

Abstract
Pyruvate carboxylase (PC) is a biotin-containing mitochondrial enzyme that catalyzes the conversion of pyruvate to oxaloacetate, thereby being involved in gluconeogenesis and in energy production through replenishment of the tricarboxylic acid (TCA) cycle with oxaloacetate. PC deficiency is a very rare metabolic disorder. We report on a new patient affected by the moderate form (the American type A). Diagnosis was nearly fortuitous, resulting from the revision of an initial diagnosis of mitochondrial complex IV (C IV) defect. The patient presented with severe lactic acidosis and pronounced ketonuria, associated with lethargy at age 23 months. Intellectual disability was noted at this time. Amino acids in plasma and organic acids in urine did not show patterns of interest for the diagnostic work-up. In skin fibroblasts PC showed no detectable activity whereas biotinidase activity was normal. We had previously reported another patient with the severe form of PC deficiency and we show that she also had secondary C IV deficiency in fibroblasts. Different anaplerotic treatments in vivo and in vitro were tested using fibroblasts of both patients with 2 different types of PC deficiency, type A (patient 1) and type B (patient 2). Neither clinical nor biological effects in vivo and in vitro were observed using citrate, aspartate, oxoglutarate and bezafibrate. In conclusion, this case report suggests that the moderate form of PC deficiency may be underdiagnosed and illustrates the challenges raised by energetic disorders in terms of diagnostic work-up and therapeutical strategy even in a moderate form.
Keywords: PC deficiency, Lactic acidosis, Secondary mitochondrial respiratory chain defects, Bezafibrate
1. Introduction
Pyruvate carboxylase (PC; EC: 6.4.1.1) is a biotin-containing mitochondrial enzyme composed of four functional domains: the N-terminal biotin carboxylase (BC) domain, the central carboxyl transferase (CT) domain, the tetramerization domain (PT) and the C-terminal biotin carboxyl carrier protein (BCCP). It is organized as a homotetramer. PC catalyzes the conversion of pyruvate to oxaloacetate, and is involved in gluconeogenesis and energy production through replenishment of the tricarboxylic acid (TCA) cycle with oxaloacetate. Adequate energy production via the TCA requires not only a constant supply of acetylCoA but also a fairly constant pool of the catalytic intermediates of the Krebs cycle including oxaloacetate, the key intermediate that condenses with acetylCoA to “initiate” the cycle. As shown in Fig. 1, PC is also closely linked to the urea cycle, because aspartate, the citrulline cosubstrate of argininosuccinate synthetase, is produced from oxaloacetate through transamination.
Fig. 1.

Functions of pyruvate carboxylase (PC), and its close relation to the urea cycle and the Krebs cycle.
Adapted from [2].
In striking contrast to genetic defects affecting the mitochondrial respiratory chain (MRC), PC deficiency (OMIM 266150) is very rare [1] as its estimated incidence of 1 in 250 000 births (http://ghr.nlm.nih.gov). PC deficiency is an autosomal recessive disorder with three nosological forms reviewed in 2010 by Marin-Valencia et al. [2]. These forms differ in the severity of clinical and biochemical manifestations: the B or “French” phenotype with neonatal onset and severe outcome, the A form or “North American phenotype” with mild to moderate hyperlactacidemia and longer survival, and the C form with episodic metabolic acidosis and ketoacidosis during metabolic stresses. PC deficiency can also be part of multicarboxylase deficiency secondary to deficiency of biotinidase or holocarboxylase.
Here we report on one patient with the A form of PC deficiency and mitochondrial complex IV (C IV) deficiency in cultured skin fibroblasts, illustrating the complexity of the diagnostic work-up of energy disorders. In vivo and in vitro therapeutic assays were devised for this patient and in fibroblasts of another previously reported patient with the type B form of the disease.
2. Patients and methods
2.1. Patients
Patient 1 was the second boy of healthy non-consanguineous Caucasian parents, born after an uneventful pregnancy and a normal birth weight. Developmental progress was apparently considered as normal until the age of 23 months when he presented a severe lactic acidosis with failure to thrive, vomiting and lethargy. As shown in Table 1, laboratory investigations found lactic acidosis (pH 6.98, blood lactate level 11.3 mmol/L, normal range < 2 mmol/L) associated with pronounced ketonuria but normal glucose plasma levels. Lactate to pyruvate ratio was elevated in cerebrospinal fluid (24, normal range 6–14) and 3-hydroxybutyrate to acetoacetate ratio was in the lower range (0.8, normal range 0.8–1). Plasma ammonia was 70 μmol/L (normal range 15–45 μmol/L). Plasma amino acid analysis showed normal alanine (298 μmol/L; normal range: 174–375 μmol/L), glycine (159 μmol/L; normal range: 160–264 μmol/L), proline (114 μmol/L; normal range: 93–233 μmol/L) and lysine levels (136 μmol/L; normal range 85–241 μmol/L) in the context of generalized trend to hypoaminoacidemia, including low citrulline (6 μmol/L, normal range: 21–38 μmol/L) and glutamine (327 μmol/L, normal range: 423–545 μmol/L). During follow-up, plasma amino acid levels were within normal range or showed mildly decreased glycine and/or mildly increased alanine. Cerebrospinal fluid amino acids at admission were normal. Urinary organic acid analysis showed very high lactate and ketone body levels at admission (lactic acid: 59 mol/mol creatinine, normal < 76 mmol/mol creatinine and 3-hydroxybutyric acid: 45 mol/mol creatinine, normal < 99 mmol/mol creatinine). Lactaturia and ketonuria completely resolved within 48 h and reappeared accompanying a decompensation episode during follow-up. Otherwise, only mildly increased lactate levels (1.5–2 fold ratios to normal upper values) were intermittently detected in urine. No hypoglycemia was observed. After the first acute episode, treated with hyperhydratation, glucose infusion and bicarbonate, the child was evaluated for cognitive functions: an intellectual disability was found (see below) and re-educations were proposed. Triheptanoin is a medium-chain triglyceride containing fatty acids with an “odd” number of carbons (seven) producing acetylCoA and propionylCoA, thereby providing anaplerotic intermediate for the TCA cycle. Although this molecule dramatically improved the neonatal distress of a previously described patient with a severe form of PC deficiency [3], it was not available anymore for our patient. Therefore, several alternative medical assays aiming to produce TCA intermediates were performed in vivo and in vitro (see below).
