

Abstract
Vici syndrome (VICIS) is a rare, autosomal recessive neurodevelopmental disorder with multisystem involvement characterized by agenesis of the corpus callosum, cataracts, cardiomyopathy, combined immunodeficiency, developmental delay, and hypopigmentation. Mutations in EPG5, a gene that encodes a key autophagy regulator, have been shown to cause VICIS, however, the precise pathomechanism underlying VICIS is yet to be clarified. Here, we describe detailed clinical (including brain MRI and muscle biopsy) and genetic features of nine Japanese patients with VICIS. Genetic dissection of these nine patients from seven families identified 14 causative mutations in EPG5. These included five nonsense, two frameshift, three splicing, one missense, and one multi-exon deletion mutations, and two initiation codon variants. Furthermore, cultured skin fibroblasts (SFs) from two affected patients demonstrated partial autophagic dysfunction. To investigate the function of EPG5, siRNA based EPG5 knock-down, and CRISPR/Cas9 mediated EPG5 knock-out HeLa cells were generated. EPG5-depleted cells exhibited a complete block of autophagic flux caused by defective autophagosome-lysosome fusion. Unexpectedly, endocytic degradation was normal in both VICIS SFs and EPG5 depleted cells, suggesting that EPG5 function is limited to the regulation of autophagosome-lysosome fusion.
Introduction
Vici syndrome (VICIS, OMIM #242840) is a rare, autosomal recessive neurodevelopmental disorder characterized by agenesis of the corpus callosum (ACC), cataracts, cardiomyopathy, combined immunodeficiency, developmental delay, and hypopigmentation1. Since first described in 1988 by Dionisi Vici et al.1, multiple case reports that describe multisystem involvement suggest that a fundamental molecular defect in cell physiology underlies the disorder2–11. Mutations in the EPG5 gene on chromosome 18q12.3 were recently identified to cause VICIS12–19. EPG5 is the human homolog of the metazoan-specific autophagy gene epg-5, encoding a key autophagy regulator (ectopic P-granules autophagy protein 5)20. Autophagy is an evolutionary conserved lysosomal degradation process essential for cell homeostasis21–23. The function of epg-5 was initially reported in C. elegans, where it is required for the formation of degradative autolysosomes20. Using Epg5 knock-out (KO) mice, Zhao et al.24, 25 demonstrated that Epg5 was involved in the maturation of autophagosomes into degradative autolysosomes, as well as endocytic degradation and recycling. However, the precise mechanism of Epg5 remains to be elucidated, and its role in human cells has not been intensively studied.
Here, we studied nine patients with VICIS from seven families and identified 14 causative mutations in EPG5. Skin fibroblasts (SFs) from VICIS patients and EPG5 knock-down (KD) and KO HeLa cells demonstrated normal endocytic function, but impaired autophagic function due to defects in autophagosome-lysosome fusion.
Results
Clinical features
We genetically identified nine patients (three males and six females) with VICIS, including two pairs of siblings, from seven Japanese families. Table 1 provides a summary of the clinical features of our nine patients and reported patients in the literature1–19. Typical clinical features are illustrated in Supplementary Fig. S1. No consanguinity was noted in any of the families. All patients had a normal gestation and delivery, with normal birth weight, length and head circumference. All patients presented with developmental delay, hypotonia, and recurrent infections. Developmental delay was profound and no patients acquired meaningful words. Most patients developed progressive microcephaly and failed to thrive (Supplementary Table S1). Two patients died; patient 2.1 at the age of 14 years due to respiratory arrest, and patient 5.1 at the age of one year due to cardiomyopathy. Cardiomyopathy and cataracts, both initially described as principle features in VICIS1, were notably uncommon (3/9 cases) or absent (0/9 cases), respectively, in these patients. Most of the patients (8/9) showed intractable epilepsy with epileptic spasms, tonic and myoclonic seizure types, and one patient exhibiting symptomatic West syndrome. Seizures started at the median age of one year and two months. Although cataracts were not present in any patient, abnormal ophthalmologic findings were identified in seven patients; two with optic disc pallor, two with erratic eye movement and five with nystagmus. Complete ACC was observed in all patients. Additional CNS abnormalities included paucity of white matter, irregularity of the ventricular wall, ventricular dilation, delayed myelination, pontine hypoplasia, cerebellar hypoplasia and cerebral atrophy while typical Probst bundle was not detected in any patients (Supplementary Fig. S2). Increased serum aspartate transaminase (AST) and alanine transaminase (ALT) was shown in all patients and increased serum creatine kinase was present in seven patients. Muscle biopsies were performed in four patients and showed only mild myopathic changes including fiber-type disproportion with type 2 atrophy (Supplementary Fig. S3a–f). Electron microscopy was performed on one patient biopsy (2.1) and revealed only slight abnormalities of autophagosome vacuoles (Supplementary Fig. S3g and h).
Table 1.
Clinical features of nine patients with Vici syndrome in current study and summary of previous reports.
| Family | 1 | 2 | 3 | 4 | 5 | 6 | 7 | Summary of our 9 patients | Summary of previously reported 67 patients1–19 | Total summary of all 76 patients | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Subject | 1.1 | 1.2 | 2.1 | 2.2 | 3.1 | 4.1 | 5.1 | 6.1 | 7.1 | |||
| Sex | F | F | M | F | M | M | F | F | F | 3 male | 13 male | 16 male |
| 6 female | 12 female | 18 female | ||||||||||
| 42 NA | 42 NA | |||||||||||
| Status | Alive | Alive | Deada | Alive | Alive | Alive | Deadb | Alive | Alive | 7 Alive | 14 Alive | 21 Alive |
| 7y | 2y | 14y | 15y | 5y | 7y | 1y | 4y | 2y | 2 Dead | 20 Dead | 22 Dead | |
| 33 NA | 33NA | |||||||||||
| Developmental delay | + | + | + | + | + | + | + | + | + | 9/9 (100%) | 58/58 (100%) | 67/67 (100%) |
| Hypotonia | + | + | + | + | + | + | + | + | + | 9/9 (100%) | 25/25 (100%) | 34/34 (100%) |
| Hypopigmentation | + | + | − | − | + | + | + | + | + | 7/9 (78%) | 65/67 (97%) | 72/76 (95%) |
