

Abstract
Stimulation of β‐adrenergic receptors (βARs) provides the most efficient physiological mechanism to enhance contraction and relaxation of the heart. Activation of βARs allows rapid enhancement of myocardial function in order to fuel the muscles for running and fighting in a fight‐or‐flight response. Likewise, βARs become activated during cardiovascular disease in an attempt to counteract the restrictions of cardiac output. However, long‐term stimulation of βARs increases the likelihood of cardiac arrhythmias, adverse ventricular remodelling, decline of cardiac performance and premature death, thereby limiting the use of βAR agonists in the treatment of heart failure. Recently the endogenous Raf kinase inhibitor protein (RKIP) was found to activate βAR signalling of the heart without adverse effects. This review will summarize the current knowledge on RKIP‐driven compared to receptor‐mediated signalling in cardiomyocytes. Emphasis is given to the differential effects of RKIP on β1‐ and β2‐ARs and their downstream targets, the regulation of myocyte calcium cycling and myofilament activity.
Keywords: beta‐adrenergic receptors, heart failure, RKIP
Abbreviations
- AAV
adeno‐associated virus
- AC6
adenylyl cyclase 6
- βAR
β‐adrenergic receptor
- βARKct
β‐adrenergic receptor kinase, C‐terminus
- CaMKII
Ca2+/calmodulin‐dependent protein kinase II
- cAMP
cyclic adenosine monophosphate
- cMyBPC
cardiac myosin binding protein C
- Epac
exchange protein directly activated by cAMP
- ERK1/2
extracellular signal‐regulated protein kinases 1 and 2
- GPCR
G‐protein‐coupled receptor
- GRK
G‐protein‐coupled receptor kinase
- Gi
inhibitory G‐protein
- Gs
stimulatory G‐protein
- KO
knock‐out
- LTCC
L‐type Ca2+ channel
- MEK
mitogen‐activated protein kinase kinase
- NCX
sodium–calcium exchanger
- PDEIII
phosphodiesterase III
- PEBP
phosphatidylethanolamine‐binding protein
- PKA
protein kinase A
- PKC
protein kinase C
- PLN
phospholamban
- PMCA
sarcolemmal Ca2+‐ATPase
- RKIP
Raf kinase inhibitor protein
- RyR2
ryanodine receptor 2
- S100A1
S100 calcium binding protein A1
- SERCA2a
sarco‐/endoplasmic reticulum Ca2+‐ATPase
- SR
sarcoplasmic reticulum
- TnI
troponin I
Introduction
Heart failure occurs if cardiac output is reduced to an extent that it cannot meet the body's needs. It represents one of the leading causes of morbidity and mortality in developed countries and results from the loss and/or dysfunction of cardiomyocytes due to literally any insult to the heart, most frequently chronic arterial hypertension, myocardial infarction, aortic stenosis or infectious diseases. The activation of the sympathetic nervous system via β‐adrenergic receptors (βARs) is the most important compensatory mechanism in heart failure, acting to stabilize the hemodynamic situation by accelerating cardiac contraction and relaxation (Ponikowski et al. 2016).
Free calcium ions (Ca2+) are the critical intermediary that translates sympathetic activity into myofilament movement. Changes of beat‐to‐beat myocyte Ca2+ cycling are also among the hallmarks of heart failure. Further, local Ca2+ release events (Ca2+ sparks) and/or altered Ca2+ sensitivity of cardiomyocytes contribute to contractile dysfunction and increase the risk of cardiac arrhythmias in failing hearts as well as in diseases like inherited cardiomyopathy or early after myocardial infarction (Cho et al. 2016). The profound effects on cardiac function of even small modifications to elements of the βAR signalling cascade, the receptor cascade that controls Ca2+ handling to the myofilaments, necessitates precise regulation of the entire system.