Table 1.
Clinical, biochemical and genetic findings at diagnosis in patient 1 and patient 2.
| Patient 1 | Patient 2 | |
|---|---|---|
| Age of onset | 18 months | Neonatal |
| Clinical findings | Lactic acidosis following gastro-enteritis Developmental delay |
Lactic acidosis Neurological distress Hepatic failure |
| Laboratory investigation | pH = 6.98 Lactate: 11.3 mmol/L Increased plasma L/P ratio LCR 3-OHB/AcAc ratio in the lower range Ammonemia: 70 μmol/L |
pH = 7.15 Lactate = 17 mmol/L Increased plasma L/P ratio Decreased plasma 3-OHB/AcAc ratio Ammonemia: 268 μmol/L |
| Plasma amino acids: Alanine: 298 μmol/L Proline: 114 μmol/L Lysine: 136 μmol/L Glutamine: 327 μmol/L Citrulline: 6 μmol/L |
Plasma amino acids: Alanine: 958 μmol/L Proline: 801 μmol/L Lysine: 713 μmol/L Glutamine: 264 μmol/L Citrulline: 158 μmol/L |
|
| Urinary organic acids: Lactic acid: 59 mol/mol creatinine 3-OHB: 45 mol/mol creatinine Complex IV: 180 nmol/min/mg proteins |
Urinary organic acids: Lactic acid: > 75 mol/mol creatinine 3-OHB: 8359 mmol/mol creatinine Complex IV: 223 nmol/min/mg proteins |
|
| Genetics | c. 808C>T; p.Arg270Trp c. 1892G>A; p.Tyr631Gln |
c. 1023-1G>T (IVS7-1G>T); p.Asp341GlufsX351 c.911A>G; p.Tyr304Cys |
| Outcome | 8 year-old: needs specialized school | Death at 6 months |
L: lactate; P: pyruvate; 3-OHB: 3-hydroxybutyrate; AcAc: aceto-acetate; normal range: plasma ammonemia: 15–45 μmol/L; alanine: 174–375 μmol/L; proline: 93–233 μmol/L; lysine: 85–241 μmol/L; glutamine: 423–545 μmol/L; citrulline: 21–38 μmol/L; lactic acid: < 76 mmol/mol creatinine; 3-hydroxybutyrate: < 99 mmol/mol creatinine; complex IV: 308–457 nmol/min/mg of protein.
For in vitro therapeutic trials (see below), fibroblasts of a second patient (patient 2) were used.
We previously reported patient 2 who had a typical severe phenotype of PC deficiency [3]. Briefly, she presented at birth with axial hypotonia but normal vigilance, a severe lactic acidosis associated with pronounced ketonuria and hyperammonemia, and liver failure. Plasma amino acid analysis revealed elevated lysine (713 μmol/L), alanine (958 μmol/L), proline (801 μmol/L) and citrulline (158 μmol/L) and low glutamine (264 μmol/L). This pattern is highly suggestive of the French form of PC deficiency. We had obtained a transient but spectacular clinical and biological improvement under triheptanoin administration [3]. The patient died at 6 months of age after developing severe infection resulting in fatal acute ketoacidosic distress.
2.2. Biochemical investigations
Lactate and pyruvate levels were determined in plasma by enzymatic methods. Plasma amino acids were assayed by nihydrin colorimetry (Jeol AminoTac Analyzers) and urinary organic acids by gas chromatography–mass spectrometry (Varian Saturn-2000). Samples for organic acid analysis were, whenever possible, from first morning urine.
2.3. Enzyme assays of PC and mitochondrial respiratory complexes
PC measurement in cultured fibroblasts was performed as previously described [4] as well as polarographic and spectrophotometric assay of mitochondrial respiratory complexes in cultured fibroblasts [5].
2.4. Molecular investigation of PC gene
Mutations in PC gene had been previously reported for both patients [1].
2.5. In vivo and in vitro drug assays
In vivo treatments were tested for patient 1, while in vitro assays were performed on cultured fibroblasts of both patients 1 and 2.
2.5.1. In vivo treatments for patient 1
Fig. 2 shows all the treatments proposed to patient 1. Vitamin therapy (thiamine, biotin, cobalamine, carnitine and riboflavin) was initiated prior to any diagnosis. Biotin (10 mg/day) was continued after the diagnosis of PC deficiency was performed. At 26 months of age, aspartate (sargenor®) at a dose of 0.14 g/kg/day and citrate of sodium and potassium (foncitril®) at a dose of 0.25 g/kg/day were started (Fig. 2). Two months later, citrate of sodium and potassium was replaced by citrate of betaine at a dose of 0.38 g/kg/day during 10 months. Aspartate was stopped after 8 months, at the time of a severe metabolic distress. Oxoglutarate (cetornan®) was initiated at age of 34 months (dose 20 g/day) as well as cornstarch at age 40 months (2 g/kg/day at bedtime) to maintain PDH activity during the night. Citrate of betaine® was stopped (because of poor compliance) at age of 38 months. Bezafibrate 0.2 g/day was introduced at the age of 5 years and 5 months.
