

Abstract
Although atrial fibrillation (AF) is the most commonly encountered cardiac arrhythmia, the basic mechanisms underlying this disorder remain incompletely understood. During the past decade or so, it has become clear that alterations in intracellular Ca2+ handling may play a role in the pathogenesis of AF. Studies in small and large animal models, as well as atrial samples from patients with different forms of AF, have implicated ryanodine receptor type 2 (RyR2) dysfunction and enhanced spontaneous Ca2+ release events from the sarcoplasmic reticulum (SR) as a potential cause of proarrhythmic cellular ectopic (triggered) activity in AF. The molecular mechanisms leading to RyR2 dysfunction and SR Ca2+ leak depend on the clinical stage of AF or specific animal model studied. This review focuses on the mechanisms and role of calcium‐mediated cellular triggered activity in AF, and addresses some of the current controversies in the field.
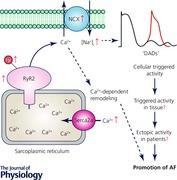
Keywords: arrhythmia, atrial fibrillation, calcium, ryanodine receptor, triggered activity
Abbreviations
- AF
atrial fibrillation
- cAF
long‐standing persistent (chronic) AF
- CaMKII
Ca2+/calmodulin‐dependent protein kinase II
- CPVT
catecholaminergic polymorphic ventricular tachycardia
- DAD
delayed afterdepolarization
- EAD
early afterdepolarization
- JPH2
junctophilin 2
- LTCC
L‐type Ca2+ channel
- NCX
Na+/Ca2+‐exchanger
- pAF
paroxysmal AF
- PP1
protein phosphatase 1
- PKA
protein kinase A
- RyR2
ryanodine receptor type 2
- SERCA2a
sarco/endoplasmic reticulum Ca2+‐ATPase 2a
- SR
sarcoplasmic reticulum
Atrial fibrillation (AF) is the most common type of cardiac arrhythmia with a rising incidence and prevalence in the developed world (Andrade et al. 2014). AF is a progressive disease that can be classified into paroxysmal AF (pAF), which converts spontaneously to sinus rhythm within 7 days (usually less than 48 h), persistent AF, which lasts for more than 7 days, long‐standing persistent (chronic) AF (cAF), which lasts for over 1 year, and permanent forms, for which no further attempts are made to restore sinus rhythm (Kirchhof et al. 2016). Although our knowledge about the pathophysiology of AF has expanded greatly during the last 20 years, particularly about the cellular and molecular basis of AF‐promoting atrial remodelling, the precise mechanisms leading to AF have remained elusive (Heijman et al. 2014a).
Independent of the underlying cause, which may be clinically diverse, it is believed that abnormal impulse formation (focal ectopic activity) and reentry are the two major determinants of AF initiation and maintenance, although the precise clinical correlates of these fundamental mechanisms are not uniformly validated and are somewhat controversial (Schotten et al. 2016). In the clinical setting atrial ectopy may occur as a result of autonomic nervous system imbalance/altered innervation, acute ischaemia, wall stress (mechanosensing), and other macroscopic determinants of arrhythmogenesis (Andrade et al. 2014). A growing body of evidence suggests that abnormal intracellular Ca2+ handling may play a role in both the initiation of AF episodes and cellular remodelling processes that drive AF progression to more persistent forms. This review article will provide a critical examination of the experimental evidence that cellular ryanodine receptor type 2 (RyR2)‐mediated triggered activity may contribute to the pathophysiology of AF.
Basic mechanisms of triggered activity in AF
Triggered activity can be caused at the cellular level by early or delayed afterdepolarizations (EADs and DADs, respectively). EADs involve a secondary depolarization before final repolarization of the primary action potential as a result of reopening of L‐type Ca2+ channels and in rare cases by reactivation of fast Na+ currents. On the other hand, DADs occur after completion of repolarization of the primary action potential as a result of spontaneous Ca2+ release events from the sarcoplasmic reticulum (SR). These SR Ca2+ release events through ryanodine receptor/intracellular Ca2+ release channels (RyR2) activate the Na+/Ca2+‐exchanger (NCX), which brings three Na+ ions in for each Ca2+ ion pumped out of the cell, producing a depolarizing transient inward current that can cause a triggered action potential leading to focal ectopic firing (Voigt et al. 2012; Heijman et al. 2014a). Repetitive focal ectopic activity may sustain AF even in the absence of an AF‐maintaining substrate. On the other hand, focal ectopic firing can also initiate a reentrant circuit that can maintain AF. Reentry is considered the main mechanism for AF maintenance, and can occur around anatomical obstacles or can be functional in nature (Heijman et al., 2012, 2016).
