in Treating Patients with Cervical Cancer | Stage IB/IIA/IIB/IIIA/IIIB cervical cancer | AZD1775 | Cisplatin | |
| 4. | NCT02508246 | WEE1 Inhibitor MK-1775, Docetaxel, and Cisplatin Before Surgery in Treating Patients with Borderline Resectable Stage III-IVB Squamous Cell Carcinoma of the Head and Neck | Stage III/IVA/IVB laryngeal squamous cell carcinoma; stage III/IVA/IVB oral cavity squamous cell carcinoma; stage III/IVA/IVB oropharyngeal squamous cell carcinoma | AZD1775 | Cisplatin | Docetaxel |
| 5. | NCT01164995 | Study with Wee-1 Inhibitor MK-1775 and Carboplatin to Treat p53 Mutated Refractory and Resistant Ovarian Cancer | Epithelial ovarian cancer | AZD1775 | Carboplatin | |
| 6. | NCT02585973 | Dose-escalating AZD1775 + Concurrent Radiation + Cisplatin for Intermediate/High Risk HNSCC | Squamous cell carcinoma of head and neck | AZD1775 | Cisplatin | |
| 7. | NCT02087241 | Phase II Trial of Carboplatin and Pemetrexed with or without AZD1775 for Untreated Lung Cancer | Lung cancer – non-squamous non-small cell stage IV previously untreated | AZD1775 | Carboplatin | Pemetrexed |
| 8. | NCT02272790 | AZD1775 Plus Chemotherapy in Patients with Platinum-Resistant Ovarian, Fallopian Tube, or Primary Peritoneal Cancer. | Ovarian, fallopian tube and peritoneal cancer; P53 mutation | AZD1775 | Carboplatin | |
| 9. | NCT02341456 | Phase Ib Study AZD1775 in Combination with Carboplatin and Paclitaxel in Adult Asian Patients with Solid Tumors | Advanced solid tumors | AZD1775 | Carboplatin | Paclitaxel |
| Clinical Trials for Chk1-2 inhibitors | ||||||
| 1. | NCT01139775 | A Study in Non-Small Cell Lung Cancer | Lung cancer – non-small cell | LY2603618 | Cisplatin | Pemetrexed |
| 2. | NCT02124148 | A Study of LY2606368 with Chemotherapy or Targeted Agents in Participants with Advanced Cancer | Metastatic neoplasm; colorectal neoplasms | LY2606368 | Cisplatin | Cetuximab, pemetrexed, fluorouracil, LY3023414, leucovorin |
| 3. | NCT02555644 | A Study of LY2606368 with Chemotherapy and Radiation in Participants with Head and Neck Cancer | Head and neck neoplasms | LY2606368 | Cisplatin | Cetuximab |
Source: www.clinicaltrials.gov
SUMMARY: Therapeutic rationale for combination/maintenance therapy using ICL agent―induced crosslinking and DDR-targeting agents
The major concept highlighted in this review is that platinum-based agents can be considered as an induction therapy for DDR-targeting agents. Our concept and rationale are depicted in Figure 3. Platinum-based agents perform their antitumor effects mainly via DNA adducts. Some of these events could induce specific lesions in genes involved in DNA repair, including those of the BER, NER and HR repair pathways. Platinum-based agents could be used as mediators leading the cell to a synthetic lethality stage favorable for DDR-targeting agents. This molecular landscape could potentiate and/or maintain the initial effects of platinum chemotherapy. PARP inhibitors, in particular, have multiple mechanisms of action that support this concept. Catalytic inhibition of PARP enzymes, trapping of PARP on the DNA strand, and PARP-mediated transcriptional regulation are the relevant mechanisms through which PARP inhibitors can exert their therapeutic activities. Additionally, the role of PARP in DNA transcription is not limited only to DDR genes but is also implicated in other signaling pathways, expanding the effect of these agents at the cellular level. A greater understanding of these mechanisms may lead to more specific and productive mechanistic rationales for combination therapy approaches for advanced malignancies.
Consideration of the combination of platinum-based agents with DDR-targeting therapies should not be limited to PARP inhibitors. New inhibitors that interfere with the activities of important genes and enzymes of the DNA damage repair pathways (i.e., ATR, WEE-1, and CHK-1) could also be used for the same purposes. As discussed above, most of these agents have not been tested in large clinical trials (see Table 2), and their results have not been analyzed in detail. There remains much to learn about the potential of these DDR-targeting agents and their potential combination with platinum-based agents. In light of the promising results of PARP inhibitors used to treat ovarian cancers with a BRCAness phenotype, it is important to pursue various DDR-targeting therapies combined with platinum-based agents.
Acknowledgments
All individuals who are listed as authors have contributed substantially to the design, performance, and/or reporting of this work. We thank Michael Worley and the Department of Scientific Publications at MD Anderson Cancer Center for their editorial assistance with the manuscript. We also acknowledge the support of grants from the National Cancer Institute, NCI P50CA140388, and NCI P30CA016672.
ABBREVIATIONS AND ACRONYMS
- AR
Androgen receptor
- ATM
Ataxia telangiectasia mutated
- ATR
Ataxia telangiectasia and Rad3 related
- BER
Base excision repair
- BRCA
Breast cancer susceptibility gene
- CTR1
Copper transport protein 1
- DDR
DNA damage repair
- DSB
Double-strand breakage
- ERCC
Excision repair cross-complementation group 1 factor
- HR
Homologous recombination
- ICL
Interstrand crosslinking agents
- MM
RMismatch repair
- NER
Nucleotide excision repair
- NHEJ
Non-homologous end-joining
- PARP
Poly(ADP-ribose) polymerase
- RPA
Replication protein A
- SNP
Single nucleotide polymorphism
- SSB
Single-strand breakage
- ssDN
ASingle-stranded DNA
- TF
Transcription factor
- TOPBP1
Topoisomerase (DNA) II binding protein 1
- XRCC1/4
X-ray repair cross-complementing protein 1 & 4
Footnotes
CONFLICT OF INTEREST
The authors declare no conflicts of interest.