Fig. 2.

Treatments and main biochemical data during follow-up in patient 1.
Top panel, serum lactate levels (y-axis) against age. Three age intervals are defined, based on different combinations of proposed treatments. Mean lactate levels (m1–m3) and the t-test p-values of the comparisons between the corresponding groups are indicated above the panel. Middle panel, the proposed therapies over different age intervals as indicated by the boxes. Bottom panel, selected organic acid levels (y-axis: millimoles/mole of creatinine).
2.5.2. In vitro treatments for patient 1
PC-deficient skin fibroblasts were grown in RPMI 1640 containing glucose (2 g/L) supplemented with 10% fetal calf serum (FCS) with different conditions (citrate 1 mM, aspartate 1 mM, biotin 40 μl/mL, nonanoate 20 mM) during 48 to 72 h. Cells were incubated at 37 °C under a humidified atmosphere containing 5% CO2. At the end of incubation, cells were removed by trypsinization and cell pellet was aliquoted into Eppendorf tubes and directly used for polarographic test and enzymatic measurements [6]. The supernatant of the different culture flasks was stored at − 20 °C until gas chromatography–mass spectrometry analysis.
For bezafibrate treatment, patient 1 and control fibroblasts were first grown in OptiMEM supplemented with Ultroser G and 3% FCS, then in Ham F10 supplemented with 12% FCS. At 90% confluency, they were treated with 400 μM bezafibrate during 72 h. Cells were removed by trypsinization and cell pellets were stored at − 80 °C until PC activity measurement.
2.5.3. In vitro treatment for patient 2
Therapeutic assays for patient 2 fibroblasts were the same as for patient 1 fibroblasts, except for bezafibrate.
3. Results
3.1. Pyruvate carboxylase deficiency
For patient 1, PC assay in cultured fibroblasts was performed as part of the systematic investigation of lactic acidosis, even though a biochemical diagnosis of MRC deficiency had been proposed initially (see below). By enzyme assays PC enzyme activity was overtly deficient (no detectable activity; normal range = 0.10–0.80 nmol/mn/mg of proteins; control = 0.30) with normal propionylCoA carboxylase (0.16 nmol/mn/mg of proteins; normal range = 0.10–0.90; control = 0.20) and normal biotinidase activities. We concluded to a clinical and biochemical moderate form (group A) of PC deficiency.
By contrast, patient 2 was highly suspected of the disorder because of characteristic features including a typical plasma amino acid profile at birth ([3] and see Discussion). Enzyme assays in fibroblasts confirmed PC deficiency (type B).
Molecular genetic investigation confirmed primary PC deficiency in both patients. As described in Table 1, patient 1 was a compound heterozygote for two missense mutations, c.808C>T (p.Arg270Trp) and c.1892G>A (p.Arg631Gln). Patient 2 was a compound heterozygote for a frameshift mutation leading to a premature codon stop in intron 7 acceptor splice site c.1023-1G>T (p.Asp341GlufsX351) and to the absence of functional protein, and for a missense mutation c.911A>G (p.Tyr304Cys). The missense mutations involved evolutionary conserved residues. Arg 270 and Tyr 304 are situated in the BC domain. Arg 270 is within a motif previously involved in bicarbonate binding [7], and in a region involved in interactions between the BC and the BCCP domain. Tyr 304 is localized close to the ATP binding site. Arg 631 is localized at the surface of the CT domain, close to the putative allosteric binding site [1].
3.2. Secondary mitochondrial respiratory chain defect
Spectrophotometric assay of mitochondrial respiratory complexes was performed for patient 1 because of inconclusive plasma amino acid chromatography. An isolated partial C IV deficiency was identified in cultured fibroblasts of both patients (patient's 1 fibroblasts: 180 nmol/min/mg of protein; normal control range between 308 and 457; patient's 2 fibroblasts: 223 nmol/min/mg of protein). In a subsequent assay, a decreased complex IV (C IV) to complex II + III (C II + III) ratio was found in both patient fibroblasts (respectively 1.8 and 1.75 for patients 1 and 2; normal range: 2.67–3.14).
3.3. Clinical follow-up
Except for a second episode of metabolic decompensation occurring at 38 months of age, patient 1 remained metabolically stable and had intellectual disability (IQ 69, verbal IQ 63 and performance IQ 83). At 5 years of age, he attained high section of preschool and needed some orthophonist reeducation. We found no regression event and he currently continues to make progress. At age 7 years, he required specialized school. No clinical difference was observed with any medication and all except biotin have been stopped.
3.4. Biochemical follow-up
The various therapeutic protocols did not led to any change in plasma lactate and amino acid levels and in organic acids in cultured fibroblasts whatever the severity of the disease (data not shown). Concomitant with both acute decompensations of patient 1, mild lactic acidosis was recurrently noted between 2.2 and 5 mmol/L along with increased urinary lactate and alpha-oxoglutarate. Fig. 2 shows patient 1 plasma lactate levels and selected urine organic acids during follow-up, along with the tested therapies (see Patients and Methods). Organic acids (Fig. 1, bottom panel) were significantly altered only during the single decompensation episode. Lactate levels were significantly (p = 0.032) though moderately reduced under bezafibrate therapy (from 2.75 to 2.14 mM; Fig. 1, top panel), whereas all other drugs alone or in combination did not show any obvious effect. Fig. 3 shows that patient 1 fibroblast PC activity remained unchanged and very low following 72 h treatment with 400 μM bezafibrate. Nevertheless in control fibroblasts, PC activity was induced two-fold by this drug.