Role of calcium in triggered activity in AF
Contraction of the atria is initiated by electrical depolarization of the myocytes, which initiates opening of voltage‐dependent L‐type Ca2+ channels. The influx of Ca2+ into the cytosol then triggers an amplified release of Ca2+ from the SR via RyR2 Ca2+ release channels (McCauley & Wehrens, 2011). The elevated cytosolic Ca2+ levels induce sarcomere shortening and atrial contraction. Relaxation occurs when cytosolic Ca2+ concentrations are brought back to diastolic levels by pumping Ca2+ back into the SR via the SR Ca2+‐ATPase 2a (SERCA2a), or by extruding Ca2+ through NCX on the plasma membrane. The amplitude of Ca2+‐induced Ca2+ release depends on proper alignment of L‐type Ca2+ channel (LTCC) on the plasma membrane and RyR2 on the SR by a structural protein known as junctophilin‐2 (JPH2) (Landstrom et al. 2014). In addition, JPH2 binding to RyR2 prevents abnormal diastolic SR Ca2+ leak (Beavers et al. 2013). The open probability of RyR2 is also regulated by phosphorylation by protein kinase A (PKA) and Ca2+/calmodulin‐dependent protein kinase II (CaMKII) and dephosphorylation by protein phosphatase 1 (PP1) in atrial myocytes.
Abnormal SR Ca2+ leak has emerged as a potential contributor to cellular triggered activity in right atrial cardiomyocytes of both pAF and cAF patients (Voigt et al. 2012, 2014; Heijman et al. 2014b; Abstract figure). In patients with pAF, an increased frequency of spontaneous SR Ca2+ release events was observed even though sinus rhythm was present at the time of tissue collection excluding rapid atrial rate‐induced remodelling as an underlying cause (Beavers et al. 2013; Voigt et al. 2014). The underlying molecular substrate was found to involve enhanced SR Ca2+ loading resulting from increased SERCA2a activity (Voigt et al. 2014). In addition, increased protein expression and open probability of RyR2 channels contributed to the increased frequency of spontaneous SR Ca2+ release events and related DADs in pAF (Voigt et al. 2014). Increased expression of RyR2 has been attributed to reduced levels of the inhibitory microRNA‐106b‐25 cluster in pAF patients (Chiang et al. 2014a). Moreover, a reduction in the ratio between RyR2‐stabilizing protein JPH2 and RyR2 may contribute to an increased RyR2 channel open probability, although the precise mechanisms of SR Ca2+ leak in pAF patients remain incompletely understood (Beavers et al. 2013). Further research is needed to elucidate the precise molecular mechanisms underlying the dysfunction of RyR2 and the related propensity to cellular triggered activity in pAF patients, and to validate whether these potentially proarrhythmic cellular events indeed contribute to the initiation and recurrence of AF episodes in pAF patients in vivo.
In patients with cAF, Ca2+‐handling abnormalities and DAD‐mediated cellular triggered activity have also been demonstrated in right atrial cardiomyocytes from patients undergoing cardiac surgery (Hove‐Madsen et al. 2004; Liang et al. 2009; Neef et al. 2010; Voigt et al. 2012). At the molecular level, enhanced phosphorylation or oxidation of RyR2 along with an increased RyR2 open probability have been proposed to cause Ca2+‐handling abnormalities in cAF patients (Vest et al. 2005; Voigt et al. 2012; Fischer et al. 2015; Xie et al. 2015). Studies in a mouse model of AF revealed a potential role for CaMKII in the development of triggered activity in AF (Chelu et al. 2009). The critical role of dysfunctional RyR2 for cellular triggered activity was confirmed in several other small and large animal models (Neef et al. 2010; Nishida et al. 2011; Li et al. 2012; Shan et al. 2012; Kettlewell et al. 2013; Purohit et al. 2013; Faggioni et al. 2014; Guo et al. 2014; Macquaide et al. 2015; Zhang et al. 2015), as well as human atrial cardiomyocytes isolated from cAF patients (Neef et al. 2010; Voigt et al. 2012). In addition, RyR2 may be more leaky due to increased PKA‐mediated phosphorylation although there is less evidence supporting this notion (Vest et al. 2005, Voigt et al. 2012; Li et al. 2012). Finally, reduced activity of protein phosphatases that dephosphorylate RyR2 can also contribute to enhanced phosphorylation of RyR2 and increased SR Ca2+ leak (Chiang et al. 2014b, 2015). Thus, extensive evidence from animal models and patients support a potential role of SR Ca2+ leak and associated cellular DAD‐mediated triggered activity in patients with persistent AF.
Finally, there is emerging evidence that SR Ca2+ leak may directly promote the progression of AF to more persistent forms. In a mouse model of spontaneous progressive AF, inhibition of RyR2 phosphorylation (and thus, enhanced RyR2‐mediated SR Ca2+ leak) was shown to prevent the development of a substrate for AF maintenance despite the presence of atrial triggered activity (Li et al. 2014). These findings suggest that RyR2‐mediated SR Ca2+ leak drives Ca2+‐dependent remodelling pathways (such as the calcineurin/NFAT pathway) that contribute to the formation of AF‐promoting atrial remodelling and an arrhythmogenic substrate for AF persistence. The role of Ca2+‐dependent remodelling in AF progression has been discussed in greater detail in another recent review and still needs to be evaluated in large animal models and patients (Heijman et al. 2014a).