In the treatment of heart failure, pharmacological activation of βARs is beneficial in acute situations due to its ability to rapidly increase cardiac output. However, sustained activation of βARs is detrimental to the heart; it promotes cardiomyocyte death and myocardial fibrosis and increases patient mortality (Engelhardt et al. 1999; Tacon et al. 2012; Ponikowski et al. 2016). A strategy that would increase cardiac output, but without the adverse effects of chronic βAR stimulation, is still lacking. This review will discuss novel therapeutic approaches aimed at selective activation of specific components of βAR signalling with a main focus on the Raf kinase inhibitor protein (RKIP). Upon phosphorylation by protein kinase C (PKC), RKIP potentiates βAR signalling through inhibition of receptor desensitization, which has proven beneficial effects on myocyte Ca2+ regulation and murine heart failure (Lorenz et al. 2003; Schmid et al. 2015).
βAR signalling in the heart
Sympathetic activity is transmitted to cardiac muscle via neuronally and circulating catecholamines that predominantly activate βARs on cardiomyocytes as those are the receptor subtypes with the highest density in the ventricular myocardium. Two different βAR subtypes are expressed in the heart: β1‐ and β2‐adrenergic receptors, at a ratio of 80:20. Stimulation of cardiac βARs mediates an increase in contractile force (positive inotropy), speed of relaxation (positive lusitropy), atrioventricular conduction (positive dromotropy) and heart rate (positive chronotropy) (Bristow et al. 1986; Brodde 1991; Jensen et al. 2009). β3ARs are a third subtype of cardiomyocyte βAR. Their role in the heart, however, is still largely unclear. They induce distinct intracellular signalling pathways and a negative inotropic effect. Since their expression is increased in several subtypes of human cardiomyopathy, they may have a potential role in heart failure. Mice with cardiac β3AR overexpression showed reduced hypertrophic remodelling through nitric oxide synthase activation (Balligand 2013; Belge et al. 2014).
β1‐ and β2ARs couple to stimulatory G‐proteins (Gs) that stimulate adenylyl cyclases to produce the second messenger cyclic adenosine monophosphate (cAMP), which in turn activates the cAMP‐dependent protein kinase A (PKA). In cardiomyocytes, regulators of beat‐to‐beat Ca2+ cycling and sarcomere proteins represent major substrates of PKA. Activation of PKA causes phosphorylation of L‐type Ca2+ channels (LTCCs). This increases Ca2+ influx; phosphorylation of phospholamban (PLN), which accelerates the reuptake of Ca2+ into the sarcoplasmatic reticulum (SR); phosphorylation of ryanodine receptors 2 (RyR2), which increases SR Ca2+ release; phosphorylation of troponin I (TnI) and of cardiac myosin binding protein C (cMyBPC), which decreases myofilament Ca2+ sensitivity; and phosphorylation of titin, which reduces the sarcomeric passive stiffness (Lefkowitz et al. 2002; Rockman et al. 2002; Krüger & Linke, 2006; Baker, 2014; Najafi et al. 2016). Taken together, these PKA‐mediated phosphorylation events enhance Ca2+ cycling and reduce myofilament Ca2+ sensitivity in cardiomyocytes, leading to the increases in force and increases in the rates of contraction and of relaxation. Myocyte Ca2+ is also important for the formation of the Ca2+–calmodulin complex, which activates Ca2+/calmodulin‐dependent protein kinase II (CaMKII), a kinase that also impacts on Ca2+ homeostasis by phosphorylation of RyR2, PLN or cMyBPC, thereby further potentiating βAR‐mediated cardiac contraction and relaxation (Fig. 1; Rockman et al. 2002; Maier & Bers, 2007; Lehnart et al. 2009; Sadayappan et al. 2011; Uchinoumi et al. 2016). In addition, βARs activate an exchange protein directly activated by cAMP (Epac). Epac1 seems to contribute to cardiac hypertrophy and is upregulated in heart failure, whereas Epac2 seems to be involved in CaMKII‐induced SR Ca2+ leak and arrhythmia. Despite these detrimental effects, Epac has also been reported to promote cardiomyocyte survival in heart failure (Métrich et al. 2008, 2009; Pereira et al. 2013).
Figure 1. Acute dobutamine application induces positive inotropy; chronic dobutamine application deteriorates cardiac function.