| Cardiomyopathy | + | − | − | − | + | − | + | − | − | 3/9 (33%) | 49/59 (83%) | 52/68 (76%) |
| Seizures | + | + | + | + | + | + | + | + | − | 8/9 (89%) | 17/29 (59%) | 25/38 (66%) |
| Seizure onset | 1y4m | 2m | 1y10m | 5m | 1y5m | 1y5m | 5m | 1y0m | ||||
| Seizure type | Spasm | Spasm | Tonic | Tonic | West | Tonic | Spasm | Myoclonic tonic | ||||
| Ophthalmologic finding | ||||||||||||
| Cataracts | − | − | − | − | − | − | − | − | − | 0/9 (0%) | 48/63 (76%) | 48/72 (67%) |
| Optic disc pallor or atrophy | − | − | − | − | + | − | − | + | − | 2/9 (22%) | 9/9 (100%) | 11/18 (61%) |
| Erratic eye movement | − | − | − | − | − | + | + | − | − | 2/9 (22%) | NA | 2/9 (22%) |
| Nystagmus | − | − | + | + | + | − | + | + | − | 5/9 (56%) | 11/21 (52%) | 16/30 (53%) |
| Hypopigmented iris | − | − | − | − | − | − | − | − | + | 1/9 (11%) | 8/10 (80%) | 9/19 (47%) |
| Recurrent infection | + | + | + | + | + | + | + | + | + | 9/9 (100%) | 62/64 (97%) | 71/73 (97%) |
| Microcephaly | + | − | + | + | + | + | + | − | + | 7/9 (78%) | 43/48 (90%) | 50/57 (88%) |
| High-arched palate | + | + | + | + | + | + | + | + | + | 9/9 (100%) | 15/24 (63%) | 24/33 (73%) |
| Central nervous system anomalies | ||||||||||||
| ACC | + | + | + | + | + | + | + | + | + | 9/9 (100%) | 65/65 (100%) | 74/74 (100%) |
| Paucity of white matter | + | − | + | + | + | − | − | + | NA | 5/8 (63%) | 5/6 (83%) | 10/14 (71%) |
| Irregularity of the ventricular wall | + | − | + | + | + | + | + | + | NA | 7/8 (88%) | NA | 7/8 (88%) |
| Ventricular dilation | + | + | + | + | + | − | + | + | + | 8/9 (89%) | 8/11 (73%) | 16/20 (80%) |
| Delayed myelination | − | − | − | − | + | + | − | − | NA | 2/8 (25%) | 4/6 (67%) | 6/14 (43%) |
| Pontine hypoplasia | + | + | + | + | + | + | + | + | + | 9/9 (100%) | 5/9 (56%) | 14/18 (78%) |
| Hypoplasia of the medulla oblongata | − | − | − | − | − | − | − | − | − | 0/9 (0%) | NA | 0/9 (0%) |
| Cerebellar hypoplasia | − | − | + | + | + | − | − | + | NA | 4/8 (50%) | 13/16 (81%) | 17/24 (71%) |
| Cerebral atrophy | + | ± | + | + | + | + | − | + | NA | 6/8 (75%) | 5/9 (56%) | 11/17 (65%) |
| Probst bundles | − | ± | − | − | − | − | − | − | NA | 0/8 (0%) | 1/4 (25%) | 1/12 (8%) |
| Elevated CK | + | + | + | + | − | + | − | + | + | 7/9 (78%) | 11/13 (85%) | 18/22 (82%) |
| Elevated AST/ALT | + | + | + | + | + | + | + | + | + | 9/9 (100%) | 12/13 (92%) | 21/22 (95%) |
| Abnormal immunology | − | − | − | − | + | + | − | − | − | 2/9 (22%) | 15/23 (65%) | 17/32 (53%) |
| Abnormal muscle biopsy | − | NA | − | − | NA | − | NA | NA | NA | 0/4 (0%) | 20/21 (95%) | 20/25 (80%) |
F = female, M = male, ACC = agenesis of the corpus callosum, NA = not available, CK = creatine kinase, AST = aspartate transaminase, ALT = alanine transaminase, aPatient 2.1 died at the age of 14 years due to respiratory arrest. bPatient 5.1 died at the age of one year due to cardiomyopathy.
Molecular genetic analyses
Of the nine patients with VICIS, eight patients from six families were identified by whole exome sequencing, while one patient (patient 7.1) was identified by the target panel analysis. Compound heterozygous mutations in EPG5 were identified in all patients (Table 2). The 14 EPG5 mutations identified were all novel and comprised five nonsense, two frameshift, three splicing, one missense, one multi-exon deletion, and two initiation codon variants (Fig. 1a). The single missense mutation (p.Ala1015Val) was predicted to be damaging by SIFT (score 0.00), PolyPhen-2 (HumVar score 0.656) and MutationTaster (probability value 0.999). One splicing mutation (c.6766 + 1 G > C), found in patient 6.1, was located at the canonical +1 splice site. The two remaining splicing mutations (c.3582 G > A and c.2598 A > G) found in patients 3.1 and 5.1, respectively, were confirmed as pathogenic by reverse transcriptase-polymerase chain reaction (RT-PCR). The c.3582 G > A mutation was located in the last codon of exon 19. RT-PCR showed that removal of exon 19 resulted in an in-frame deletion of 66 amino acid residues (p.Ala1129_Lys1194del) (Fig. 1b). The c.2598 A > G mutation generated a cryptic splicing site, which introduced a 45-bp in-frame deletion of 15 amino acid residues (p.Val852_Gln866del) (Fig. 1c). A multi-exon deletion of exons 17 to 21 in entirety and the 5’-end of exon 22 was identified in patient 7.1. Cloning the breakpoint identified a 7,380 bp deletion and an 8 bp insertion (Fig. 1d). Both parents from five families (Family1–4, 6) were revealed as carriers. One of the mutations in patient 5.1 arose de novo (c.1188delC; p.Tyr396*) on the paternal allele, while the other mutation was maternally inherited (c.2598 A > G). We verified the paternity in family 5 by confirming the segregation of SNPs. The parents’ samples from patient 7.1 were not available.
Table 2.
Mutations identified in the nine Vici syndrome patients.
| Family | Subject | Allele 1 | Allele 2 | ||||
|---|---|---|---|---|---|---|---|
| Nucleotide | Amino acid | Origin | Nucleotide | Amino acid | Origin | ||
| 1 | 1.1 | c.2 T > C | p.Met1? | M | c.5792delT | p.Leu1931Trpfs*5 | P |
| 1.2 | c.2 T > C | p.Met1? | M | c.5792delT | p.Leu1931Trpfs*5 | P | |
| 2 | 2.1 | c.3152 C > G | p.Ser1051* | P | c.4230 G > A | p.Trp1410* | M |
| 2.2 | c.3152 C > G | p.Ser1051* | P | c.4230 G > A | p.Trp1410* | M | |
| 3 | 3.1 | c.2461 C > T | p.Arg821* | P | c.3582 G > A | splicing (p.Ala1129_Lys1194del) | M |
| 4 | 4.1 | c.1 A > G | p.Met1? | M | c.4108delC | p.Leu1370Serfs*22 | P |
| 5 | 5.1 | c.1188delC | p.Tyr396* | P (de novo) | c.2598 A > G | splicing (p.Val852_Gln866del) | M |
| 6 | 6.1 | c.3044 C > T | p.Ala1015Val | P | c.6766 + 1 G > C | — | M |
| 7 | 7.1 | c.2863 C > T | p.Arg955* | NT | multi-exon deletion (g.52621_60000del7380insCAACATCC) | NT | |
The numbering of the mutations and alterations is relative to NM_020964.2 (gene) and NP_066015.2 (protein), respectively. With regard to multi-exon deletions, the variant’s description is relative to genomic sequence records for NG_042838.1. Synonymous mutations in patient 3.1 and patient 5.1 were found to cause an in-frame deletion on the basis of cDNA sequence data derived from affected individuals. ?protein has not been analysed, an effect is expected but difficult to predict, *=the protein coding sequence ends at a translation termination codon, P = paternal, M = maternal, NT = not tested.