Fig. 3.
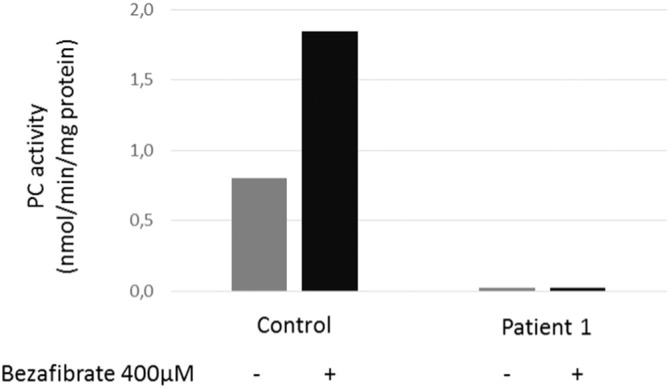
Effect of bezafibrate treatment on PC activity.
PC activity was measured in patient 1 fibroblasts under basal conditions (gray bars) or after a 72 h treatment with 400 μM bezafibrate (black bars). Y-axis: PC activity in nmol/min/mg proteins.
4. Discussion
We report on a moderate form (the American type A) of pyruvate carboxylase (patient 1) in which diagnosis was essentially fortuitous, revising an initial diagnosis of mitochondrial C IV defect. We compare this patient to a previous case of severe PC deficiency (type B) previously described in our unit (patient 2) [3] and we retrospectively reveal secondary C IV deficiency in this patient as well, as shown in Table 1. In an attempt to identify anaplerotic surrogates to triheptanoin, different drug regimens were carried with patient 1 and several assays were carried in vitro for both patients. Indeed, triheptanoin is a triglyceride containing a 7 carbon fatty acid and was found to be transiently effective in patient 2, as described previously [3], [8]. It was unfortunately no longer available to us.
Patient 1 had a non-specific clinical presentation of lactic acidosis and intellectual disability consistent with either type A of PC deficiency or a mitochondrial respiratory chain (MRC) defect. The oxidoreduction markers and amino acids in plasma did not enable us to discriminate between these diseases. Indeed, oxidoreduction analysis was not informative (there was no decrease in the 3-hydroxybutyrate/acetoacetate ratio associated with increased lactate/pyruvate ratio), and the characteristic amino acid profile of PC deficiency group B was absent: the patient had neither hypercitrullinemia nor hyperlysinemia. Conversely, plasma amino acid profiles were highly suggestive of a neonatal form of PC deficiency in patient 2. Indeed, this patient presented with low plasma glutamine level (related to the impaired anaplerosis of the Krebs cycle) along with a secondary urea cycle dysfunction biochemical phenotype including elevated ammonemia and plasma citrulline levels, resulting from decreased cytosolic aspartate. Similarly, in line with the French form of PC deficiency, patient 2 presented with signs of “reduced cytosol” and “oxidized mitochondria”. “Reduced cytosol” is due to low oxaloacetate level and is associated with an increased NADH/NAD ratio, accounting for elevated lactate to pyruvate ratio as detected in plasma. The “oxidized mitochondria” with low NADH/NAD ratio in mitochondria is related to the decreased 3OHbutyrate to acetoacetate ratio [2].
Patient 1 was initially diagnosed with a mitochondrial respiratory C IV defect, which was subsequently recognized as a secondary consequence of the PC defect. This illustrates the difficulty to establish a diagnosis of energetic disorders based on the combination of clinical findings and intermediate activities of the MRC enzyme complexes. Clinical and biochemical features are diverse and display mild specificity [6] including the pattern of lactate and associated amino acid alterations (high alanine and proline) in blood and CSF as previously reported in the “American form” of PC deficiency [9]. Our results raise the possibility that the moderate (American) type of PC deficiency may be underdiagnosed and should be considered for patients with unexplained lactic acidosis. Intermediate activities of MRC enzymes should be interpreted with caution and may be seen in a variety of other mitochondrial disorders [10]. The modified Walker diagnostic criteria suggested that MRC complex activity should be less than 20% of control citrate synthase or complex II activity in a tissue in order to be considered a convincing diagnostic criterion of primary MRC defect [11]. Until now, PC deficiency has not been described as a possible cause of secondary MRC deficiency except in one case of holocarboxylase synthetase deficiency [12]. However other diseases with secondary MRC defect have been described including pyruvate dehydrogenase (PDHc) deficiency (OMIM 312170), which is the most frequently reported one [12], fatty acid oxidation defects [13], [14], [15], [16], neonatal hemochromatosis (OMIM 231100) [12], [17], pantothenate kinase deficiency (OMIM 606157) [12], holocarboxylase synthetase deficiency (OMIM 253270) [12], molybdenum cofactor deficiency [9], spino-cerebellar ataxia type 7 (OMIM 164500) [18], [19], Menkes disease (OMIM 309400) [20], [21], Wilson disease (OMIM 277900) [17], OTC deficiency (OMIM 311250) [17], progressive familial intrahepatic cholestasis type 2 (OMIM 601847) [22], hereditary spastic paraparesis type 7 (OMIM 607259) [23], Fanconi–Bickel syndrome (OMIM 227810) [24], autism spectrum disorder (ASD) [25], [26] and more recently organic acidurias [27], [28].