References
- 1.Lindahl T, Barnes DE. Repair of endogenous DNA damage. Cold Spring Harbor symposia on quantitative biology. 2000;65:127–133. doi: 10.1101/sqb.2000.65.127. [DOI] [PubMed] [Google Scholar]
- 2.Friedberg EC. Out of the shadows and into the light: the emergence of DNA repair. Trends in biochemical sciences. 1995;20(10):381. doi: 10.1016/s0968-0004(00)89082-9. [DOI] [PubMed] [Google Scholar]
- 3.Noll DM, Mason TM, Miller PS. Formation and repair of interstrand cross-links in DNA. Chemical reviews. 2006;106(2):277–301. doi: 10.1021/cr040478b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Goodman LS, Wintrobe MM, et al. Nitrogen mustard therapy; use of methyl-bis (beta-chloroethyl) amine hydrochloride and tris (beta-chloroethyl) amine hydrochloride for Hodgkin’s disease, lymphosarcoma, leukemia and certain allied and miscellaneous disorders. Journal of the American Medical Association. 1946;132:126–132. doi: 10.1001/jama.1946.02870380008004. [DOI] [PubMed] [Google Scholar]
- 5.Rosenberg B, VanCamp L, Trosko JE, Mansour VH. Platinum compounds: a new class of potent antitumour agents. Nature. 1969;222(5191):385–386. doi: 10.1038/222385a0. [DOI] [PubMed] [Google Scholar]
- 6.Kelland L. The resurgence of platinum-based cancer chemotherapy. Nat Rev Cancer. 2007;7(8):573–584. doi: 10.1038/nrc2167. [DOI] [PubMed] [Google Scholar]
- 7.Tanida S, Mizoshita T, Ozeki K, Tsukamoto H, Kamiya T, Kataoka H, Sakamuro D, Joh T. Mechanisms of Cisplatin-Induced Apoptosis and of Cisplatin Sensitivity: Potential of BIN1 to Act as a Potent Predictor of Cisplatin Sensitivity in Gastric Cancer Treatment. Int J Surg Oncol. 2012;2012:862879. doi: 10.1155/2012/862879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jamieson ER, Lippard SJ. Structure, Recognition, and Processing of Cisplatin-DNA Adducts. Chemical reviews. 1999;99(9):2467–2498. doi: 10.1021/cr980421n. [DOI] [PubMed] [Google Scholar]
- 9.Dronkert ML, Kanaar R. Repair of DNA interstrand cross-links. Mutation research. 2001;486(4):217–247. doi: 10.1016/s0921-8777(01)00092-1. [DOI] [PubMed] [Google Scholar]
- 10.Lord CJ, Tutt AN, Ashworth A. Synthetic lethality and cancer therapy: lessons learned from the development of PARP inhibitors. Annu Rev Med. 2015;66:455–470. doi: 10.1146/annurev-med-050913-022545. [DOI] [PubMed] [Google Scholar]
- 11.Dobzhansky T. Genetics of Natural Populations. Xiii. Recombination and Variability in Populations of Drosophila Pseudoobscura. Genetics. 1946;31(3):269–290. doi: 10.1093/genetics/31.3.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lord CJ, Ashworth A. BRCAness revisited. Nat Rev Cancer. 2016;16(2):110–120. doi: 10.1038/nrc.2015.21. [DOI] [PubMed] [Google Scholar]
- 13.Amable L. Cisplatin resistance and opportunities for precision medicine. Pharmacological research. 2016 doi: 10.1016/j.phrs.2016.01.001. [DOI] [PubMed] [Google Scholar]
- 14.Rabik CA, Dolan ME. Molecular mechanisms of resistance and toxicity associated with platinating agents. Cancer treatment reviews. 2007;33(1):9–23. doi: 10.1016/j.ctrv.2006.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wisnovsky SP, Wilson JJ, Radford RJ, Pereira MP, Chan MR, Laposa RR, Lippard SJ, Kelley SO. Targeting mitochondrial DNA with a platinum-based anticancer agent. Chemistry & biology. 2013;20(11):1323–1328. doi: 10.1016/j.chembiol.2013.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yang XL, Wang AH. Structural studies of atom-specific anticancer drugs acting on DNA. Pharmacology & therapeutics. 1999;83(3):181–215. doi: 10.1016/s0163-7258(99)00020-0. [DOI] [PubMed] [Google Scholar]
- 17.Fuertes MA, Alonso C, Perez JM. Biochemical modulation of Cisplatin mechanisms of action: enhancement of antitumor activity and circumvention of drug resistance. Chemical reviews. 2003;103(3):645–662. doi: 10.1021/cr020010d. [DOI] [PubMed] [Google Scholar]
- 18.Eastman A. The formation, isolation and characterization of DNA adducts produced by anticancer platinum complexes. Pharmacology & therapeutics. 1987;34(2):155–166. doi: 10.1016/0163-7258(87)90009-x. [DOI] [PubMed] [Google Scholar]
- 19.Huang Y, Li L. DNA crosslinking damage and cancer – a tale of friend and foe. Translational cancer research. 2013;2(3):144–154. doi: 10.3978/j.issn.2218-676X.2013.03.01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li X, Heyer WD. Homologous recombination in DNA repair and DNA damage tolerance. Cell research. 2008;18(1):99–113. doi: 10.1038/cr.2008.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sorenson CM, Eastman A. Influence of cis-diamminedichloroplatinum(II) on DNA synthesis and cell cycle progression in excision repair proficient and deficient Chinese hamster ovary cells. Cancer research. 1988;48(23):6703–6707. [PubMed] [Google Scholar]