Figure 1.
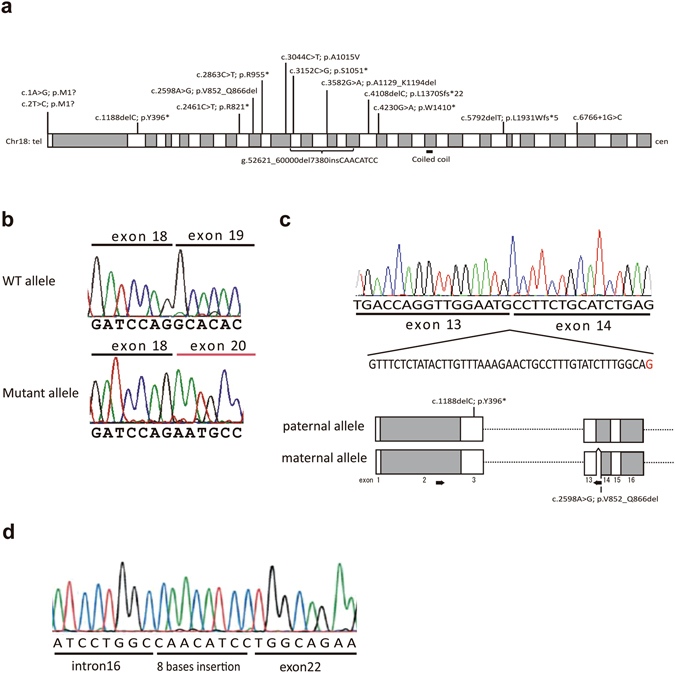
EPG5 mutations identified in nine Vici syndrome patients. (a) Schematic of the EPG5 gene located in 18q12.3 illustrating mutation locations. White and gray rectangles represent exons (1 through 44). (b) Aberrant splicing caused by the synonymous mutation in patient 3.1 (c.3582 G > A) identified the removal of exon 19. WT, wild type. (c) Aberrant splicing caused by the synonymous mutation in patient 5.1 (c.2598 A > G). Above; RT-PCR identified a 45-bp in-frame deletion in exon 14 induced by a cryptic splicing acceptor site created by 2598 A > G (shown in red). The mother of patient 5.1 also had this mutation. Below; Confirmation of compound heterozygosity. The paternal allele was amplified by PCR using a reverse primer that binds to the region deleted in the maternal allele. The paternal PCR product contained the c.1188delC mutation, indicating that patient 5.1 was a compound heterozygote. The location of the mutation in patient 5.1 is shown. The arrows represent PCR primers. White and gray rectangles represent exons. (d) A multi-exon deletion identified in patient 7.1. Cloning and sequencing of the genomic breakpoint and flanking regions revealed an 8-bp insertion of unknown origin at the breakpoint.
Accumulation of autophagosomes in VICIS SFs
Since EPG5 has been reported to function in autophagic and endocytic pathways12, 20, 24, 25, we analyzed these functions in patient SFs. Cultured SFs isolated from two patients (patient 1.2 and 4.1) were treated with nutrient-rich or starvation medium for 6 hours with/without the lysosomal inhibitor Bafilomycin A1 (BafA1), then LC3 expression determined by western blot. LC3 is one of the core autophagy genes and a mammalian homologue of yeast Atg826. LC3-I localizes to the cytosol, and is converted into the membrane-bound form LC3-II as autophagy progresses, which specifically localizes on the autophagosome membrane. LC3-II is continuously generated and degraded during the autophagic degradation pathway. The lysososmal inhibitor BafA1 blocks autophagic degradation and causes an accumulation of LC3-II compared with BafA1 non-treated cells. The differences in LC3-II levels in the presence and absence of BafA1 indicated “autophagic flux”, an estimation of the amount of autophagosomes that should have been degraded during that period. Immunoblotting showed a slight accumulation of LC3-II in VICIS SFs under starved conditions without BafA1 (Fig. 2a). Under starved conditions, autophagic flux was reduced in VICIS SFs (Fig. 2a). Immunofluorescence analysis confirmed the accumulation of LC3-containing vacuoles in VICIS SFs under starved conditions (Fig. 2b). These results indicate that the autophagic degradation pathway is slowed in VICIS SFs leading to accumulation of un-degraded autophagic vacuoles within cells.
Figure 2.
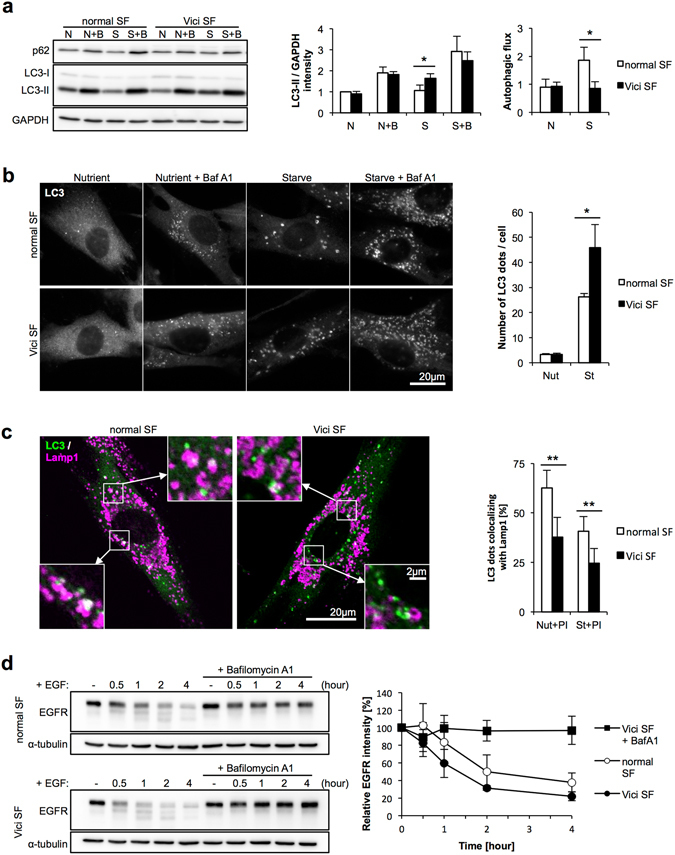
Mild autophagic impairment in VICIS SFs. (a) Normal and VICIS SFs were incubated in nutrient-rich or starved conditions with/without BafA1 for 6 hours. A representative western blot of LC3 and p62, and quantitative analysis of band intensities are shown. LC3 intensities were standardized by GAPDH intensities, and values relative to that of normal cells in nutrient-rich conditions without BafA1 are shown. Autophagic flux was calculated by subtracting values for BafA1 untreated samples from treated samples in each condition. Upon starvation, VICIS SFs exhibited mild accumulation of LC3-II and reduced autophagic flux. p62 is another substrate which is degraded by autophagy, but it showed little variation in expression. (b) SFs were incubated in nutrient-rich or starved conditions with/without BafA1 for 2 hours, followed by LC3 immunostaining. Representative images and quantified numbers of punctate LC3 staining per cell are shown. Under starved conditions, LC3 punctate staining was higher in VICIS SFs compared with normal SFs, which is consistent with the increased intensity of the LC3-II band in VICIS SFs under the starved conditions shown in (a). (c) SFs were incubated in nutrient-rich or starved conditions with/without protease inhibitors for 2 hours, followed by LC3 and Lamp1 immunostaining. Representative images and calculated percentages of punctate LC3 staining colocalized with Lamp1 are shown. Decreased colocalization between LC3 and Lamp1 staining in VICIS SFs indicates reduced autophagosome-lysosome fusion. (d) Conventional endocytic pathway activity assessed by EGFR degradation was normal in VICIS SFs. N or Nut, nutrient-rich conditions; S or St, starved conditions; B or BafA1, Bafilomycin A1; PI, protease inhibitors (E64d+ pepstatin A). Mean and SD from at least three independent experiments with 10 images assessed per treatment condition. Statistics by two-tailed Student’s t-test; *p < 0.05, **p < 0.01.