Controversies regarding the role of SR Ca2+ leak in AF pathophysiology
An ideal way to test the hypothesis that SR Ca2+ leak though RyR2 is responsible for triggered activity in patients with AF would be to conduct a clinical trial in which highly specific pharmacological RyR2 channel modulators would be tested. To the best of our knowledge, such clinical trials have not been conducted to date. Several groups have developed RyR2 channel modulators – some more RyR2‐specific than others – that have been tested in animal models and tissue samples from patients with heart disease (Nakaya et al. 2000; Kumagai et al. 2003; Wehrens et al. 2004; Loughrey et al. 2007; Toischer et al. 2010; Sadrpour et al. 2015). For example, 1,4‐benzothiazepine derivative K201 (JT519) was shown to terminate atrial flutter and AF in canine models of sterile pericarditis (Kumagai et al. 2003; Sadrpour et al. 2015). This particular compound has many off‐target effects in addition to RyR2 inhibition. Further studies are needed to test whether the aforementioned and newer generation RyR2 inhibitors can actually suppress triggered activity in patients with AF.
There is strong clinical evidence that genetic gain‐of‐function defects in RyR2 – in patients with ‘catecholaminergic polymorphic ventricular tachycardia’ (CPVT) and otherwise normal hearts – develop cardiac arrhythmias including AF (Paavola et al. 2007). Although initially neglected, it is now established that individuals with CPVT have a higher propensity to both ventricular and atrial arrhythmias including AF. Most important, DADs were recorded in a patient with CPVT, which demonstrated for the first time that genetic RyR2 dysfunction might directly cause atrial triggered activity in the clinical setting (Paavola et al. 2007). Future studies are needed to elucidate whether gain‐of‐function defects of RyR2 in patients with non‐genetic forms of AF also contribute to DADs and ectopic activity.
Because of better clinical availability, most of the work dealing with RyR2 dysfunction was performed in right atrial cardiomyocytes from patients with AF (Hove‐Madsen et al. 2004; Vest et al. 2005; Neef et al. 2010; Voigt et al. 2012; Beavers et al. 2013; Voigt et al. 2014). Although it has been a widely accepted notion that the right atrium is not a major source for AF induction and maintenance, this assumption may be incorrect. Right atrium‐located AF drivers/rotors are detectable in up to 30–50% of patients with AF and in ex vivo perfused human hearts and in some patients only ablation of such right atrium‐located drivers stops the arrhythmia (Hocini et al. 2010; Hasebe et al. 2016; Li et al. 2016; Spitzer et al. 2017). This is solid evidence that the right atrium can be a relevant source for clinical AF and accordingly studies performed in human right atrial tissue clearly provide valuable insights into the molecular basis of AF. In addition, RyR2‐mediated SR Ca2+ leak was recently demonstrated in left atrial cardiomyocytes from AF patients (Fischer et al. 2015), making it likely that the RyR2 abnormalities detected in right atrial cardiomyocytes from patients with AF are not restricted to the right atrium only (Hove‐Madsen et al. 2004; Vest et al. 2005; Neef et al. 2010; Voigt et al. 2012; Beavers et al. 2013; Voigt et al. 2014). Additional work in human left atrial tissue is clearly needed to further validate the role of RyR2 abnormalities for AF pathophysiology.
A few recent studies of human tissue samples and animal models have suggested that silencing of intracellular Ca2+ handling rather than increased SR Ca2+ leak contribute to AF pathogenesis (Greiser et al. 2014). Greiser et al. (2014) showed that there was no increase in SR Ca2+ leak despite PKA hyperphosphorylation of serine 2808 on RyR2 in rabbits with atrial tachycardia remodelling but otherwise normal atria. In their model, 5 days of atrial tachypacing at 10 Hz led to a severe downregulation of RyR2 protein levels, which according to computational modelling might offset the effects of RyR2 hyperphosphorylation on SR Ca2+ leak. However, such a downregulation of RyR2 has not been reported in AF patients, suggesting that this model has limited clinical relevance or represents a disease stage not previously characterized in patients or healthy individuals. In addition, the reduction in intracellular [Na+] observed in this rabbit model and dogs with atrial tachycardia (Akar et al. 2003) is inconsistent with findings in human atrial cardiomyocytes from cAF patients, in which a significantly (3‐fold) elevated intracellular [Na+] has been reported (Hammer et al. 2016), although this finding needs further independent validation. Finally, rabbits with chronic myocardial infarction‐induced atrial remodelling and dogs with congestive heart failure show DAD‐mediated triggered activity and AF (Yeh et al. 2008; Kettlewell et al. 2013), which is in line with the phenotype of patients with AF outlined above and the emerging evidence that atrial cardiomyopathy is a critical determinant of the increased propensity to pAF and more persistent forms of AF (Goette et al. 2017).