- 22.Osborn MF, White JD, Haley MM, DeRose VJ. Platinum-RNA modifications following drug treatment in S. cerevisiae identified by click chemistry and enzymatic mapping. ACS chemical biology. 2014;9(10):2404–2411. doi: 10.1021/cb500395z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Karasawa T, Sibrian-Vazquez M, Strongin RM, Steyger PS. Identification of cisplatin-binding proteins using agarose conjugates of platinum compounds. PloS one. 2013;8(6):e66220. doi: 10.1371/journal.pone.0066220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hoeijmakers JH. Genome maintenance mechanisms for preventing cancer. Nature. 2001;411(6835):366–374. doi: 10.1038/35077232. [DOI] [PubMed] [Google Scholar]
- 25.Rosell R, Mendez P, Isla D, Taron M. Platinum resistance related to a functional NER pathway. J Thorac Oncol. 2007;2(12):1063–1066. doi: 10.1097/JTO.0b013e31815ba2a1. [DOI] [PubMed] [Google Scholar]
- 26.de Laat WL, Jaspers NG, Hoeijmakers JH. Molecular mechanism of nucleotide excision repair. Genes Dev. 1999;13(7):768–785. doi: 10.1101/gad.13.7.768. [DOI] [PubMed] [Google Scholar]
- 27.Marteijn JA, Lans H, Vermeulen W, Hoeijmakers JH. Understanding nucleotide excision repair and its roles in cancer and ageing. Nat Rev Mol Cell Biol. 2014;15(7):465–481. doi: 10.1038/nrm3822. [DOI] [PubMed] [Google Scholar]
- 28.Sugasawa K, Okamoto T, Shimizu Y, Masutani C, Iwai S, Hanaoka F. A multistep damage recognition mechanism for global genomic nucleotide excision repair. Genes Dev. 2001;15(5):507–521. doi: 10.1101/gad.866301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Earley JN, Turchi JJ. Interrogation of nucleotide excision repair capacity: impact on platinum-based cancer therapy. Antioxid Redox Signal. 2011;14(12):2465–2477. doi: 10.1089/ars.2010.3369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhao W, Hu L, Xu J, Shen H, Hu Z, Ma H, Shu Y, Shao Y, Yin Y. Polymorphisms in the base excision repair pathway modulate prognosis of platinum-based chemotherapy in advanced non-small cell lung cancer. Cancer Chemother Pharmacol. 2013;71(5):1287–1295. doi: 10.1007/s00280-013-2127-8. [DOI] [PubMed] [Google Scholar]
- 31.Aebi S, Kurdi-Haidar B, Gordon R, Cenni B, Zheng H, Fink D, Christen RD, Boland CR, Koi M, Fishel R, Howell SB. Loss of DNA mismatch repair in acquired resistance to cisplatin. Cancer research. 1996;56(13):3087–3090. [PubMed] [Google Scholar]
- 32.Yang W. Structure and mechanism for DNA lesion recognition. Cell research. 2008;18(1):184–197. doi: 10.1038/cr.2007.116. [DOI] [PubMed] [Google Scholar]
- 33.Duckett DR, Drummond JT, Murchie AI, Reardon JT, Sancar A, Lilley DM, Modrich P. Human MutSalpha recognizes damaged DNA base pairs containing O6-methylguanine, O4-methylthymine, or the cisplatin-d(GpG) adduct. Proceedings of the National Academy of Sciences of the United States of America. 1996;93(13):6443–6447. doi: 10.1073/pnas.93.13.6443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Peltomaki P. Role of DNA mismatch repair defects in the pathogenesis of human cancer. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2003;21(6):1174–1179. doi: 10.1200/JCO.2003.04.060. [DOI] [PubMed] [Google Scholar]
- 35.Couch FJ, Nathanson KL, Offit K. Two decades after BRCA: setting paradigms in personalized cancer care and prevention. Science. 2014;343(6178):1466–1470. doi: 10.1126/science.1251827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.D’Andrea AD, Grompe M. The Fanconi anaemia/BRCA pathway. Nat Rev Cancer. 2003;3(1):23–34. doi: 10.1038/nrc970. [DOI] [PubMed] [Google Scholar]
- 37.Prakash R, Zhang Y, Feng W, Jasin M. Homologous recombination and human health: the roles of BRCA1, BRCA2, and associated proteins. Cold Spring Harbor perspectives in biology. 2015;7(4):a016600. doi: 10.1101/cshperspect.a016600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Haber JE. Partners and pathwaysrepairing a double-strand break. Trends Genet. 2000;16(6):259–264. doi: 10.1016/s0168-9525(00)02022-9. [DOI] [PubMed] [Google Scholar]
- 39.Johnson RD, Jasin M. Double-strand-break-induced homologous recombination in mammalian cells. Biochem Soc Trans. 2001;29(Pt 2):196–201. doi: 10.1042/0300-5127:0290196. [DOI] [PubMed] [Google Scholar]
- 40.Sung P, Klein H. Mechanism of homologous recombination: mediators and helicases take on regulatory functions. Nat Rev Mol Cell Biol. 2006;7(10):739–750. doi: 10.1038/nrm2008. [DOI] [PubMed] [Google Scholar]