Reduced autophagosome-lysosome fusion in VICIS SFs
Next we investigated which process of autophagy is impaired in VICIS. Lack of LC3-I accumulation in VICIS SFs (Fig. 2a) suggested an impairment of autophagosome degradation rather than impairment of an initial step of autophagy, such as the conversion of LC3-I to LC3-II. Immunofluorescence analysis of LC3 (Fig. 2b) supported a defective clearance of autophagic vacuoles. Therefore, we next investigated the fusion of autophagosomes with lysosomes, which results in degradation of cytoplasmic material by the acidic lysosomal environment and hydrolases. In order to directly observe colocalization of autophagosomes and lysosomes, we performed co-immunostaining of LC3 and the lysosomal marker Lamp1 in SFs in the presence of lysosomal PI (protease inhibitor) that prevents lysosomal degradation of LC3 protein. VICIS SFs showed a reduced ratio of colocalization both in nutrient-rich and starved conditions (Fig. 2c), suggesting an impairment of autophagosome-lysosome fusion in VICIS SFs.
Normal lysosomal function and endocytic degradation in VICIS SFs
It has been previously shown that a KO of mouse Epg5 causes defective degradation via the endocytic pathway25. To assess endocytic degradation in VICIS SFs, we firstly measured the activity of eight lysosomal enzymes (Supplementary Fig. S4a) and processing of cathepsin D (Supplementary Fig. S4b). The results indicated that lysosomal proteolytic activity was normal in VICIS SFs. Next, SFs were treated with DQ-BSA reagent, which is taken up by endocytosis and generates fluorescence after being cleaved within lysosomes. After six hours of treatment there were similar levels of fluorescence in normal and VICIS SFs, indicating comparable activities of endocytic degradation (Supplementary Fig. S4c). Uptake of BSA conjugate was normalized using a second fluorescence (AlexaFluor 488)-conjugated BSA, which does not require cleavage to fluoresce. These results indicated normal endocytic uptake and normal lysosomal degradation in VICIS SFs. We also assessed endocytic degradation using EGFR (epidermal growth factor receptor) dynamics. Upon EGF stimulation, EGF-bound EGFR on the cell surface is internalized by endocytosis. The majority of EGFR is subsequently transported to lysosomes and degraded. A time course of EGFR degradation in VICIS SFs after EGF treatment showed comparable EGFR degradation to EGF-treated normal SFs (Fig. 2d), confirming normal endocytic uptake and lysosomal degradation.
Defective autophagy in EPG5 depleted cell lines caused by impaired autophagosome-lysosome fusion
Since VICIS SFs showed mild cellular phenotypes involving autophagy, we decided to analyze EPG5 function in EPG5 depleted cell lines. In HeLa cells, we performed EPG5 KD using siRNA, and generated an EPG5 KO cell line using CRISPR/Cas9 system (Fig. 3a). EPG5 KD and KO cells showed a complete block of autophagic flux (Fig. 3b and c) and accumulation of LC3-II, indicating a fundamental role of EPG5 in autophagy. Next, we analyzed autophagosome-lysosome fusion in EPG5 KO cells using two approaches: tandem-fluorescent (RFP and GFP) tagged LC3 (tfLC3) and colocalization of LC3 and Lamp1. The tfLC3 construct (mRFP-GFP-LC3) was firstly transfected into EPG5 KO cells27. As LC3-II localizes on the autophagosomal membrane, both RFP and GFP signals can be detected in autophagosomes. When fused with lysosomes, autophagosomes become autolysosomes and the acidic environment quenches the GFP signal while the RFP signal remains relatively stable. As a result, autolysosomes exhibit an RFP signal only, enabling distinction between autophagosomes and autolysosomes. The tfLC3 assay exhibited an almost complete merge of GFP and RFP signals in EPG5 KO cells suggesting accumulation of autophagosomes before fusion (Fig. 3d). The ratio of colocalization between LC3 and Lamp1 decreased in EPG5 KO cells both in nutrient-rich and starved conditions (Fig. 3e). These results clearly indicate that EPG5 functions in autophagosome-lysosome fusion.
Figure 3.
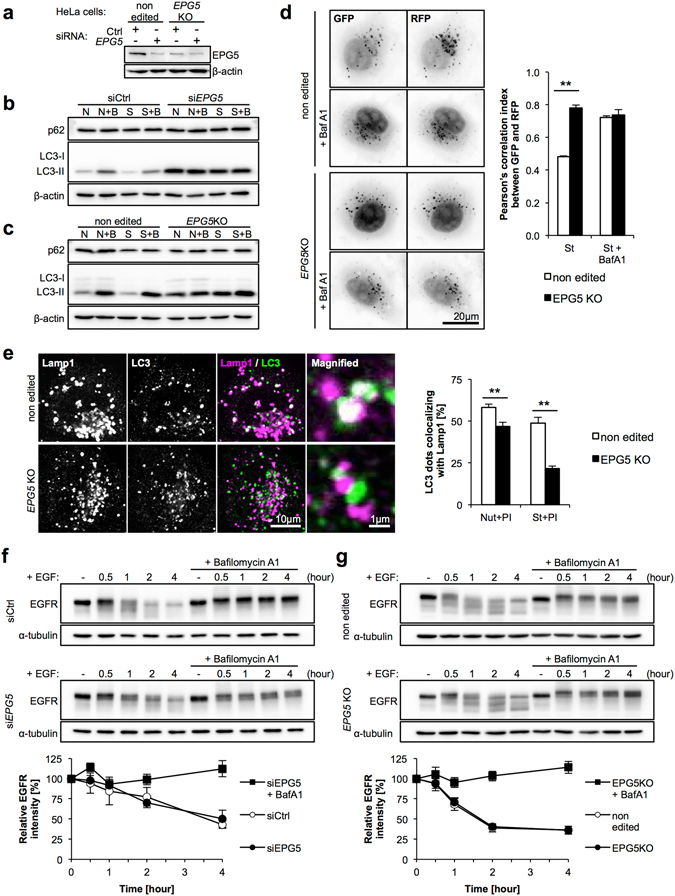
Autophagic impairment is caused by reduced fusion between autophagosomes and lysosomes in EPG5 KD and KO cells. (a) EPG5 depletion by siRNA treatment. (b,c) LC3-II has accumulated and autophagic flux is stopped in EPG5 KD and KO HeLa cells. (d) Tandem fluorescent-tagged LC3 (tf-LC3) assay indicated reduced autophagosome-lysosome fusion. EPG5 KO cells were infected with a retroviral mRFP-GFP-LC3 construct, then incubated in nutrient-rich or starved media with/without BafA1 for two hours. BafA1 inhibited lysosomal V-ATPase and led to disruption of the lysosomal acidic environment, as well as inhibition of autophagosome-lysosome fusion, which we used as the negative control for this experiment. RFP and GFP fluorescence images were obtained, and Pearson’s correlation index was calculated to show colocalization between RFP and GFP. Higher colocalization indicates accumulation of autophagosomes. (e) Colocalization of punctate LC3 staining with Lamp1 in the presence of protease inhibitors was lower in EPG5 KO cells under both nutrient rich and starved conditions. (f,g) Endocytic degradation of EGFR was normal in EPG5 KD and KO cells. N or Nut, nutrient-rich conditions; S or St, starved conditions; B or BafA1, Bafilomycin A1; PI, protease inhibitors (E64d+ pepstatin A). The mean and SD are from at least three independent experiments, with 10 images assessed per treatment condition. Statistical analysis was performed using a two-tailed Student’s t-test; **p < 0.01.