Some authors have reported reduced, rather than enhanced, spontaneous arrhythmic activity in multicellular atrial trabeculae from patients with cAF compared with patients with sinus rhythm (Sossalla et al. 2010; Christ et al. 2014). However, the study of Sossalla et al. (2010) was not specifically designed to study DAD‐mediated triggered activity and one possible explanation for the opposite results of Christ et al. (2014) is that they incubated the trabeculae with phenoxybenzamine, a non‐selective, irreversible α‐adrenoceptor blocker which inhibits calmodulin and L‐type Ca2+ channels, among other things (Gengo et al. 1984; Cimino & Weiss, 1988). Irreversible inhibition of calmodulin could impact intracellular Ca2+ release due to inhibition of various channels, including L‐type Ca2+ channels, Na+ channels and RyR2. Therefore, the fact that abnormal aftercontractions (as used as an index of arrhythmias) were not more prevalent in atrial trabeculae from cAF patients as compared to sinus rhythm patients does not disprove the importance of DAD‐mediated triggered activity (Christ et al. 2014). If anything, it is very likely that these experiments rather proved that calmodulin inhibition is a more effective antiarrhythmic strategy in multicellular preparations from cAF patients compared to patients in sinus rhythm. Moreover, contraction of multicellular atrial preparations is probably not a suitable readout for proarrhythmic events, particularly when atrial contraction itself is strongly impaired as commonly observed in AF patients (Schotten et al. 2001; Wettwer et al. 2004).
In their study, Christ et al. (2014) did not observe an increased rate of spontaneous SR Ca2+ release events between the regular L‐type Ca2+‐current (I Ca,L)‐stimulated Ca2+ transients (CaTs), even under conditions of increased SR Ca2+ load with β‐adrenoceptor activation, an observation which is in line with the results of Voigt et al. (2012) and Beavers et al. (2013). In order to increase the susceptibility to spontaneous CaT generation, Voigt et al. (2012) used 5 mm instead of 2 mm extracellular Ca2+ (which increases SR Ca2+ load) and clamped the cardiomyocytes at −80 mV (mimicking diastolic conditions) after SR Ca2+ loading with regular I Ca,L stimulation. These conditions – which simulate the situation of a pause after a run of atrial tachycardia – unmasked the higher propensity to spontaneous SR Ca2+ release events in atrial cardiomyocytes of cAF patients compared to controls. Moreover these findings were also validated in the current‐clamp configuration (with physiological extracellular Ca2+) where DADs more frequently occurred in atrial cardiomyocytes from cAF patients compared to controls. Clearly independent verification in additional cohorts of AF patients using similar patch clamp protocols is needed to further validate the presence and role of cellular triggered activity in human AF.
Conclusions
There is a growing body of evidence that abnormal RyR2 activity and enhanced SR Ca2+ leak may cause DADs and cellular triggered activity in animal models of and patients with AF. The molecular mechanisms underlying SR Ca2+ leak, in particular RyR2 dysfunction, appear to depend on the clinical stage of AF or the specific animal model studied. Further assessment of single RyR2 channel properties, comprehensive mapping of post‐translational modifications of RyR2, recording of cellular DADs and evaluation of multicellular preparations from patients with different forms of AF are needed to gain a better understanding of the molecular basis of triggered activity in AF and to validate the role and importance of focal ectopic firing for atrial arrhythmogenesis. Ultimately, the only way to test the contribution of dysfunctional RyR2 channels to macroscopic AF mechanisms in suitable cohorts of AF patients is to employ highly selective RyR2 inhibitors, which are being developed by several groups but to the best of our knowledge have not been tested yet in patients with AF. Work in perfused intact human atria as successfully employed in a very recent study could be the next intermediate step in the translation of findings obtained in a cellular context to the clinical setting (Li et al. 2016).
Additional information
Competing interests
X.H.T.W. is a founding partner of Elex Biotech, a start‐up company that develops drug molecules that target ryanodine receptors for the treatment of cardiac arrhythmia disorders. D.D. is consultant for OMEICOS Therapeutics, which develops drug molecules targeting the ω‐fatty acid metabolism as an antiarrhythmic therapeutic strategy.
Author contributions
Both authors have approved the final version of the manuscript and agree to be accountable for all aspects of the work. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
D.D. is supported by DZHK (German Centre for Cardiovascular Research) the German Research Foundation (DO 769/4‐1), and a National Institutes of Health grant (NIH R01‐HL131517); X.H.T.W. is supported National Institutes of Health grants R01‐HL089598, R01‐HL091947, R01‐HL117641 and R41‐HL129570, and American Heart Association grant 13EIA14560061.