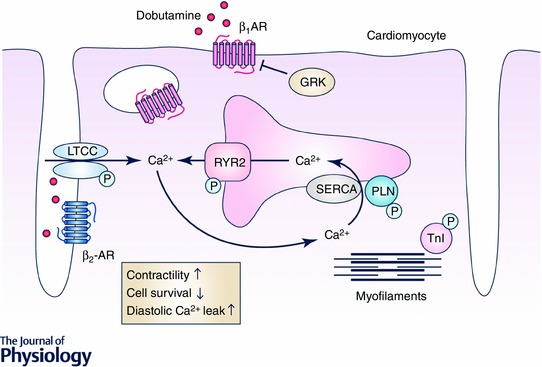
Dobutamine activates β1‐ and β2‐adrenergic receptors (β1AR and β2AR). Activated βARs increase contractility and relaxation of cardiomyocytes via the activation of stimulatory G‐proteins (Gs), which in turn activate protein kinase A and Ca2+/calmodulin‐dependent protein kinase II. These kinases increase Ca2+ cycling: upon phosphorylation, phospholamban (PLN) dissociates from sarco‐/endoplasmatic reticulum Ca2+‐ATPase (SERCA2a). This leads to increased SERCA2a‐mediated Ca2+ re‐uptake into the sarcoplasmatic reticulum and cardiomyocyte contractility. Phosphorylation of troponin I (TnI) decreases Ca2+ sensitivity and thereby increases cardiomyocyte relaxation. However, G‐protein‐coupled receptor kinase (GRK) phosphorylates activated G‐protein‐coupled receptors (GPCR) as for example β1AR and β2AR, which induces receptor desensitization and internalization. This blunts βAR signalling and the initial increase in cardiomyocyte contractility upon dobutamine application. Further, chronic βAR stimulation induces apoptosis, fibrosis and arrhythmia, in particular via hyperphosphorylation of the ryanodine receptor 2 (RyR2) and L‐type Ca2+ channels (LTCC), thereby leading to increased diastolic Ca2+ leak.
Thus, βARs, as major drivers of heart rate, contractile force, speed of contraction and relaxation, play an important role in so‐called fight‐or‐flight situations or whenever cardiac output needs to be enhanced. Analogously, βAR agonists such as adrenaline, dobutamine and dopamine are used to stabilize patients in acute cardiac failure (Felker, 2001; Tacon et al. 2012; Ponikowski et al. 2016). However, long‐lasting application of these β‐AR agonists for several days or even weeks induces structural cardiac damage, including cardiac hypertrophy, cardiomyocyte apoptosis and interstitial fibrosis (Fig. 1; Engelhardt et al. 1999; O'Connor, et al. 1999; Felker, 2001; Tacon et al. 2012; Vidal et al. 2012; Ponikowski et al. 2016). As a natural defence against this damage, prolonged activation of βAR leads to receptor desensitization via phosphorylation by G‐protein‐coupled receptor kinases (GRK), thereby protecting the heart from long‐term sympathetic overdrive. The predominant GRK subtype in the heart is GRK2. Phosphorylation of βARs by GRK2 increases the affinity of the receptor for β‐arrestin, a protein that blocks G‐protein coupling upon receptor stimulation and promotes receptor internalization and degradation, rendering myocytes less responsive to agonist binding of βARs (Rockman et al. 2002). However, such loss of βAR function also promotes contractile decline of failing hearts. This dilemma between the need for positive inotropy of failing hearts on one side and receptor desensitization to prevent cardiac damage upon sustained βAR activation on the other side pushes for a well‐synchronized, well‐balanced and fine‐tuned way to regulate βAR signalling in the heart.
βAR signalling in heart failure
βAR agonists augment cardiac contraction at the beginning of treatment. In contrast, sustained βAR stimulation is cardiotoxic, consistent with the finding that noradrenaline plasma levels correlate with the degree of cardiac dysfunction and mortality of heart failure patients (Thomas & Marks, 1987; Cohn et al. 1993; Zhang et al. 2013). Under conditions of increased sympathetic nervous system activation or chronic βAR agonist treatment, both βAR density at the surface of the cell membrane and the responsiveness of the remaining receptors are reduced. These molecular characteristics of failing hearts correlate well with the stage of heart failure independent of the underlying cause of the disease (Ohsuzu et al. 1994). The pattern of βAR subtype downregulation, however, seems to depend on the aetiology of heart failure: β1ARs but not β2ARs are downregulated in the majority of heart failure cases, but in mitral valve disease and ischaemic cardiomyopathy both βAR subtypes are affected to a similar extent (Brodde et al. 1986; Bristow et al. 1991; Steinfath et al. 1991, 1992).