Normal endocytic degradation in EPG5 depleted cell lines
To confirm the normal endocytic degradation observed in VICIS SFs, we performed the same assay for EGFR degradation in EPG5 KD and KO cells. Despite EPG5 KD and KO cells demonstrating clear defects in autophagic function, endocytic function, as measured by EGFR degradation, was completely normal (Fig. 3f and g), indicating EPG5 is not involved in endocytosis.
Discussion
We have identified nine patients from seven families with genetically confirmed VICIS. ACC, cataracts, hypopigmentation, cardiomyopathy, and immune dysfunction have been indicated as the five principle features of VICIS1. In our study, all patients had ACC and recurrent infections, seven patients had hypopigmentation, three patients had cardiac involvement, and no patients had cataracts. Thus, the proposed principle features of VICIS were not consistently present in our patient. Two Japanese VICIS patients previously reported also had no cataracts4. These findings suggest that the frequency of cataracts may be affected by race. Recently Byrne et al.18 proposed that profound developmental delay, progressive microcephaly, and a failure to thrive, are equally consistent features of VICIS. In our cohort, all patients demonstrated profound developmental delay, while progressive microcephaly and failure to thrive were exhibited in most patients. Thus, our findings support those of Byrne et al.18. Moreover, we propose three additional features of VICIS; epilepsy, high-arched palate and elevated AST/ALT, based upon their presence in all or most of our patients. MRI findings in our study were basically comparable to those in previous reports18. Complete ACC and pontine hypoplasia were consistently present while paucity of white matter, irregularity of the ventricular wall, ventricular dilation, and cerebral atrophy were present in most patients. It is of note that Probst bundle was not detected in any patients. It may reflect a failure to form commissural axons28. Previously, it was reported that muscle biopsies in patients with VICIS were characterized by increased variability in fiber size, increase in internal and centralized nuclei, abnormal glycogen accumulation on light microscopy6, 7, 12, 18. There might be some histological overlap between VICIS and a number of primary neuromuscular disorders where aberrant autophagy was implicated as important secondary mechanisms, including vacuolar myopathies, centronuclear myopathies and glycogen storage disorders29–31. In our study, muscle biopsies were non-specific and not diagnostic, indicating that a negative result did not exclude a VICIS diagnosis. Because the precise mechanism for the abnormal muscle histopathologies remains to be clarified, further investigation should be needed. In conclusion, VICIS should be suspected in any child with ACC and profound developmental delay, even if some of the principle features of VICIS are absent. It should be noted that while the type of complications in other organs differed between patients, the severity of developmental delay was uniformly profound and indicated a consistently severe dysfunction of brain development in our patients.
The majority of mutations identified in this study produced truncations and/or caused profound dysfunction. Five nonsense mutations, two frameshift mutations, and one multi-exon deletion are likely to produce a non-functional protein, and are thus essentially null alleles. Two of the three splicing mutations were assessed by RT-PCR to locate mutations leading to in-frame deletions of 66 and 15 amino acid residues, respectively, which could potentially cause significant functional defects. The canonical +1 splicing mutation (c.6766 + 1 G > C) was not assessed due to lack of RNA. Nonetheless, it is likely to cause aberrant splicing that induces major functional defects in the translated protein. It is of considerable interest that two EPG5 variants resided in the initial start codon. Since these mutations are likely to alter the translation start site, mutated EPG5 protein would have a truncated N-terminal structure, resulting in substantial functional defects. One missense mutation (c.3044 C > T) was predicted to be pathogenic. A recent report by Kane et al.32 demonstrated aberrant splicing was induced by a missense mutation c.1007 A > G in EPG5, however we were not able to test if this occurred with c.3044 C > T because RNA was not available.
Previous reports have demonstrated that most EPG5 mutations identified in VICIS appear to be null alleles18. Very few missense mutations have been identified18, and of these, some may induce aberrant splicing. Furthermore, Epg5 KO mice are viable and show an overlapping phenotype with VICIS patients, although with some distinct features24. This suggests VICIS pathogenesis is likely driven by a significant functional or even complete defect in EPG5.
The two patients for whom we analyzed SFs (patient 1.2 and 4.1) exhibited typical clinical VICIS symptoms including ACC, profound developmental delay and early onset seizure, with one patient also showing cardiomyopathy. Genetically, these patients harbored variants in the initiation codon and frameshift mutation (patient 1.2 and 4.1). Functional studies using their SFs however, demonstrated only a partial defect in autophagy compared to EPG5 KD and KO cells. The discrepancy between clinical severity and cellular phenotype may arise from differences in cell types or, in this case, from retained function of the truncated EPG5 protein induced by variants in the initial codon. Compound heterozygous mutations of null alleles would most likely cause more profound functional defects in SFs, however we were not able to test this hypothesis. Therefore, further studies are needed to elucidate the mechanisms underlying the neurodevelopmental features in VICIS, which are possibly common to other congenital neuronal disorders associated with autophagy33.
In this study, we demonstrated defects in autophagosome-lysosome fusion in VICIS SFs and EPG5 depleted cells. Several mechanisms have been proposed for EPG5 function. Consistent with our findings, the work that first described an association between VICIS and EPG5 observed an accumulation of autophagosomes, indicative of impaired autophagosome-lysosome fusion in VICIS12. Another group reported EPG5 functions in autophagosome maturation, and endocytic degradation and recycling20, 24, 25 where loss of EPG5/Epg5 impaired autolysosomal proteolysis activity, and delayed endosomal trafficking. The same group recently showed EPG5 functions as a tethering factor that facilitates the fusion between endosomes/lysosomes and autophagosomes34. This is supported by our data where autophagy was significantly impaired in EPG5 KD and KO cells, yet endocytic degradation remained normal. Thus, we propose the function of EPG5 is limited to autophagosome-lysosome fusion.