Biography
Xander Wehrens did his MD and PhD degrees at Maastricht University in the Netherlands, and a postdoctoral fellowship with Andrew Marks at Columbia University. In 2005, he moved as a faculty member to Baylor College of Medicine in Houston, TX, where has been a full professor since 2011, and director of the Cardiovascular Research Institute since 2012. His research lab focuses on mechanisms underlying altered intracellular calcium handling in heart disease. Dobromir Dobrev did his MD degree at Department of Pharmacology, Dresden University of Technology, Germany. In 2010, he moved to Heidelberg University (Germany) to become full professor and chair of Division of Experimental Cardiology. Since 2012 he is full professor of pharmacology and toxicology and director of Institute of Pharmacology, West German Heart and Vascular Center, University Duisburg‐Essen, Essen, Germany. His research focuses on the molecular mechanisms of arrhythmias, particularly of atrial fibrillation.

Both authors contributed equally.
Contributor Information
Dobromir Dobrev, Email: dobromir.dobrev@uk-essen.de.
Xander H. T. Wehrens, Email: wehrens@bcm.edu.
References
- Akar JG, Everett TH, Ho R, Craft J, Haines DE, Somlyo AP & Somlyo AV (2003). Intracellular chloride accumulation and subcellular elemental distribution during atrial fibrillation. Circulation 107, 1810–1815. [DOI] [PubMed] [Google Scholar]
- Andrade J, Khairy P, Dobrev D & Nattel S (2014). The clinical profile and pathophysiology of atrial fibrillation: relationships among clinical features, epidemiology, and mechanisms. Circ Res 114, 1453–1468. [DOI] [PubMed] [Google Scholar]
- Beavers DL, Wang W, Ather S, Voigt N, Garbino A, Dixit SS, Landstrom AP, Li N, Wang Q, Olivotto I, Dobrev D, Ackerman MJ & Wehrens XH (2013). Mutation E169K in junctophilin‐2 causes atrial fibrillation due to impaired RyR2 stabilization. J Am Coll Cardiol 62, 2010–2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chelu MG, Sarma S, Sood S, Wang S, van Oort RJ, Skapura DG, Li N, Santonastasi M, Muller FU, Schmitz W, Schotten U, Anderson ME, Valderrabano M, Dobrev D & Wehrens XH (2009). Calmodulin kinase II‐mediated sarcoplasmic reticulum Ca2+ leak promotes atrial fibrillation in mice. J Clin Invest 119, 1940–1951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang DY, Kongchan N, Beavers DL, Alsina KM, Voigt N, Neilson JR, Jakob H, Martin JF, Dobrev D, Wehrens XH & Li N (2014a). Loss of microRNA‐106b‐25 cluster promotes atrial fibrillation by enhancing ryanodine receptor type‐2 expression and calcium release. Circ Arrhythm Electrophysiol 7, 1214–1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang DY, Lebesgue N, Beavers DL, Alsina KM, Damen JM, Voigt N, Dobrev D, Wehrens XH & Scholten A (2015). Alterations in the interactome of serine/threonine protein phosphatase type‐1 in atrial fibrillation patients. J Am Coll Cardiol 65, 163–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang DY, Li N, Wang Q, Alsina KM, Quick AP, Reynolds JO, Wang G, Skapura D, Voigt N, Dobrev D & Wehrens XH (2014b). Impaired local regulation of ryanodine receptor type 2 by protein phosphatase 1 promotes atrial fibrillation. Cardiovasc Res 103, 178–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christ T, Rozmaritsa N, Engel A, Berk E, Knaut M, Metzner K, Canteras M, Ravens U & Kaumann A (2014). Arrhythmias, elicited by catecholamines and serotonin, vanish in human chronic atrial fibrillation. Proc Natl Acad Sci USA 111, 11193–11198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cimino M & Weiss B (1988). Characteristics of the binding of phenoxybenzamine to calmodulin. Biochem Pharmacol 37, 2739–2745. [DOI] [PubMed] [Google Scholar]
- Faggioni M, Savio‐Galimberti E, Venkataraman R, Hwang HS, Kannankeril PJ, Darbar D & Knollmann BC (2014). Suppression of spontaneous Ca elevations prevents atrial fibrillation in calsequestrin 2‐null hearts. Circ Arrhythm Electrophysiol 7, 313–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer TH, Herting J, Mason FE, Hartmann N, Watanabe S, Nikolaev VO, Sprenger JU, Fan P, Yao L, Popov AF, Danner BC, Schondube F, Belardinelli L, Hasenfuss G, Maier LS & Sossalla S (2015). Late I Na increases diastolic SR‐Ca2+‐leak in atrial myocardium by activating PKA and CaMKII. Cardiovasc Res 107, 184–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gengo PJ, Yousif F, Janis RA & Triggle DJ (1984). Interaction of phenoxybenzamine with muscarinic receptors and calcium channels. Biochem Pharmacol 33, 3445–3449. [DOI] [PubMed] [Google Scholar]