The pathogenic mechanism of secondary MRC deficiency may result from secondary down-regulation due to redox imbalance and decreased entry of substrate to the MRC [12], i.e. low levels of oxaloacetate in PC deficiency resulting in diminished flux through the TCA cycle and impaired ability of the TCA cycle to produce adequate reducing power for electron transport. Accumulation of toxic metabolites in the inner mitochondrial membrane has also been proposed, which directly inhibits MRC enzymes or acts as a “detergent-like” on biomembranes, such as acyl-CoA metabolites in fatty acid oxidation deficiency [12], [13], [29], [30], [31], [32]. Because PC is localized within the mitochondria matrix, abnormal import and/or assembly of the MRC complex subunits are possible – as described with PDHc, beta-oxidation pathway and TCA cycle enzymes – due to the presence of macromolecular complexes involving direct interactions between these enzymatic components [33]. Finally, PC deficiency could interfere with MRC regulation [18] as mitochondrial transcription rates correlate directly with the ability to produce ATP and ATP levels [34], [35]. Inversely, a primary MRC defect can also induce a secondary TCA cycle defect and/or fatty acid oxidation enzyme inhibition defects by accumulation of NADH, hence an increased NADH/NAD-ratio [28], [36], [37], [38].
In addition to genetic counseling, an efficient diagnostic protocol for TCA cycle defects could be put forward as it could involve potential therapies. Several trials have been led (see [2] for review). Indeed, anaplerotic substrates may increase availability of Krebs cycle intermediate compounds [39] and replenish the oxaloacetate pool, as observed albeit only transiently in one patient under triheptanoin [8]. The C5 ketone bodies, which are “odd” carbon number compounds derived from the oxidation of triheptanoin, provide the anaplerotic propionylCoA substrate, as well as acetylCoA, and can fuel the TCA cycle. Triheptanoin was found to be transiently effective in patient 2 as previously described; however, a recent report reported that triheptanoin was ineffective in two patients with type B PC deficiency [40]. Indeed, these patients showed persistent hyperlactatemia, episodes of severe ketoacidosis, renal tubular acidosis and little neurodevelopmental progress. They died at the age of 7 and 8 months respectively. Triheptanoin is also used in the treatment of other inherited metabolic diseases such as GLUT1 deficiency syndrome (GLUT1DS; OMIM 606777) or in β-oxidation defects. In GLUT1DS, the compound was found to decrease spike-wave seizures and improved neuropsychological performances [41]. In three patients with VLCAD deficiency (OMIM 201475), triheptanoin led to clinical improvement with decrease of rhabdomyolysis crises and muscle weakness. Cardiomyopathy disappeared in one of them [42]. In seven patients with myopathic CPT2 deficiency (OMIM 255110), triheptanoin reduced muscle pain [43]. However triheptanoin was impossible to obtain for our patient. Other substrates provide interesting alternatives including dicarboxylic acids such as azelate [44], a precursor of succinic acid and acetyl-CoA. However, biochemical investigations performed by incubating the patient fibroblasts with four different compounds (citrate, aspartate, biotin and nonanoate) did not show any effect (data not shown). Moreover, the clinical evolution and the biological parameters (plasma lactate) were unmodified except for a partial, possible effect of bezafibrate in patient 1. In VLCAD and CPT2 deficient fibroblasts, bezafibrate was shown to increase mRNA expression and correct deficiency [45], [46]. In adipocytes, PC mRNA and protein levels correlate with PPAR-γ expression, and a PPAR response element in the PC gene promoter has been identified [47]. A 1.4 fold increase of PC mRNA expression was found in β-cells of rat pancreatic islets after an 8 h treatment by 300 μM bezafibrate and persists after a 48 h treatment [48]. We suggested the possibility that in patient 1, who showed a slight decrease of lactatemia under bezafibrate, this molecule may act by increasing PC mRNA expression thereby resulting in greater residual activity of the mutant PC enzyme. As a consequence, we decided to test bezafibrate effect on PC activity in patient 1 fibroblasts. We found a two-fold increase of PC activity in control fibroblasts after a 72 h treatment with 400 μM bezafibrate. Unfortunately, this treatment was unable to increase PC activity in patient 1 (i.e., enzyme activity did not rise above background despite the moderate type A of the disease). In patient 1, p.Arg270Trp may have an impact on bicarbonate binding, biotin binding and may destabilize the interdomain interactions. p.Arg631Gln might alter interactions between CT and BC domains and disturb the transmission of the regulatory signal delivered by acetyl-coA [1]. The increase in mRNA and protein levels might not compensate for the functional consequences of these mutations. Nevertheless, these results raise the possibility that, for some mutations leading to milder type C of the disease, in the presence of sufficient residual activity, bezafibrate might be beneficial. In patient 1, the moderate decrease of lactatemia under bezafibrate might be a consequence of respiratory chain stimulation [49]. Nevertheless, it is important to note that the liver is the true tissue of interest in PC deficiency, and the fibroblasts a surrogate. Also bezafibrate action could vary in the liver and fibroblasts in the same patient, and might possibly explain the decrease of lactate in the patient in spite of unchanged enzyme activity in fibroblasts. The parents eventually stopped all medications except biotin.