- 41.Mimitou EP, Symington LS. Nucleases and helicases take center stage in homologous recombination. Trends in biochemical sciences. 2009;34(5):264–272. doi: 10.1016/j.tibs.2009.01.010. [DOI] [PubMed] [Google Scholar]
- 42.Moynahan ME, Jasin M. Mitotic homologous recombination maintains genomic stability and suppresses tumorigenesis. Nat Rev Mol Cell Biol. 2010;11(3):196–207. doi: 10.1038/nrm2851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cass I, Baldwin RL, Varkey T, Moslehi R, Narod SA, Karlan BY. Improved survival in women with BRCA-associated ovarian carcinoma. Cancer. 2003;97(9):2187–2195. doi: 10.1002/cncr.11310. [DOI] [PubMed] [Google Scholar]
- 44.Cheng HH, Pritchard CC, Boyd T, Nelson PS, Montgomery B. Biallelic Inactivation of BRCA2 in Platinum-sensitive Metastatic Castration-resistant Prostate Cancer. Eur Urol. 2016;69(6):992–995. doi: 10.1016/j.eururo.2015.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Deans AJ, West SC. DNA interstrand crosslink repair and cancer. Nat Rev Cancer. 2011;11(7):467–480. doi: 10.1038/nrc3088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Peng G, Lin SY. Exploiting the homologous recombination DNA repair network for targeted cancer therapy. World J Clin Oncol. 2011;2(2):73–79. doi: 10.5306/wjco.v2.i2.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ishida S, McCormick F, Smith-McCune K, Hanahan D. Enhancing tumor-specific uptake of the anticancer drug cisplatin with a copper chelator. Cancer Cell. 2010;17(6):574–583. doi: 10.1016/j.ccr.2010.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liang ZD, Long Y, Tsai WB, Fu S, Kurzrock R, Gagea-Iurascu M, Zhang F, Chen HH, Hennessy BT, Mills GB, Savaraj N, Kuo MT. Mechanistic basis for overcoming platinum resistance using copper chelating agents. Mol Cancer Ther. 2012;11(11):2483–2494. doi: 10.1158/1535-7163.MCT-12-0580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kim ES, Lee JJ, He G, Chow CW, Fujimoto J, Kalhor N, Swisher SG, Wistuba II, Stewart DJ, Siddik ZH. Tissue platinum concentration and tumor response in non-small-cell lung cancer. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2012;30(27):3345–3352. doi: 10.1200/JCO.2011.40.8120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jansen BA, Brouwer J, Reedijk J. Glutathione induces cellular resistance against cationic dinuclear platinum anticancer drugs. J Inorg Biochem. 2002;89(3–4):197–202. doi: 10.1016/s0162-0134(02)00381-1. [DOI] [PubMed] [Google Scholar]
- 51.Dabholkar M, Vionnet J, Bostick-Bruton F, Yu JJ, Reed E. Messenger RNA levels of XPAC and ERCC1 in ovarian cancer tissue correlate with response to platinum-based chemotherapy. The Journal of clinical investigation. 1994;94(2):703–708. doi: 10.1172/JCI117388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li Q, Yu JJ, Mu C, Yunmbam MK, Slavsky D, Cross CL, Bostick-Bruton F, Reed E. Association between the level of ERCC-1 expression and the repair of cisplatin-induced DNA damage in human ovarian cancer cells. Anticancer research. 2000;20(2A):645–652. [PubMed] [Google Scholar]
- 53.Costa RM, Chigancas V, Galhardo Rda S, Carvalho H, Menck CF. The eukaryotic nucleotide excision repair pathway. Biochimie. 2003;85(11):1083–1099. doi: 10.1016/j.biochi.2003.10.017. [DOI] [PubMed] [Google Scholar]
- 54.Kirschner K, Melton DW. Multiple roles of the ERCC1-XPF endonuclease in DNA repair and resistance to anticancer drugs. Anticancer research. 2010;30(9):3223–3232. [PubMed] [Google Scholar]
- 55.Deloia JA, Bhagwat NR, Darcy KM, Strange M, Tian C, Nuttall K, Krivak TC, Niedernhofer LJ. Comparison of ERCC1/XPF genetic variation, mRNA and protein levels in women with advanced stage ovarian cancer treated with intraperitoneal platinum. Gynecologic oncology. 2012;126(3):448–454. doi: 10.1016/j.ygyno.2012.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bai ZL, Wang YY, Zhe H, He JL, Hai P. ERCC1 mRNA levels can predict the response to cisplatin-based concurrent chemoradiotherapy of locally advanced cervical squamous cell carcinoma. Radiation oncology. 2012;7:221. doi: 10.1186/1748-717X-7-221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tiseo M, Bordi P, Bortesi B, Boni L, Boni C, Baldini E, Grossi F, Recchia F, Zanelli F, Fontanini G, Naldi N, Campanini N, Azzoni C, Bordi C, Ardizzoni A;, Bio, F.t.g. ERCC1/BRCA1 expression and gene polymorphisms as prognostic and predictive factors in advanced NSCLC treated with or without cisplatin. British journal of cancer. 2013;108(8):1695–1703. doi: 10.1038/bjc.2013.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ueda S, Shirabe K, Morita K, Umeda K, Kayashima H, Uchiyama H, Soejima Y, Taketomi A, Maehara Y. Evaluation of ERCC1 expression for cisplatin sensitivity in human hepatocellular carcinoma. Annals of surgical oncology. 2011;18(4):1204–1211. doi: 10.1245/s10434-010-1414-4. [DOI] [PubMed] [Google Scholar]