- Goette A, Kalman JM, Aguinaga L, Akar J, Cabrera JA, Chen SA, Chugh SS, Corradi D, D'Avila A, Dobrev D, Fenelon G, Gonzalez M, Hatem SN, Helm R, Hindricks G, Ho SY, Hoit B, Jalife J, Kim YH, Lip GY, Ma CS, Marcus GM, Murray K, Nogami A, Sanders P, Uribe W, Van Wagoner DR & Nattel S (2017). EHRA/HRS/APHRS/SOLAECE expert consensus on atrial cardiomyopathies: Definition, characterization, and clinical implication. Heart Rhythm 14, e3–e40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greiser M, Kerfant BG, Williams GS, Voigt N, Harks E, Dibb KM, Giese A, Meszaros J, Verheule S, Ravens U, Allessie MA, Gammie JS, van der Velden J, Lederer WJ, Dobrev D & Schotten U (2014). Tachycardia‐induced silencing of subcellular Ca2+ signalling in atrial myocytes. J Clin Invest 124, 4759–4772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo X, Yuan S, Liu Z & Fang Q (2014). Oxidation‐ and CaMKII‐mediated sarcoplasmic reticulum Ca2+ leak triggers atrial fibrillation in aging. J Cardiovasc Electrophysiol 25, 645–652. [DOI] [PubMed] [Google Scholar]
- Hammer K, Maier A, Winkelmann H, Wagner S & Maier LS (2016). Intracellular Na+ is increased in atrial myocytes from patients with atrial fibrillation. Clin Res Cardiol 105, doi 10.1007/s00392-016-0967-z (abstract). [Google Scholar]
- Hasebe H, Yoshida K, Iida M, Hatano N, Muramatsu T & Aonuma K (2016). Right‐to‐left frequency gradient during atrial fibrillation initiated by right atrial ectopies and its augmentation by adenosine triphosphate: Implications of right atrial fibrillation. Heart Rhythm 13, 354–363. [DOI] [PubMed] [Google Scholar]
- Heijman J, Algalarrondo V, Voigt N, Melka J, Wehrens XH, Dobrev D & Nattel S (2016). The value of basic research insights into atrial fibrillation mechanisms as a guide to therapeutic innovation: a critical analysis. Cardiovasc Res 109, 467–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heijman J, Voigt N, Nattel S & Dobrev D (2012). Calcium handling and atrial fibrillation. Wien Med Wochenschr 162, 287–291. [DOI] [PubMed] [Google Scholar]
- Heijman J, Voigt N, Nattel S & Dobrev D (2014a). Cellular and molecular electrophysiology of atrial fibrillation initiation, maintenance, and progression. Circ Res 114, 1483–1499. [DOI] [PubMed] [Google Scholar]
- Heijman J, Voigt N, Wehrens XH & Dobrev D (2014b). Calcium dysregulation in atrial fibrillation: the role of CaMKII. Front Pharmacol 5, 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hocini M, Nault I, Wright M, Veenhuyzen G, Narayan SM, Jais P, Lim KT, Knecht S, Matsuo S, Forclaz A, Miyazaki S, Jadidi A, O'Neill MD, Sacher F, Clementy J & Haissaguerre M (2010). Disparate evolution of right and left atrial rate during ablation of long‐lasting persistent atrial fibrillation. J Am Coll Cardiol 55, 1007–1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hove‐Madsen L, Llach A, Bayes‐Genis A, Roura S, Rodriguez Font E, Aris A & Cinca J (2004). Atrial fibrillation is associated with increased spontaneous calcium release from the sarcoplasmic reticulum in human atrial myocytes. Circulation 110, 1358–1363. [DOI] [PubMed] [Google Scholar]
- Kettlewell S, Burton FL, Smith GL & Workman AJ (2013). Chronic myocardial infarction promotes atrial action potential alternans, afterdepolarizations, and fibrillation. Cardiovasc Res 99, 215–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirchhof P, Benussi S, Kotecha D, Ahlsson A, Atar D, Casadei B, Castella M, Diener HC, Heidbuchel H, Hendriks J, Hindricks G, Manolis AS, Oldgren J, Popescu BA, Schotten U, Van Putte B, Vardas P, Agewall S, Camm J, Baron Esquivias G, Budts W, Carerj S, Casselman F, Coca A, De Caterina R, Deftereos S, Dobrev D, Ferro JM, Filippatos G, Fitzsimons D, Gorenek B, Guenoun M, Hohnloser SH, Kolh P, Lip GY, Manolis A, McMurray J, Ponikowski P, Rosenhek R, Ruschitzka F, Savelieva I, Sharma S, Suwalski P, Tamargo JL, Taylor CJ, Van Gelder IC, Voors AA, Windecker S, Zamorano JL & Zeppenfeld K (2016). 2016 ESC Guidelines for the management of atrial fibrillation developed in collaboration with EACTS. Europace 18, 1609–1678. [DOI] [PubMed] [Google Scholar]