In summary, we report on one patient with the moderate form of PC deficiency and a secondary C IV deficiency in fibroblasts. Based on the non-specific metabolic profile of our patient, we suggest that PC defects may currently be underdiagnosed. Anaplerotic substrates may represent a therapeutic option for these severe energetic diseases, but further investigations are necessary as our in vitro results have been disappointing with several of these compounds. A possibly more effective anaplerotic drug, triheptanoin, is no longer available. Depending on the causative mutation and on the severity of the deficit, bezafibrate might represent an interesting option to increase residual activity.
References
- 1.Monnot S., Serre V., Chadefaux-Vekemans B., Aupetit J., Romano S., De Lonlay P. Structural insights on pathogenic effects of novel mutations causing pyruvate carboxylase deficiency. Hum. Mutat. May 2009;30(5):734–740. doi: 10.1002/humu.20908. [DOI] [PubMed] [Google Scholar]
- 2.Marin-Valencia I., Roe C., Pascual J.M. Pyruvate carboxylase deficiency: mechanisms, mimics and anaplerosis. 2010;101:9–17. doi: 10.1016/j.ymgme.2010.05.004. [DOI] [PubMed] [Google Scholar]
- 3.Mochel F. Pyruvate carboxylase deficiency: clinical and biochemical response to anaplerotic diet therapy. Mol. Genet. Metab. Apr 2005;84(4):305–312. doi: 10.1016/j.ymgme.2004.09.007. [DOI] [PubMed] [Google Scholar]
- 4.Wang D. The molecular basis of pyruvate carboxylase deficiency: mosaicism correlates with prolonged survival. Mol. Genet. Metab. 2008;95(1–2):31–38. doi: 10.1016/j.ymgme.2008.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chrétien D., Rustin P., Bourgeron T., Rötig A., Saudubray J., Munnich A. Reference charts for respiratory chain activities in human tissues. Clin. Chim. Acta. 1994;228(1):53–70. doi: 10.1016/0009-8981(94)90056-6. [DOI] [PubMed] [Google Scholar]
- 6.Munnich A., Rustin P. Clinical spectrum and diagnosis of mitochondrial disorders. Am. J. Med. Genet. Spring 2001;106(1):4–17. doi: 10.1002/ajmg.1391. [DOI] [PubMed] [Google Scholar]
- 7.Li S., Cronan J. The gene encoding the biotin carboxylase subunit of Escherichia coli acetyl-coA carboxylase. J. Biol. Chem. 1992;267:855–863. [PubMed] [Google Scholar]
- 8.Roe C., Mochel F. Anaplerotic diet therapy in inherited metabolic disease: therapeutic potential. J. Inherit. Metab. Dis. Jun 2006;29(2–3):332–340. doi: 10.1007/s10545-006-0290-3. [DOI] [PubMed] [Google Scholar]
- 9.Kang P., Hunter J., Kaye E. Lactic acid elevation in extramitochondrial childhood neurodegenerative diseases. J. Child Neurol. Sep 2001;16(9):657–660. doi: 10.1177/088307380101600906. [DOI] [PubMed] [Google Scholar]
- 10.Thorburn D., Smeitink J. Diagnosis of mitochondrial disorders: clinical and biochemical approach. J. Inherit. Metab. Dis. Apr 2001;24(2):312–316. doi: 10.1023/a:1010347808082. [DOI] [PubMed] [Google Scholar]
- 11.Bernier F., Boneh A., Dennett X., Chow C., Cleary M., Thorburn D. Diagnostic criteria for respiratory chain disorders in adults and children. Neurology. Nov. 12 2002;59(9):1406–1411. doi: 10.1212/01.wnl.0000033795.17156.00. [DOI] [PubMed] [Google Scholar]
- 12.Hui J., Kirby D., Thorburn D., Boneh A. Decreased activities of mitochondrial respiratory chain complexes in non-mitochondrial respiratory chain diseases. Dev. Med. Child Neurol. Feb 2006;48(2):132–136. doi: 10.1017/S0012162206000284. [DOI] [PubMed] [Google Scholar]
- 13.Das A., Fingerhut R., Wanders R., Ullrich K. Secondary respiratory chain defect in a boy with long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency: possible diagnostic pitfalls. Eur. J. Pediatr. Apr 2000;159(4):243–246. doi: 10.1007/s004310050063. [DOI] [PubMed] [Google Scholar]
- 14.Reichmann H., Scheel H., Bier B., Ketelsen U., Zabransky S. Cytochrome c oxidase deficiency and long-chain acyl coenzyme A dehydrogenase deficiency with Leigh's subacute necrotizing encephalomyelopathy. Ann. Neurol. Jan 1992;31(1):107–109. doi: 10.1002/ana.410310120. [DOI] [PubMed] [Google Scholar]
- 15.Tyni T., Majander A., Kalimo H., Rapola J., Pihko H. Pathology of skeletal muscle and impaired respiratory chain function in long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency with the G1528C mutation. Neuromuscul. Disord. Oct 1996;6(5):327–337. doi: 10.1016/0960-8966(96)00352-5. [DOI] [PubMed] [Google Scholar]
- 16.Röschinger W., Muntau A., Duran M., Dorland L., IJlst L., Wanders R. Carnitine-acylcarnitine translocase deficiency: metabolic consequences of an impaired mitochondrial carnitine cycle. Clin. Chim. Acta. Aug 2000;298(1–2):55–68. doi: 10.1016/s0009-8981(00)00268-0. [DOI] [PubMed] [Google Scholar]