- 59.Metzger R, Leichman CG, Danenberg KD, Danenberg PV, Lenz HJ, Hayashi K, Groshen S, Salonga D, Cohen H, Laine L, Crookes P, Silberman H, Baranda J, Konda B, Leichman L. ERCC1 mRNA levels complement thymidylate synthase mRNA levels in predicting response and survival for gastric cancer patients receiving combination cisplatin and fluorouracil chemotherapy. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 1998;16(1):309–316. doi: 10.1200/JCO.1998.16.1.309. [DOI] [PubMed] [Google Scholar]
- 60.Cai Y, Yan X, Zhang G, Zhao W, Jiao S. The predictive value of ERCC1 and p53 for the effect of panobinostat and cisplatin combination treatment in NSCLC. Oncotarget. 2015;6(22):18997–19005. doi: 10.18632/oncotarget.3620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Reles A, Wen WH, Schmider A, Gee C, Runnebaum IB, Kilian U, Jones LA, El-Naggar A, Minguillon C, Schonborn I, Reich O, Kreienberg R, Lichtenegger W, Press MF. Correlation of p53 mutations with resistance to platinum-based chemotherapy and shortened survival in ovarian cancer. Clin Cancer Res. 2001;7(10):2984–2997. [PubMed] [Google Scholar]
- 62.Tan DS, Rothermundt C, Thomas K, Bancroft E, Eeles R, Shanley S, Ardern-Jones A, Norman A, Kaye SB, Gore ME. “BRCAness” syndrome in ovarian cancer: a case-control study describing the clinical features and outcome of patients with epithelial ovarian cancer associated with BRCA1 and BRCA2 mutations. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2008;26(34):5530–5536. doi: 10.1200/JCO.2008.16.1703. [DOI] [PubMed] [Google Scholar]
- 63.Konstantinopoulos PA, Spentzos D, Karlan BY, Taniguchi T, Fountzilas E, Francoeur N, Levine DA, Cannistra SA. Gene expression profile of BRCAness that correlates with responsiveness to chemotherapy and with outcome in patients with epithelial ovarian cancer. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2010;28(22):3555–3561. doi: 10.1200/JCO.2009.27.5719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Skidmore CJ, Davies MI, Goodwin PM, Halldorsson H, Lewis PJ, Shall S, Zia’ee AA. The involvement of poly(ADP-ribose) polymerase in the degradation of NAD caused by gamma-radiation and N-methyl-N-nitrosourea. Eur J Biochem. 1979;101(1):135–142. doi: 10.1111/j.1432-1033.1979.tb04225.x. [DOI] [PubMed] [Google Scholar]
- 65.Morales J, Li L, Fattah FJ, Dong Y, Bey EA, Patel M, Gao J, Boothman DA. Review of poly (ADP-ribose) polymerase (PARP) mechanisms of action and rationale for targeting in cancer and other diseases. Crit Rev Eukaryot Gene Expr. 2014;24(1):15–28. doi: 10.1615/critreveukaryotgeneexpr.2013006875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Flohr C, Burkle A, Radicella JP, Epe B. Poly(ADP-ribosyl)ation accelerates DNA repair in a pathway dependent on Cockayne syndrome B protein. Nucleic Acids Res. 2003;31(18):5332–5337. doi: 10.1093/nar/gkg715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Caldecott KW, Aoufouchi S, Johnson P, Shall S. XRCC1 polypeptide interacts with DNA polymerase beta and possibly poly (ADP-ribose) polymerase, and DNA ligase III is a novel molecular ’nick-sensor’ in vitro. Nucleic Acids Res. 1996;24(22):4387–4394. doi: 10.1093/nar/24.22.4387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Krishnakumar R, Kraus WL. The PARP side of the nucleus: molecular actions, physiological outcomes, and clinical targets. Molecular cell. 2010;39(1):8–24. doi: 10.1016/j.molcel.2010.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Rouleau M, Patel A, Hendzel MJ, Kaufmann SH, Poirier GG. PARP inhibition: PARP1 and beyond. Nat Rev Cancer. 2010;10(4):293–301. doi: 10.1038/nrc2812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ame JC, Spenlehauer C, de Murcia G. The PARP superfamily. BioEssays : news and reviews in molecular, cellular and developmental biology. 2004;26(8):882–893. doi: 10.1002/bies.20085. [DOI] [PubMed] [Google Scholar]
- 71.Malanga M, Althaus FR. The role of poly(ADP-ribose) in the DNA damage signaling network. Biochemistry and cell biology = Biochimie et biologie cellulaire. 2005;83(3):354–364. doi: 10.1139/o05-038. [DOI] [PubMed] [Google Scholar]
- 72.King BS, Cooper KL, Liu KJ, Hudson LG. Poly(ADP-ribose) contributes to an association between poly(ADP-ribose) polymerase-1 and xeroderma pigmentosum complementation group A in nucleotide excision repair. J Biol Chem. 2012;287(47):39824–39833. doi: 10.1074/jbc.M112.393504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.D’Amours D, Desnoyers S, D’Silva I, Poirier GG. Poly(ADP-ribosyl)ation reactions in the regulation of nuclear functions. Biochem J. 1999;342(Pt 2):249–268. [PMC free article] [PubMed] [Google Scholar]