In conclusion, we have identified novel, compound heterozygous mutations in patients with VICIS, and demonstrated partial impairment of autophagosome-lysosome fusion in VICIS SFs, as well as complete impairment in EPG5 KD and KO cells. Our study indicates that precise regulation of autophagy is crucial for neuronal development, as well as the normal function of other tissues.
Materials and Methods
Patients
We reviewed the clinicopathological features of nine affected patients (three male/six female) from seven families with genetically confirmed VICIS. Eight patients were not clinically diagnosed with VICIS but were categorized as having undiagnosed neurological disorders and DNA of parent-patient trios subjected to whole exome sequencing. One patient (7.1) was clinically diagnosed with VICIS, and their DNA was subjected to targeted gene panel sequencing analysis for EPG5. This study was approved by the institutional review board in Nagoya City University Graduate School of Medical Sciences (#169), and were carried out in accordance with the approved guidelines. Patients’ parents provided written informed consent for genetic analysis and for the usage of recognizable clinical photographs for the online open-access publication.
Sequence analyses
Whole exome sequencing was performed on eight parent-patient trios (families 1–6). Genomic DNA was captured using the SureSelect XT Human All Exon V5 capture library (Agilent Technologies, Santa Clara, CA, USA), and sequenced using the Illumina HiSeq 2000 (Illumina, San Diego, CA, USA) with 100 bp paired-end reads. Exome data processing, variant calling and variant annotation were performed as described previously35, 36. Multiplex targeted sequencing analysis was performed on one additional patient (patient 7.1). Amplicon libraries corresponding to the 44 exons of the EPG5 gene were prepared with an Ion AmpliSeq Custom Panel (Thermo Fisher Scientific, Waltham, MA, USA), and sequenced with an Ion Torrent Personal Genome Machine (PGM) system37. Sequence data was analyzed using a CLC Genomics Workbench 7.0 (CLC bio, Aarhus, Denmark) and Ion reporter (Thermo Fisher Scientific). Identified mutations were validated in all nine patients by Sanger sequencing. The breakpoints in patient 7.1 were PCR amplified and sequenced. The ClinVar accession numbers for the DNA variants reported in this paper are SCV000328413, SCV000328414, SCV000328415, SCV000328416, SCV000328417, SCV000328418, and SCV000328419 (ClinVar, https://www.ncbi.nlm.nih.gov/clinvar/).
Muscle biopsy
Muscle biopsy was performed in four patients. Biopsy specimens were either frozen in liquid nitrogen-cooled isopentane for histochemistry or fixed with glutaraldehyde for electron microscopy. Transverse serial frozen sections of 10-μm thickness were stained with hematoxylin and eosin, modified Gomori trichrome, and a battery of histochemical methods. For electron microscopy, biopsy specimens were fixed in buffered 2% isotonic glutaraldehyde at pH 7.4, post-fixed in osmium tetroxide, and embedded in epoxide resin. Ultrathin sections were stained with uranyl acetate and lead nitrate and examined with an H-700 electron microscope (Hitachi, Tokyo, Japan).
Cell culture and generation of EPG5 KD and KO lines
Cultured SFs from two affected individuals (patient 1.2 and 4.1) were established by conventional methods38. Representative data from patient 4.1 SF are shown in the figures, with comparable phenotypes observed in SFs from patient 1.2. Normal SFs were purchased (Gibco, Waltham, MA, USA, Lonza, Basel, Switzerland, and Kurabo, Osaka, Japan). For the KD of EPG5, we transfected EPG5 siRNA20 with Lipofectamine RNAiMax (Thermo Fisher Scientific) into HeLa cells for 96 hours (transfected twice, every 48 hours). EPG5 KO cells were generated with HeLa cells by cloning guide RNA within the exon 13 of EPG5 (designed using CRISPR design tool, http://crispr.mit.edu) into the pSpCas9(BB)-2A-GFP (PX458) vector (Addgene, Cambridge, Massachusetts, USA) that uses a hU6 promoter. We transfected the cloned vector into HeLa cells for 48 hours, then GFP expressing cells collected by fluorescence activated cell sorting (FACS). Single clones were isolated by diluting the cell suspension and culturing in 96-well plates. Genomic modification of EPG5 was confirmed with Sanger sequencing. In this study we used a clone with homozygous mutation of c.2512_2515delCTTC (p.Leu838Trpfs*34). Cells were cultured and maintained in complete medium (Dulbecco’s Modified Eagle Medium, DMEM) supplemented with 10% fetal bovine serum (FBS).
Assays for autophagy and endocytosis
For the nutrient-rich and starved conditions, cells were incubated with complete medium or Earle’s Balanced Salt Solution (EBSS), respectively, following two phosphate-buffered saline (PBS) washes. To measure autophagosome-lysosome fusion, the tandem fluorescent-tagged LC3 (tfLC3) construct (described previously27) was transfected into cells 48 hours prior to measuring GFP and RFP fluorescence. Specific activities of lysosomal enzymes were measured with total cell lysates using artificial 4-Methylumbelliferone (4-MU) fluorescence substrates39. Sonicated cell lysates were incubated with each of the artificial 4-MU substrates (M1633, M2133, M3633, M3567, M8527, M9766, M7633, M9130; Sigma-Aldrich, St. Louis, Missouri, USA) in acidic acetate buffer or citrate-phosphate buffer for 3 hours at 37 °C and the reaction stopped by adding glycine-NaOH buffer (0.2 M Glycine, 0.2 M NaOH). Fluorescence at Em 450 nm/Ex 365 nm was measured with a microplate reader (Corona, Hitachinaka, Ibaraki, Japan). The standard, 4-MU, (M1381; Sigma-Aldrich) was also measured in the same buffer, and enzyme activities were calculated as nmol/protein mg/h. To assess endocytic activity, DQ-Red BSA and AlexaFluor488-BSA (Invitrogen, Waltham, Massachusetts, USA) were incubated in culture medium at 50 μg/ml for 6 hours. Cells were fixed and fluorescence intensity was quantified with fluorescence microscope and ImageJ software (NIH). For the EGFR degradation assay, cultured cells were washed twice with warm serum-free DMEM, and pre-incubated in serum-free DMEM for two hours before adding EGF. BafA1 (bafilomycin A1) was added 1 hour before EGF. EGF (53003018, Thermo Fisher Scientific) was added to the medium (100 ng/ml) at time 0, and cells were washed once with PBS and collected at the indicated time points. Samples were subsequently lysed and analyzed by immunoblotting for EGFR and α-tubulin.
Antibodies and reagents
Primary antibodies for western blotting (WB) and immunocytochemistry (ICC) were purchased and used at the indicated dilution; anti-LC3 (PM036, MBL; WB 1:3000; ICC 1:1000), anti-p62 (PM045, MBL; WB 1:5000), anti-Lamp1 (clone H4A3, sc-20011, Santa Cruz Biotechnology; ICC 1:1000), anti-EGFR (MI-12-1, MBL; WB 1:500), anti-cathepsin D (sc-6486, Santa Cruz Biotechnology; WB 1:1000), anti-EPG5 (sc-85198, Santa Cruz Biotechnology; WB 1:200), anti-GAPDH (MAb314, Chemicon; WB 1:5000), anti-α-tubulin (clone B-5-1-2, T5168, Sigma-Aldrich; WB 1:5000), anti-β-actin (clone 6D1, M177-3, MBL; WB 1:3000). BafA1 was used at 125 nM. PI comprised 10 μg/ml E64d and 10 μg/ml pepstatin A.