- Kumagai K, Nakashima H, Gondo N & Saku K (2003). Antiarrhythmic effects of JTV‐519, a novel cardioprotective drug, on atrial fibrillation/flutter in a canine sterile pericarditis model. J Cardiovasc Electrophysiol 14, 880–884. [DOI] [PubMed] [Google Scholar]
- Landstrom AP, Beavers DL & Wehrens XH (2014). The junctophilin family of proteins: from bench to bedside. Trends Mol Med 20, 353–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li N, Chiang DY, Wang S, Wang Q, Sun L, Voigt N, Respress JL, Ather S, Skapura DG, Jordan VK, Horrigan FT, Schmitz W, Muller FU, Valderrabano M, Nattel S, Dobrev D & Wehrens XH (2014). Ryanodine‐receptor mediated calcium leak drives progressive development of an atrial fibrillation substrate in a transgenic mouse model. Circulation 129, 1276–1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li N, Csepe TA, Hansen BJ, Sul LV, Kalyanasundaram A, Zakharkin SO, Zhao J, Guha A, Van Wagoner DR, Kilic A, Mohler PJ, Janssen PM, Biesiadecki BJ, Hummel JD, Weiss R & Fedorov VV (2016). Adenosine‐induced atrial fibrillation: localized reentrant drivers in lateral right atria due to heterogeneous expression of adenosine A1 receptors and GIRK4 subunits in the human heart. Circulation 134, 486–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li N, Wang T, Wang W, Cutler MJ, Wang Q, Voigt N, Rosenbaum DS, Dobrev D & Wehrens XH (2012). Inhibition of CaMKII phosphorylation of RyR2 prevents induction of atrial fibrillation in FKBP12.6 knockout mice. Circ Res 110, 465–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang X, Xie H, Zhu PH, Hu J, Zhao Q, Wang CS & Yang C (2009). Enhanced activity of inositol‐1,4,5‐trisphosphate receptors in atrial myocytes of atrial fibrillation patients. Cardiology 114, 180–191. [DOI] [PubMed] [Google Scholar]
- Loughrey CM, Otani N, Seidler T, Craig MA, Matsuda R, Kaneko N & Smith GL (2007). K201 modulates excitation‐contraction coupling and spontaneous Ca2+ release in normal adult rabbit ventricular cardiomyocytes. Cardiovasc Res 76, 236–246. [DOI] [PubMed] [Google Scholar]
- Macquaide N, Tuan HT, Hotta J, Sempels W, Lenaerts I, Holemans P, Hofkens J, Jafri MS, Willems R & Sipido KR (2015). Ryanodine receptor cluster fragmentation and redistribution in persistent atrial fibrillation enhance calcium release. Cardiovasc Res 108, 387–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCauley MD & Wehrens XH (2011). Targeting ryanodine receptors for anti‐arrhythmic therapy. Acta Pharmacol Sin 32, 749–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakaya H, Furusawa Y, Ogura T, Tamagawa M & Uemura H (2000). Inhibitory effects of JTV‐519, a novel cardioprotective drug, on potassium currents and experimental atrial fibrillation in guinea‐pig hearts. Br J Pharmacol 131, 1363–1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neef S, Dybkova N, Sossalla S, Ort KR, Fluschnik N, Neumann K, Seipelt R, Schondube FA, Hasenfuss G & Maier LS (2010). CaMKII‐dependent diastolic SR Ca2+ leak and elevated diastolic Ca2+ levels in right atrial myocardium of patients with atrial fibrillation. Circ Res 106, 1134–1144. [DOI] [PubMed] [Google Scholar]
- Nishida K, Qi XY, Wakili R, Comtois P, Chartier D, Harada M, Iwasaki YK, Romeo P, Maguy A, Dobrev D, Michael G, Talajic M & Nattel S (2011). Mechanisms of atrial tachyarrhythmias associated with coronary artery occlusion in a chronic canine model. Circulation 123, 137–146. [DOI] [PubMed] [Google Scholar]
- Paavola J, Viitasalo M, Laitinen‐Forsblom PJ, Pasternack M, Swan H, Tikkanen I, Toivonen L, Kontula K & Laine M (2007). Mutant ryanodine receptors in catecholaminergic polymorphic ventricular tachycardia generate delayed afterdepolarizations due to increased propensity to Ca2+ waves. Eur Heart J 28, 1135–1142. [DOI] [PubMed] [Google Scholar]
- Purohit A, Rokita AG, Guan X, Chen B, Koval OM, Voigt N, Neef S, Sowa T, Gao Z, Luczak ED, Stefansdottir H, Behunin AC, Li N, El‐Accaoui RN, Yang B, Swaminathan PD, Weiss RM, Wehrens XH, Song LS, Dobrev D, Maier LS & Anderson ME (2013). Oxidized Ca2+/calmodulin‐dependent protein kinase II triggers atrial fibrillation. Circulation 128, 1748–1757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadrpour SA, Serhal M, Khrestian CM, Lee S, Fields T, Dittrich HC & Waldo AL (2015). Termination of atrial flutter and fibrillation by K201's metabolite M‐II: studies in the canine sterile pericarditis model. J Cardiovasc Pharmacol 65, 494–499. [DOI] [PubMed] [Google Scholar]