- 74.Shall S, de Murcia G. Poly(ADP-ribose) polymerase-1: what have we learned from the deficient mouse model? Mutation research. 2000;460(1):1–15. doi: 10.1016/s0921-8777(00)00016-1. [DOI] [PubMed] [Google Scholar]
- 75.Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, Santarosa M, Dillon KJ, Hickson I, Knights C, Martin NM, Jackson SP, Smith GC, Ashworth A. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434(7035):917–921. doi: 10.1038/nature03445. [DOI] [PubMed] [Google Scholar]
- 76.Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, Kyle S, Meuth M, Curtin NJ, Helleday T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005;434(7035):913–917. doi: 10.1038/nature03443. [DOI] [PubMed] [Google Scholar]
- 77.Fong PC, Boss DS, Yap TA, Tutt A, Wu P, Mergui-Roelvink M, Mortimer P, Swaisland H, Lau A, O’Connor MJ, Ashworth A, Carmichael J, Kaye SB, Schellens JH, de Bono JS. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med. 2009;361(2):123–134. doi: 10.1056/NEJMoa0900212. [DOI] [PubMed] [Google Scholar]
- 78.Grasso CS, Wu YM, Robinson DR, Cao X, Dhanasekaran SM, Khan AP, Quist MJ, Jing X, Lonigro RJ, Brenner JC, Asangani IA, Ateeq B, Chun SY, Siddiqui J, Sam L, Anstett M, Mehra R, Prensner JR, Palanisamy N, Ryslik GA, Vandin F, Raphael BJ, Kunju LP, Rhodes DR, Pienta KJ, Chinnaiyan AM, Tomlins SA. The mutational landscape of lethal castration-resistant prostate cancer. Nature. 2012;487(7406):239–243. doi: 10.1038/nature11125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Robinson D, Van Allen EM, Wu YM, Schultz N, Lonigro RJ, Mosquera JM, Montgomery B, Taplin ME, Pritchard CC, Attard G, Beltran H, Abida W, Bradley RK, Vinson J, Cao X, Vats P, Kunju LP, Hussain M, Feng FY, Tomlins SA, Cooney KA, Smith DC, Brennan C, Siddiqui J, Mehra R, Chen Y, Rathkopf DE, Morris MJ, Solomon SB, Durack JC, Reuter VE, Gopalan A, Gao J, Loda M, Lis RT, Bowden M, Balk SP, Gaviola G, Sougnez C, Gupta M, Yu EY, Mostaghel EA, Cheng HH, Mulcahy H, True LD, Plymate SR, Dvinge H, Ferraldeschi R, Flohr P, Miranda S, Zafeiriou Z, Tunariu N, Mateo J, Perez-Lopez R, Demichelis F, Robinson BD, Schiffman M, Nanus DM, Tagawa ST, Sigaras A, Eng KW, Elemento O, Sboner A, Heath EI, Scher HI, Pienta KJ, Kantoff P, de Bono JS, Rubin MA, Nelson PS, Garraway LA, Sawyers CL, Chinnaiyan AM. Integrative clinical genomics of advanced prostate cancer. Cell. 2015;161(5):1215–1228. doi: 10.1016/j.cell.2015.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Mateo J, Carreira S, Sandhu S, Miranda S, Mossop H, Perez-Lopez R, Nava Rodrigues D, Robinson D, Omlin A, Tunariu N, Boysen G, Porta N, Flohr P, Gillman A, Figueiredo I, Paulding C, Seed G, Jain S, Ralph C, Protheroe A, Hussain S, Jones R, Elliott T, McGovern U, Bianchini D, Goodall J, Zafeiriou Z, Williamson CT, Ferraldeschi R, Riisnaes R, Ebbs B, Fowler G, Roda D, Yuan W, Wu YM, Cao X, Brough R, Pemberton H, A’Hern R, Swain A, Kunju LP, Eeles R, Attard G, Lord CJ, Ashworth A, Rubin MA, Knudsen KE, Feng FY, Chinnaiyan AM, Hall E, de Bono JS. DNA-Repair Defects and Olaparib in Metastatic Prostate Cancer. N Engl J Med. 2015;373(18):1697–1708. doi: 10.1056/NEJMoa1506859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kim G, Ison G, McKee AE, Zhang H, Tang S, Gwise T, Sridhara R, Lee E, Tzou A, Philip R, Chiu HJ, Ricks TK, Palmby T, Russell AM, Ladouceur G, Pfuma E, Li H, Zhao L, Liu Q, Venugopal R, Ibrahim A, Pazdur R. FDA Approval Summary: Olaparib Monotherapy in Patients with Deleterious Germline BRCA-Mutated Advanced Ovarian Cancer Treated with Three or More Lines of Chemotherapy. Clin Cancer Res. 2015;21(19):4257–4261. doi: 10.1158/1078-0432.CCR-15-0887. [DOI] [PubMed] [Google Scholar]
- 82.Kaufman B, Shapira-Frommer R, Schmutzler RK, Audeh MW, Friedlander M, Balmana J, Mitchell G, Fried G, Stemmer SM, Hubert A, Rosengarten O, Steiner M, Loman N, Bowen K, Fielding A, Domchek SM. Olaparib monotherapy in patients with advanced cancer and a germline BRCA1/2 mutation. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2015;33(3):244–250. doi: 10.1200/JCO.2014.56.2728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kummar S, Chen A, Parchment RE, Kinders RJ, Ji J, Tomaszewski JE, Doroshow JH. Advances in using PARP inhibitors to treat cancer. BMC medicine. 