- 17.Murayama K., Ohtake A. Children's toxicology from bench to bed—liver injury (4): mitochondrial respiratory chain disorder and liver disease in children. J. Toxicol. Sci. 2009;34(Suppl. 2):SP237–SP243. doi: 10.2131/jts.34.sp237. [DOI] [PubMed] [Google Scholar]
- 18.Sperl W. Diagnosis of mitochondrial encephalomyopathies. J. Pediatr. Jul 1992;121(1):166–168. doi: 10.1016/s0022-3476(05)82578-8. [DOI] [PubMed] [Google Scholar]
- 19.Powers J., DeCiero D., Cox C., Richfield E., Ito M., Moser A. The dorsal root ganglia in adrenomyeloneuropathy: neuronal atrophy and abnormal mitochondria. J. Neuropathol. Exp. Neurol. May 2001;60(5):493–501. doi: 10.1093/jnen/60.5.493. [DOI] [PubMed] [Google Scholar]
- 20.Kodama H., Okabe I., Yanagisawa M., Kodama Y. Copper deficiency in the mitochondria of cultured skin fibroblasts from patients with Menkes syndrome. J. Inherit. Metab. Dis. 1989;12(4):386–389. doi: 10.1007/BF01802032. [DOI] [PubMed] [Google Scholar]
- 21.Pedespan J., Jouaville L., Cances C., Letellier T., Malgat M., Guiraud P. Menkes disease: study of the mitochondrial respiratory chain in three cases. Eur. J. Pediatr. Neurol. 1999;3(4):167–170. doi: 10.1016/s1090-3798(99)90050-8. [DOI] [PubMed] [Google Scholar]
- 22.Davit-Spraul A., Beinat M., Debray D., Rötig A., Slama A., Jacquemin E. Secondary mitochondrial respiratory chain defect can delay accurate PFIC2 diagnosis. JIMD Rep. 2014;14:17–21. doi: 10.1007/8904_2013_278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Arnoldi A., Tonelli A., Crippa F., Villani G., Pacelli C., Sironi M. A clinical, genetic, and biochemical characterization of SPG7 mutations in a large cohort of patients with hereditary spastic paraplegia. Hum. Mutat. Apr 2008;29(4):522–531. doi: 10.1002/humu.20682. [DOI] [PubMed] [Google Scholar]
- 24.Odièvre M., Lombès A., Dessemme P., Santer R., Brivet M., Chevallier B. A secondary respiratory chain defect in a patient with Fanconi–Bickel syndrome. J. Inherit. Metab. Dis. Sep 2002;25(5):379–384. doi: 10.1023/a:1020147716990. [DOI] [PubMed] [Google Scholar]
- 25.Rossignol D., Frye R. Mitochondrial dysfunction in autism spectrum disorders: a systematic review of literature. Mol. Psychiatry. Mar 2012;17(3):290–314. doi: 10.1038/mp.2010.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Frye R., Rossignol D. Mitochondrial dysfunction can connect the diverse medical symptoms associated with autism spectrum disorders. Pediatr. Res. May 2011;69(5 Pt 2):41R–47R. doi: 10.1203/PDR.0b013e318212f16b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cosson M., Touati G., Lacaille F., Valayannopoulos V., Guyot C., Guest G. Liver hepatoblastoma and multiple OXPHOS deficiency in the follow-up of a patient with methylmalonic aciduria. Mol. Genet. Metab. Oct 2008;95(1–2):107–109. doi: 10.1016/j.ymgme.2008.06.007. [DOI] [PubMed] [Google Scholar]
- 28.Vockley J., Rinaldo P., Bennett M., Matern D., Vladutiu G. Synergistic heterozygosity: disease resulting from multiple partial defects in one or more metabolic pathways. Mol. Genet. Metab. Oct 2000;71(1–2):10–18. doi: 10.1006/mgme.2000.3066. [DOI] [PubMed] [Google Scholar]
- 29.McMillin Wood J. Carnitine palymityltransferase in neonatal and adult heart and liver mitochondria. Effect of phospholipase C treatment. J. Biol. Chem. Apr 25 1975;250(8):3062–3066. [PubMed] [Google Scholar]
- 30.Pande S., Blanchaer M. Reversible inhibition of mitochondrial adenosine diphosphate phosphorylation by long chain acyl coenzyme A esters. J. Biol. Chem. Jan 25 1971;246(2):402–411. [PubMed] [Google Scholar]
- 31.Ventura F., Ruiter J., IJlst L., Almeida I., Wanders R. Inhibition of oxidative phosphorylation by palmitoyl-CoA in digitonin permeabilized fibroblasts: implications for long-chain fatty acid beta-oxidation disorders. Biochim. Biophys. Acta. Aug 15 1995;1272(1):14–20. doi: 10.1016/0925-4439(95)00064-b. [DOI] [PubMed] [Google Scholar]
- 32.Wojtczak L., Schönfeld P. Effect of fatty acids on energy coupling processes in mitochondria. Biochim. Biophys. Acta. Nov 2 1993;1183(1):41–57. doi: 10.1016/0005-2728(93)90004-y. [DOI] [PubMed] [Google Scholar]
- 33.Vélot C., Srere P. Reversible transdominant inhibition of a metabolic pathway. In vivo evidence of interaction between two sequential tricarboxylic acid cycle enzymes in yeast. J. Biol. Chem. Apr 2000;275(17):12926–12933. doi: 10.1074/jbc.275.17.12926. [DOI] [PubMed] [Google Scholar]
- 34.Amiott E., Jaehning J. Mitochondrial transcription is regulated via an ATP “sensing” mechanism that couples RNA abundance to respiration. Mol. Cell. May 2006;22(3):329–338. doi: 10.1016/j.molcel.2006.03.031. [DOI] [PubMed] [Google Scholar]