Western blotting
Cells were lysed directly by sodium dodecyl sulphate (SDS) sample buffer and subjected to SDS-PAGE, followed by transfer onto PVDF membrane (Millipore, Billerica, Massachusetts, USA). The membrane was blocked with 1% skim milk (Nacalai Tesque, Kyoto, Japan) in TBST (0.1% Tween-20 in Tris-buffered saline) for 20 min at room temperature, and incubated with a primary antibody as described above in 1% skim milk TBST for 1 hour at room temperature. For EPG5 immunoblotting, the primary antibody was incubated at 4 °C overnight. The membrane was washed with TBST for at least 30 min and incubated with HRP-conjugated secondary antibody (GE Healthcare, Chicago, Illinois, USA) at room temperature for 1 hour. After the final wash with TBST, the membrane was treated with Luminata Forte western HRP substrate (GE Healthcare) and chemiluminescence was visualized with an analyzer (ChemiDoc Touch; BioRad, Hercules, California, USA). Band intensities were measured using ImageJ software.
Immunocytochemistry
Cells were grown on glass cover slips in culture dishes, treated, and fixed with 4% paraformaldehyde in PBS for 20 min at room temperature. Cells were then permeabilized with 50 μg/ml digitonin in PBS for 10 min at room temperature, and immunostained with primary antibodies in 0.2% gelatin PBS for 1 hour at room temperature. After washing cells with PBS, they were incubated with fluorescence-conjugated secondary antibodies for 1 hour at room temperature together with DAPI for nuclear staining. Images were acquired with a wide field fluorescence microscope (IX83; Olympus, Tokyo, Japan) or with a confocal laser-scanning microscope (FV1000, Olympus), and analyzed using ImageJ software.
Electronic supplementary material
Acknowledgements
We are extremely grateful to all the patients and their parents who took part in this study. This study was supported in part by grants for Research on Applying Health Technology from the Ministry of Health, Labour and Welfare of Japan (F.M., N.O., M.K., M.Y., K.Y., K.K., and S.S.); Research on Measures for Intractable Diseases (N.M.); Comprehensive Research on Disability Health and Welfare (N.M.); the Strategic Research Program for Brain Science (N.M.); the Initiative on Rare and Undiagnosed Diseases in Pediatrics from the Japanese Agency for Medical Research and Development (N.M.); the Initiative on Rare and Undiagnosed Diseases in Adults from the Japanese Agency for Medical Research and Development (N.M); a Grant-in-Aid for Scientific Research on Innovative Areas (Transcription Cycle) from the Ministry of Education, Culture, Sports, Science and Technology of Japan (N.M); Grants-in-Aid for Scientific Research (T.O.); Grants-in-Aid for Scientific Research (B) (H. Saitsu.); Grants-in-Aid for Scientific Research (C) (M. Nakashima.); the fund for the Creation of Innovation Centers for Advanced Interdisciplinary Research Areas Program in the Project for Developing Innovation Systems from the Japanese Science and Technology Agency (N.M); and the Takeda Science Foundation (H. Saitsu., N.M.); Intramural Research Grant for Neurological and Psychiatric Disorders of NCNP (I.N.); Health and Labour Sciences Research Grants for Practical Research Project for Rare/Intractable Diseases from Japan Agency for Medical Research and Development (I.N.).
Author Contributions
Conception and design: I.H., T.O., and S.S; collection and assembly of data: I.H., T.O., Y. Negishi, H. Shiraishi, Y. Nonoda, S.M., J.T., N.O., T.K., K.S., Y.Y., D.K., A.H., N.A., M.K., Y.K., and M.Y.; data analysis and interpretation: I.H., T.O., M. Nakashima, F.M., T.M., M. Nakagawa, I.N., T.T., H. Saitsu, K.K., N.M., T. Y., and S.S.; manuscript writing: I.H., T.O., and S.S; and final approval for the manuscript: all authors.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Ikumi Hori and Takanobu Otomo contributed equally to this work.
Electronic supplementary material
Supplementary information accompanies this paper at doi:10.1038/s41598-017-02840-8
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Dionisi Vici C, et al. Agenesis of the corpus callosum, combined immunodeficiency, bilateral cataract, and hypopigmentation in two brothers. Am. J. Med. Genet. 1988;29:1–8. doi: 10.1002/ajmg.1320290102. [DOI] [PubMed] [Google Scholar]
- 2.del Campo M, et al. Albinism and agenesis of the corpus callosum with profound developmental delay: Vici syndrome, evidence for autosomal recessive inheritance. Am. J. Med. Genet. 1999;85:479–485. doi: 10.1002/(SICI)1096-8628(19990827)85:5<479::AID-AJMG9>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 3.Chiyonobu T, et al. Sister and brother with Vici syndrome: agenesis of the corpus callosum, albinism, and recurrent infections. Am. J. Med. Genet. 2002;109:61–66. doi: 10.1002/ajmg.10298. [DOI] [PubMed] [Google Scholar]
- 4.Miyata R, et al. Sibling cases of Vici syndrome: sleep abnormalities and complications of renal tubular acidosis. Am. J. Med. Genet. A. 2007;143:189–194. doi: 10.1002/ajmg.a.31584. [DOI] [PubMed] [Google Scholar]
- 5.Miyata R, Hayashi M, Itoh E. Pathological changes in cardiac muscle and cerebellar cortex in Vici syndrome. Am. J. Med. Genet. A. 2014;164:3203–3205. doi: 10.1002/ajmg.a.36753. [DOI] [PubMed] [Google Scholar]
- 6.McClelland V, et al. Vici syndrome associated with sensorineural hearing loss and evidence of neuromuscular involvement on muscle biopsy. Am. J. Med. Genet. A. 2010;152:741–747. doi: 10.1002/ajmg.a.33296. [DOI] [PubMed] [Google Scholar]
- 7.Al-Owain M, et al. Vici syndrome associated with unilateral lung hypoplasia and myopathy. Am. J. Med. Genet. A. 2010;152:1849–1853. doi: 10.1002/ajmg.a.33421. [DOI] [PubMed] [Google Scholar]
- 8.Rogers RC, Aufmuth B, Monesson S. Vici syndrome: a rare autosomal recessive syndrome with brain anomalies, cardiomyopathy, and severe intellectual disability. Case Rep. Genet. 2011;2011:421582. doi: 10.1155/2011/421582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Finocchi A, et al. Immunodeficiency in Vici syndrome: a heterogeneous phenotype. Am. J. Med. Genet. A. 2012;158:434–439. doi: 10.1002/ajmg.a.34244. [DOI] [PubMed] [Google Scholar]