Toxic effects mediated through βAR activation appear to originate from β1ARs, because cardiac overexpression of β1ARs in mice led to cardiac hypertrophy, interstitial fibrosis and cardiac dysfunction (Engelhardt et al. 1999; Zhang et al. 2013). Furthermore, β1ARs mediate pro‐apoptotic signalling through the kinases PKA and CaMKII. For example, selective inhibition of β1AR resulted in protection of catecholamine‐induced apoptosis in rat ventricular myocytes (Zaugg et al. 2000; Shizukuda & Buttrick, 2002).
β2ARs in contrast, have been described as cardioprotective receptors (Liggett et al. 1998; Siedlecka et al. 2008). Cardiac overexpression of β2ARs in mice prevented myocardial remodelling and contractile dysfunction in a genetic model of heart failure generated by Gαq overexpression (Dorn et al. 1999). However, favourable effects were achieved only at relatively low levels of β2AR overexpression, whereas higher expression levels turned out deleterious, suggesting that specificity of β2AR signalling must be preserved to achieve beneficial effects via this activation. In addition, β2AR overexpression of up to 60‐fold was tolerated in ageing mouse hearts without detriment for a period of at least 1 year (Liggett et al. 2000). Further, selective β2AR activation protected from stress‐induced apoptosis in isolated cardiomyocytes as well as from myocardial dysfunction and apoptosis in a rat model of heart failure (Paur et al. 2012) and mice lacking β2ARs had a higher mortality than wild‐type mice in response to chronic isoproterenol application (Patterson et al. 2004). Also, in human heart failure an Ile164 polymorphism in the β2AR, which reduces its signalling efficiency, was found to worsen patients’ prognosis (Liggett et al. 1998). Beneficial effects of β2ARs in the heart are often associated with β2AR coupling to inhibitory G‐proteins (Gi). In line with this, the unfavourable outcome of the Ile164 polymorphism was suggested to result from the loss of β2ARs coupled to Gi and their protective effects on apoptosis (Chesley et al. 2000). On the other hand, enhanced β2AR–Gi signalling is also reported to contribute to cardiac deterioration in heart failure by further reducing cardiac contractility. Thus, several groups hypothesized that a combination of β1AR blockade with β2AR–Gs activation may be ideal for improving cardiac contractility without adverse effects (Ahmet et al. 2008; Woo & Xiao, 2012); others, however, suggested β1AR blockade combined with β2AR–Gi activation as the preferred strategy for heart failure therapy with particularly striking results in a model of Takotsubo cardiomyopathy (Siedlecka et al. 2008, clenbuterol as β2AR–Gi biased β2‐agonist; Paur et al. 2012). Takotsubo cardiomyopathy is characterized by ballooning and contractile dysfunction only of the apical portions of the heart in response to excessive emotional stress and subsequent exposure to high levels of catecholamines. Prevention of adrenaline‐mediated Gi effects increased mortality, thus providing strong evidence for the beneficial effects of Gi coupling in activating β2ARs. β2AR–Gi signalling may thus be essential to counteract hyperactivated β1AR–Gs signalling (Gorelik et al. 2013).
Finally, the general view of the β1AR as the ‘bad’ and the β2AR as the ‘good’ receptor in heart failure also has been challenged by the finding that deletion of β2AR was cardioprotective in a model of genetic cardiomyopathy. Deletion of β1AR in this particular mouse mutant was proposed to worsen the phenotype via a PKA‐independent pathway employing Epac (Fajardo et al. 2013; Zhang et al. 2013). In summary, even though chronic β1AR signalling is generally thought to be cardiotoxic and chronic β2AR signalling cardioprotective, the outcome of β1AR vs. β2AR activation depends at least partially on the underlying disease type.