2012;10:25. doi: 10.1186/1741-7015-10-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kedar PS, Stefanick DF, Horton JK, Wilson SH. Increased PARP-1 association with DNA in alkylation damaged, PARP-inhibited mouse fibroblasts. Molecular cancer research : MCR. 2012;10(3):360–368. doi: 10.1158/1541-7786.MCR-11-0477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Helleday T. The underlying mechanism for the PARP and BRCA synthetic lethality: clearing up the misunderstandings. Molecular oncology. 2011;5(4):387–393. doi: 10.1016/j.molonc.2011.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Shen Y, Rehman FL, Feng Y, Boshuizen J, Bajrami I, Elliott R, Wang B, Lord CJ, Post LE, Ashworth A. BMN 673, a novel and highly potent PARP1/2 inhibitor for the treatment of human cancers with DNA repair deficiency. Clin Cancer Res. 2013;19(18):5003–5015. doi: 10.1158/1078-0432.CCR-13-1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Murai J, Zhang Y, Morris J, Ji J, Takeda S, Doroshow JH, Pommier Y. Rationale for poly(ADP-ribose) polymerase (PARP) inhibitors in combination therapy with camptothecins or temozolomide based on PARP trapping versus catalytic inhibition. The Journal of pharmacology and experimental therapeutics. 2014;349(3):408–416. doi: 10.1124/jpet.113.210146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Murai J, Huang SY, Renaud A, Zhang Y, Ji J, Takeda S, Morris J, Teicher B, Doroshow JH, Pommier Y. Stereospecific PARP trapping by BMN 673 and comparison with olaparib and rucaparib. Mol Cancer Ther. 2014;13(2):433–443. doi: 10.1158/1535-7163.MCT-13-0803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Murai J, Huang SY, Das BB, Renaud A, Zhang Y, Doroshow JH, Ji J, Takeda S, Pommier Y. Trapping of PARP1 and PARP2 by Clinical PARP Inhibitors. Cancer research. 2012;72(21):5588–5599. doi: 10.1158/0008-5472.CAN-12-2753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lavrik OI, Prasad R, Sobol RW, Horton JK, Ackerman EJ, Wilson SH. Photoaffinity labeling of mouse fibroblast enzymes by a base excision repair intermediate. Evidence for the role of poly(ADP-ribose) polymerase-1 in DNA repair. J Biol Chem. 2001;276(27):25541–25548. doi: 10.1074/jbc.M102125200. [DOI] [PubMed] [Google Scholar]
- 91.Horton JK, Stefanick DF, Prasad R, Gassman NR, Kedar PS, Wilson SH. Base excision repair defects invoke hypersensitivity to PARP inhibition. Molecular cancer research : MCR. 2014;12(8):1128–1139. doi: 10.1158/1541-7786.MCR-13-0502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Pines A, Vrouwe MG, Marteijn JA, Typas D, Luijsterburg MS, Cansoy M, Hensbergen P, Deelder A, de Groot A, Matsumoto S, Sugasawa K, Thoma N, Vermeulen W, Vrieling H, Mullenders L. PARP1 promotes nucleotide excision repair through DDB2 stabilization and recruitment of ALC1. J Cell Biol. 2012;199(2):235–249. doi: 10.1083/jcb.201112132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Maltseva EA, Rechkunova NI, Sukhanova MV, Lavrik OI. Poly(ADP-ribose) Polymerase 1 Modulates Interaction of the Nucleotide Excision Repair Factor XPC-RAD23B with DNA via Poly(ADP-ribosyl)ation. J Biol Chem. 2015;290(36):21811–21820. doi: 10.1074/jbc.M115.646638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Michels J, Vitale I, Senovilla L, Enot DP, Garcia P, Lissa D, Olaussen KA, Brenner C, Soria JC, Castedo M, Kroemer G. Synergistic interaction between cisplatin and PARP inhibitors in non-small cell lung cancer. Cell Cycle. 2013;12(6):877–883. doi: 10.4161/cc.24034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Schreiber V, Dantzer F, Ame JC, de Murcia G. Poly(ADP-ribose): novel functions for an old molecule. Nat Rev Mol Cell Biol. 2006;7(7):517–528. doi: 10.1038/nrm1963. [DOI] [PubMed] [Google Scholar]
- 96.Lupo B, Trusolino L. Inhibition of poly(ADP-ribosyl)ation in cancer: old and new paradigms revisited. Biochim Biophys Acta. 2014;1846(1):201–215. doi: 10.1016/j.bbcan.2014.07.004. [DOI] [PubMed] [Google Scholar]
- 97.Simbulan-Rosenthal CM, Rosenthal DS, Luo R, Samara R, Espinoza LA, Hassa PO, Hottiger MO, Smulson ME. PARP-1 binds E2F-1 independently of its DNA binding and catalytic domains, and acts as a novel coactivator of E2F-1-mediated transcription during re-entry of quiescent cells into S phase. Oncogene. 2003;22(52):8460–8471. doi: 10.1038/sj.onc.1206897. [DOI] [PubMed] [Google Scholar]