- 10.Ozkale M, Erol I, Gumus A, Ozkale Y, Alehan F. Vici syndrome associated with sensorineural hearing loss and laryngomalacia. Pediatr. Neurol. 2012;47:375–378. doi: 10.1016/j.pediatrneurol.2012.07.007. [DOI] [PubMed] [Google Scholar]
- 11.Said E, Soler D, Sewry C. Vici syndrome–a rapidly progressive neurodegenerative disorder with hypopigmentation, immunodeficiency and myopathic changes on muscle biopsy. Am. J. Med. Genet. A. 2012;158:440–444. doi: 10.1002/ajmg.a.34273. [DOI] [PubMed] [Google Scholar]
- 12.Cullup T, et al. Recessive mutations in EPG5 cause Vici syndrome, a multisystem disorder with defective autophagy. Nat. Genet. 2013;45:83–87. doi: 10.1038/ng.2497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Filloux FM, Hoffman RO, Viskochil DH, Jungbluth H, Creel DJ. Ophthalmologic features of Vici syndrome. J. Pediatr. Ophthalmol. Strabismus. 2014;51:214–220. doi: 10.3928/01913913-20140423-02. [DOI] [PubMed] [Google Scholar]
- 14.Ehmke N, et al. First description of a patient with Vici syndrome due to a mutation affecting the penultimate exon of EPG5 and review of the literature. Am. J. Med. Genet. A. 2014;164:3170–3175. doi: 10.1002/ajmg.a.36772. [DOI] [PubMed] [Google Scholar]
- 15.Tasdemir S, et al. Vici syndrome in siblings born to consanguineous parents. Am. J. Med. Genet. A. 2016;170:220–225. doi: 10.1002/ajmg.a.37398. [DOI] [PubMed] [Google Scholar]
- 16.El-Kersh K, Jungbluth H, Gringras P, Senthilvel E. Severe Central Sleep Apnea in Vici Syndrome. Pediatrics. 2015;136:e1390–1394. doi: 10.1542/peds.2015-0297. [DOI] [PubMed] [Google Scholar]
- 17.Huenerberg K, et al. Two cases of Vici syndrome associated with Idiopathic Thrombocytopenic Purpura (ITP) with a review of the literature. Am. J. Med. Genet. A. 2016;170:1343–1346. doi: 10.1002/ajmg.a.37589. [DOI] [PubMed] [Google Scholar]
- 18.Byrne S, et al. EPG5-related Vici syndrome: a paradigm of neurodevelopmental disorders with defective autophagy. Brain. 2016;139:765–781. doi: 10.1093/brain/awv393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Maillard C, et al. Prenatal and postnatal presentations of corpus callosum agenesis with polymicrogyria caused by EGP5 mutation. Am. J. Med. Genet. A. 2017;173:706–711. doi: 10.1002/ajmg.a.38061. [DOI] [PubMed] [Google Scholar]
- 20.Tian Y, et al. C. elegans screen identifies autophagy genes specific to multicellular organisms. Cell. 2010;141:1042–1055. doi: 10.1016/j.cell.2010.04.034. [DOI] [PubMed] [Google Scholar]
- 21.Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature. 2008;451:1069–1075. doi: 10.1038/nature06639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mizushima N, Yoshimori T, Ohsumi Y. The role of Atg proteins in autophagosome formation. Annu. Rev. Cell Dev. Biol. 2011;27:107–132. doi: 10.1146/annurev-cellbio-092910-154005. [DOI] [PubMed] [Google Scholar]
- 23.Lamb CA, Yoshimori T, Tooze SA. The autophagosome: origins unknown, biogenesis complex. Nat. Rev. Mol. Cell Biol. 2013;14:759–774. doi: 10.1038/nrm3696. [DOI] [PubMed] [Google Scholar]
- 24.Zhao YG, Zhao H, Sun H, Zhang H. Role of Epg5 in selective neurodegeneration and Vici syndrome. Autophagy. 2013;9:1258–1262. doi: 10.4161/auto.24856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhao H, et al. Mice deficient in Epg5 exhibit selective neuronal vulnerability to degeneration. J. Cell Biol. 2013;200:731–741. doi: 10.1083/jcb.201211014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kabeya Y, et al. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000;19:5720–5728. doi: 10.1093/emboj/19.21.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kimura S, Noda T, Yoshimori T. Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. Autophagy. 2007;3:452–460. doi: 10.4161/auto.4451. [DOI] [PubMed] [Google Scholar]
- 28.Schell-Apacik CC, et al. Agenesis and dysgenesis of the corpus callosum: clinical, genetic and neuroimaging findings in a series of 41 patients. Am. J. Med. Genet. A. 2008;146:2501–2511. doi: 10.1002/ajmg.a.32476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jungbluth H, Sewry CA, Muntoni F. Core myopathies. Semin. Pediatr. Neurol. 2011;18:239–249. doi: 10.1016/j.spen.2011.10.005. [DOI] [PubMed] [Google Scholar]
- 30.Malicdan MC, Nishino I. Autophagy in lysosomal myopathies. Brain Pathol. 2012;22:82–88. doi: 10.1111/j.1750-3639.2011.00543.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Merlini L, Nishino I. 201st ENMC International Workshop: Autophagy in muscular dystrophies–translational approach, 1–3 November 2013, Bussum, The Netherlands. Neuromuscul. Disord. 2014;24:546–561. doi: 10.1016/j.nmd.2014.03.009. [DOI] [PubMed] [Google Scholar]
- 32.Kane MS, et al. Aberrant splicing induced by the most common EPG5 mutation in an individual with Vici syndrome. Brain. 2016;139:e52. doi: 10.1093/brain/aww135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ebrahimi-Fakhari D, et al. Congenital disorders of autophagy: an emerging novel class of inborn errors of neuro-metabolism. Brain. 2016;139:317–337. doi: 10.1093/brain/awv371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang Z, et al. The Vici Syndrome Protein EPG5 is a Rab7 effector that determines the fusion specificity of autophagosomes with late endosomes/lysosomes. Mol. Cell. 2016;63:781–795. doi: 10.1016/j.molcel.2016.08.021. [DOI] [PubMed] [Google Scholar]
- 35.Fujita A, et al. De novo MEIS2 mutation causes syndromic developmental delay with persistent gastro-esophageal reflux. J. Hum. Genet. 2016;61:835–838. doi: 10.1038/jhg.2016.54. [DOI] [PubMed] [Google Scholar]
- 36.Okamoto N, et al. KIF1A mutation in a patient with progressive neurodegeneration. J. Hum. Genet. 2014;59:639–641. doi: 10.1038/jhg.2014.80. [DOI] [PubMed] [Google Scholar]
- 37.Li X, et al. Towards clinical molecular diagnosis of inherited cardiac conditions: a comparison of bench-top genome DNA sequencers. PLoS One. 2013;8:e67744. doi: 10.1371/journal.pone.0067744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rittie L, Fisher GJ. Isolation and culture of skin fibroblasts. Methods Mol. Med. 2005;117:83–98. doi: 10.1385/1-59259-940-0:083. [DOI] [PubMed] [Google Scholar]
- 39.Hindman J, Cotlier E. Glycosidases in normal human leukocytes and abnormalities in GM1 -gangliosidosis. Clin. Chem. 1972;18:971–975. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.