- Schotten U, Ausma J, Stellbrink C, Sabatschus I, Vogel M, Frechen D, Schoendube F, Hanrath P & Allessie MA (2001). Cellular mechanisms of depressed atrial contractility in patients with chronic atrial fibrillation. Circulation 103, 691–698. [DOI] [PubMed] [Google Scholar]
- Schotten U, Dobrev D, Platonov PG, Kottkamp H & Hindricks G (2016). Current controversies in determining the main mechanisms of atrial fibrillation. J Intern Med 279, 428–438. [DOI] [PubMed] [Google Scholar]
- Shan J, Xie W, Betzenhauser M, Reiken S, Chen BX, Wronska A & Marks AR (2012). Calcium leak through ryanodine receptors leads to atrial fibrillation in 3 mouse models of catecholaminergic polymorphic ventricular tachycardia. Circ Res 111, 708–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sossalla S, Fluschnik N, Schotola H, Ort KR, Neef S, Schulte T, Wittkopper K, Renner A, Schmitto JD, Gummert J, El‐Armouche A, Hasenfuss G & Maier LS (2010). Inhibition of elevated Ca2+/calmodulin‐dependent protein kinase II improves contractility in human failing myocardium. Circ Res 107, 1150–1161. [DOI] [PubMed] [Google Scholar]
- Spitzer SG, Karolyi L, Rammler C, Scharfe F, Weinmann T, Zieschank M & Langbein A (2017). Treatment of recurrent non‐paroxysmal atrial fibrillation using focal impulse and rotor mapping (FIRM)‐guided rotor ablation: early recurrence and long‐term outcomes. J Cardiovasc Electrophysiol 28, 31–38. [DOI] [PubMed] [Google Scholar]
- Toischer K, Lehnart SE, Tenderich G, Milting H, Korfer R, Schmitto JD, Schondube FA, Kaneko N, Loughrey CM, Smith GL, Hasenfuss G & Seidler T (2010). K201 improves aspects of the contractile performance of human failing myocardium via reduction in Ca2+ leak from the sarcoplasmic reticulum. Basic Res Cardiol 105, 279–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vest JA, Wehrens XH, Reiken SR, Lehnart SE, Dobrev D, Chandra P, Danilo P, Ravens U, Rosen MR & Marks AR (2005). Defective cardiac ryanodine receptor regulation during atrial fibrillation. Circulation 111, 2025–2032. [DOI] [PubMed] [Google Scholar]
- Voigt N, Heijman J, Wang Q, Chiang DY, Li N, Karck M, Wehrens XH, Nattel S & Dobrev D (2014). Cellular and molecular mechanisms of atrial arrhythmogenesis in patients with paroxysmal atrial fibrillation. Circulation 129, 145–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voigt N, Li N, Wang Q, Wang W, Trafford AW, Abu‐Taha I, Sun Q, Wieland T, Ravens U, Nattel S, Wehrens XH & Dobrev D (2012). Enhanced sarcoplasmic reticulum Ca2+ leak and increased Na+‐Ca2+ exchanger function underlie delayed afterdepolarizations in patients with chronic atrial fibrillation. Circulation 125, 2059–2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wehrens XH, Lehnart SE, Reiken SR, Deng SX, Vest JA, Cervantes D, Coromilas J, Landry DW & Marks AR (2004). Protection from cardiac arrhythmia through ryanodine receptor‐stabilizing protein calstabin2. Science 304, 292–296. [DOI] [PubMed] [Google Scholar]
- Wettwer E, Hala O, Christ T, Heubach JF, Dobrev D, Knaut M, Varro A & Ravens U (2004). Role of I Kur in controlling action potential shape and contractility in the human atrium: influence of chronic atrial fibrillation. Circulation 110, 2299–2306. [DOI] [PubMed] [Google Scholar]
- Xie W, Santulli G, Reiken SR, Yuan Q, Osborne BW, Chen BX & Marks AR (2015). Mitochondrial oxidative stress promotes atrial fibrillation. Sci Rep 5, 11427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh YH, Wakili R, Qi XY, Chartier D, Boknik P, Kaab S, Ravens U, Coutu P, Dobrev D & Nattel S (2008). Calcium‐handling abnormalities underlying atrial arrhythmogenesis and contractile dysfunction in dogs with congestive heart failure. Circ Arrhythm Electrophysiol 1, 93–102. [DOI] [PubMed] [Google Scholar]
- Zhang H, Cannell MB, Kim SJ, Watson JJ, Norman R, Calaghan SC, Orchard CH & James AF (2015). Cellular hypertrophy and increased susceptibility to spontaneous calcium‐release of rat left atrial myocytes due to elevated afterload. PLoS One 10, e0144309. [DOI] [PMC free article] [PubMed] [Google Scholar]