- 98.Byers LA, Wang J, Nilsson MB, Fujimoto J, Saintigny P, Yordy J, Giri U, Peyton M, Fan YH, Diao L, Masrorpour F, Shen L, Liu W, Duchemann B, Tumula P, Bhardwaj V, Welsh J, Weber S, Glisson BS, Kalhor N, Wistuba II, Girard L, Lippman SM, Mills GB, Coombes KR, Weinstein JN, Minna JD, Heymach JV. Proteomic profiling identifies dysregulated pathways in small cell lung cancer and novel therapeutic targets including PARP1. Cancer Discov. 2012;2(9):798–811. doi: 10.1158/2159-8290.CD-12-0112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Schiewer MJ, Goodwin JF, Han S, Brenner JC, Augello MA, Dean JL, Liu F, Planck JL, Ravindranathan P, Chinnaiyan AM, McCue P, Gomella LG, Raj GV, Dicker AP, Brody JR, Pascal JM, Centenera MM, Butler LM, Tilley WD, Feng FY, Knudsen KE. Dual roles of PARP-1 promote cancer growth and progression. Cancer Discov. 2012;2(12):1134–1149. doi: 10.1158/2159-8290.CD-12-0120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Goodwin JF, Schiewer MJ, Dean JL, Schrecengost RS, de Leeuw R, Han S, Ma T, Den RB, Dicker AP, Feng FY, Knudsen KE. A hormone-DNA repair circuit governs the response to genotoxic insult. Cancer Discov. 2013;3(11):1254–1271. doi: 10.1158/2159-8290.CD-13-0108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Polkinghorn WR, Parker JS, Lee MX, Kass EM, Spratt DE, Iaquinta PJ, Arora VK, Yen WF, Cai L, Zheng D, Carver BS, Chen Y, Watson PA, Shah NP, Fujisawa S, Goglia AG, Gopalan A, Hieronymus H, Wongvipat J, Scardino PT, Zelefsky MJ, Jasin M, Chaudhuri J, Powell SN, Sawyers CL. Androgen receptor signaling regulates DNA repair in prostate cancers. Cancer Discov. 2013;3(11):1245–1253. doi: 10.1158/2159-8290.CD-13-0172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Li L, Chang W, Yang G, Ren C, Park S, Karantanos T, Karanika S, Wang J, Yin J, Shah PK, Takahiro H, Dobashi M, Zhang W, Efstathiou E, Maity SN, Aparicio AM, Li Ning Tapia EM, Troncoso P, Broom B, Xiao L, Lee HS, Lee JS, Corn PG, Navone N, Thompson TC. Targeting poly(ADP-ribose) polymerase and the c-Myb-regulated DNA damage response pathway in castration-resistant prostate cancer. Sci Signal. 2014;7(326):ra47. doi: 10.1126/scisignal.2005070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Watanabe N, Broome M, Hunter T. Regulation of the human WEE1Hu CDK tyrosine 15-kinase during the cell cycle. EMBO J. 1995;14(9):1878–1891. doi: 10.1002/j.1460-2075.1995.tb07180.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Russell P, Nurse P. Negative regulation of mitosis by wee1+, a gene encoding a protein kinase homolog. Cell. 1987;49(4):559–567. doi: 10.1016/0092-8674(87)90458-2. [DOI] [PubMed] [Google Scholar]
- 105.Guertin AD, Li J, Liu Y, Hurd MS, Schuller AG, Long B, Hirsch HA, Feldman I, Benita Y, Toniatti C, Zawel L, Fawell SE, Gilliland DG, Shumway SD. Preclinical evaluation of the WEE1 inhibitor MK-1775 as single-agent anticancer therapy. Mol Cancer Ther. 2013;12(8):1442–1452. doi: 10.1158/1535-7163.MCT-13-0025. [DOI] [PubMed] [Google Scholar]
- 106.Do K, Wilsker D, Ji J, Zlott J, Freshwater T, Kinders RJ, Collins J, Chen AP, Doroshow JH, Kummar S. Phase I Study of Single-Agent AZD1775 (MK-1775), a Wee1 Kinase Inhibitor, in Patients With Refractory Solid Tumors. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2015;33(30):3409–3415. doi: 10.1200/JCO.2014.60.4009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Weber AM, Ryan AJ. ATM and ATR as therapeutic targets in cancer. Pharmacology & therapeutics. 2015;149:124–138. doi: 10.1016/j.pharmthera.2014.12.001. [DOI] [PubMed] [Google Scholar]
- 108.Cimprich KA, Cortez D. ATR: an essential regulator of genome integrity. Nat Rev Mol Cell Biol. 2008;9(8):616–627. doi: 10.1038/nrm2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Thompson R, Eastman A. The cancer therapeutic potential of Chk1 inhibitors: how mechanistic studies impact on clinical trial design. Br J Clin Pharmacol. 2013;76(3):358–369. doi: 10.1111/bcp.12139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Calvo E, Chen VJ, Marshall M, Ohnmacht U, Hynes SM, Kumm E, Diaz HB, Barnard D, Merzoug FF, Huber L, Kays L, Iversen P, Calles A, Voss B, Lin AB, Dickgreber N, Wehler T, Sebastian M. Preclinical analyses and phase I evaluation of LY2603618 administered in combination with pemetrexed and cisplatin in patients with advanced cancer. Invest New Drugs. 2014;32(5):955–968. doi: 10.1007/s10637-014-0114-5. [DOI] [PubMed] [Google Scholar]
- 111.Hong D, Infante J, Janku F, Jones S, Nguyen LM, Burris H, Naing A, Bauer TM, Piha-Paul S, Johnson FM, Kurzrock R, Golden L, Hynes S, Lin J, Lin AB, Bendell J. Phase I Study of LY2606368, a Checkpoint Kinase 1 Inhibitor, in Patients With Advanced Cancer. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2016 doi: 10.1200/JCO.2015.64.5788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Reaper PM, Griffiths MR, Long JM, Charrier JD, Maccormick S, Charlton PA, Golec JM, Pollard JR. Selective killing of ATM- or p53-deficient cancer cells through inhibition of ATR. Nat Chem Biol. 2011;7(7):428–430. doi: 10.1038/nchembio.573. [DOI] [PubMed] [Google Scholar]