- 35.Gupta U., Katoch K., Sharma R., Singh H., Natragan M., Singh D. Analysis of quantitative relationship between viability determination in leprosy by MFP, ATP bioluminescence and gene amplification assay. Int. J. Lepr. Other Mycobact. Dis. Dec 2001;69(4):328–334. [PubMed] [Google Scholar]
- 36.Christensen E., Brandt N., Schmalbruch H., Kamieniecka Z., Hertz B., Ruitenbeek W. Muscle cytochrome c oxidase deficiency accompanied by a urinary organic acid pattern mimicking multiple acyl-CoA dehydrogenase deficiency. J. Inherit. Metab. Dis. 1993;16(3):553–556. doi: 10.1007/BF00711679. [DOI] [PubMed] [Google Scholar]
- 37.Mayatepek E., Wanders R., Becker M., Bremer H., Hoffman G. Mitochondropathy presenting with non-ketotic hypoglycaemia as 3-hydroxydicarboxylic aciduria. J. Inherit. Metab. Dis. 1995;18(2):249–252. doi: 10.1007/BF00711780. [DOI] [PubMed] [Google Scholar]
- 38.Enns G., Bennett M., Hoppel C., Goodman S., Weisiger K., Ohnstad C. Mitochondrial respiratory chain complex I deficiency with clinical and biochemical features of long-chain 3-hydroxyacyl-coenzyme A dehydrogenase deficiency. J. Pediatr. Feb 2000;136(2):251–254. doi: 10.1016/s0022-3476(00)70111-9. [DOI] [PubMed] [Google Scholar]
- 39.Weinberg J., Venkatachalam M., Roeser N., Nissim I. Mitochondrial dysfunction during hypoxia/reoxygenation and its correction by anaerobic metabolism of citric acid cycle intermediates. Proc. Natl. Acad. Sci. U. S. A. Mar 14 2000;97(6):2826–2831. doi: 10.1073/pnas.97.6.2826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Breen C., White F., Scott C.A.B., Heptinstall L., Walter J., Jones S. Unsuccessful treatment of sever carboxylase deficiency with triheptanoin. 2014;173:361–366. doi: 10.1007/s00431-013-2166-5. [DOI] [PubMed] [Google Scholar]
- 41.Pascual J.M., Liu P., Mao D., Kelly D.I., Hernandez A., Sheng M. Triheptanoin for glucose transporter type I deficiency (G1D): modulation of human ictogenesis, cerebral metabolic rate, and g = cognitive indices by a food supplement. JAMA Neurol. 2014;71(10):1255–1265. doi: 10.1001/jamaneurol.2014.1584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Roe C., Sweetman L., Roe D., David F., Brunengraber H. Treatment of cardiomyopathy and rhabdomyolysis in long-chain fat oxidation disorders using an anaplerotic odd-chain triglyceride. J. Clin. Invest. 2002;110:259–269. doi: 10.1172/JCI15311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Roe C., Yang B., Brunengraber H., Roe D., Wallace M., Garritson B. Carnitine palmitoyltransferase II deficiency: successful anaplerotic diet therapy. Neurology. 2008;71:260–264. doi: 10.1212/01.wnl.0000318283.42961.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Grego A., Mingrone G. Dicarboxylic acids, an alternate fuel substrate in parenteral nutrition: an update. Clin. Nutr. Jun 1995;14(3):143–148. doi: 10.1016/s0261-5614(95)80011-5. [DOI] [PubMed] [Google Scholar]
- 45.Djouadi F., Bonnefont J., Thuillier L., Droin V., Khadom N., Munnich A. Correction of fatty acid oxidation in carnitine palmitoyl transferase 2-deficient cultured skin fibroblasts by bezafibrate. Pediatr. Res. Oct 2003;54(4):446–451. doi: 10.1203/01.PDR.0000083001.91588.BB. [DOI] [PubMed] [Google Scholar]
- 46.Djouadi F., Aubey F., Schlemmer D., Ruiter J., Wanders R., Strauss A. Bezafibrate increases very-long-chain acyl-coA dehydrogenase protein and mRNA expression in deficient fibroblasts and is a potential therapy for fatty acid oxidation disorders. Hum. Mol. Genet. Sep 15 2005;14(18):2695–2703. doi: 10.1093/hmg/ddi303. [DOI] [PubMed] [Google Scholar]
- 47.Jitrapakdee S., Slawik M., Medina-Gomez G., Campbell M., Wallace J., Sethi J. The peroxisome proliferator-activated receptor-gamma regulates murine pyruvate carboxylase gene expression in vivo and in vitro. J. Biol. Chem. Jul 22 2005;280(29):27466–27476. doi: 10.1074/jbc.M503836200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yoshikawa H., Tajiri Y., Sako Y., Hashimoto T., Umeda F., Nawata H. Effects of bezafibrate on beta-cell function of rat pancreatic islets. Eur. J. Pharmacol. Aug 31 2001;426(3):201–206. doi: 10.1016/s0014-2999(01)01204-3. [DOI] [PubMed] [Google Scholar]
- 49.Bastin J., Aubey F., Rötig A., Munnich A., Djouadi F. Activation of peroxisome proliferator-activated receptor pathway stimulates the mitochondrial respiratory chain and can correct deficiencies in patients' cells lacking its components. J. Clin. Endocrinol. Metab. Apr 2008;93(4):1433–1441. doi: 10.1210/jc.2007-1701. [DOI] [PubMed] [Google Scholar]