Strategies in heart failure that target βAR signalling
While pharmacological stimulation of βARs is commonly used to stabilize a failing heart in an acute situation, blockage of βARs in chronic heart failure turned out to be beneficial due to disruption of the vicious circle between sympathetic overdrive and maladaptive remodelling processes. Unlike initial expectations from negative inotropic drugs, antagonists of βARs (β‐blockers) improve patients’ symptoms and significantly promote survival when applied carefully at slowly increasing dosages. Multiple studies within the last two decades have shown that β‐blockers improve survival for chronic heart failure by up to 30% (Packer et al. 1996a,b; Lechat et al. 1998). Low dosages of β‐blockers are sufficient to protect from sympathetic overdrive, thereby preventing β1AR‐mediated remodelling processes and restoring β‐adrenergic function by re‐sensitization and increased expression of βARs (Felker, 2001; Lompré et al. 2010; Tacon et al. 2012; Ponikowski et al. 2016). However, not all patients tolerate β‐blockers well and the withdrawal rate is high due to side effects like fatigue, sleep disturbance, depression, weight gain, pulmonary side effects and sexual dysfunction (Packer et al. 1996a,b). In heart failure, depression of cardiac contractility further hampers the use of β‐blockers in general or at least at the desired dose. The ideal drug in the treatment of heart failure would increase cardiac output and thereby instantly alleviate symptoms, but without the adverse effects of chronic βAR stimulation.
Several new experimental strategies have been added in recent years to increase cardiac contractility in heart failure by activation of βAR or modulation of βAR downstream signalling, particularly by targeting regulators of myocyte Ca2+ cycling. Most attempts to reconstitute βAR signalling failed, because they accelerated rather than attenuated deterioration of cardiac morphology and function. These studies evaluated the use of isoproterenol or dobutamine; the inhibition of phosphodiesterase III (PDEIII), an enzyme that degrades cAMP; activation of PKA; inhibition of protein phosphatase 1, an enzyme that reduces PKA‐mediated activation of calcium cycling proteins; or activation of CaMKII (El‐Armouche et al. 2008; Lehnart et al. 2009; Lompré et al. 2010; Tacon et al. 2012; Bers, 2014; Ponikowski et al. 2016).
Studies aiming at myocyte Ca2+ cycling yielded more promising results for the treatment of heart failure. Ca2+ coordinates myofilament activity in the contractile apparatus of the cardiac myocyte. Upon electrical stimulation, the concentration of Ca2+ in the contractile units increases at least 10‐fold, thereby inducing the formation of cross‐bridges between myofilaments. The subsequent conformational changes of the myosin head finally lead to myocardial contraction. Therefore, levels of cellular Ca2+ directly correlate with the heart's mechanical function and enhancing myocyte Ca2+ cycling increases mechanical force of the contractile units and the rate of contraction and relaxation. Potential therapeutic strategies were evaluated in animal models aiming at nodal points of the signalling cascade such as sarco‐/endoplasmatic reticulum Ca2+‐ATPase (SERCA2a), which plays an important role in diastolic Ca2+ removal. SERCA2a activity was modulated via deletion of the SERCA2a inhibitor PLN, overexpression of SERCA2a or overexpression of S100 calcium binding protein A1 (S100A1). Further, the LTCC was targeted using the Gβγ scavenger C‐terminus of the β‐adrenergic receptor kinase (βARKct), which leads to disinhibition of G‐protein (Gβγ)‐mediated inhibition of the channel (Slack et al. 2001; Schmitt et al. 2009; Pleger et al. 2011; Völkers et al. 2011; Kairouz et al. 2012). In healthy hearts, none of these strategies led to rapid deterioration of cardiac function and they all successfully rescued animal models of heart failure. The most‐progressed target, SERCA2a, was evaluated in patients with moderate to severe heart failure in the Calcium Upregulation by Percutaneous Administration of Gene Therapy in Cardiac Disease (CUPID) trial. In phase 1/2, intracoronary infusion of a recombinant adeno‐associated virus (AAV) vector for delivery of SERCA2a DNA appeared promising; however, in a follow‐up study that evaluated the effects on hospitalization and mortality, SERCA2a gene transfer turned out to be safe but did not improve the endpoints. Technical issues leading to inefficient cellular uptake of the viral vector are discussed as likely causes for the failure of the phase 2b CUPID trial. Further investigation of this trial is needed to avoid failure of future gene therapy trials (Pleger et al. 2014; Greenberg et al. 2014, 2016; Greenberg, 2015; Lother & Hein, 2016).
The underlying reasons why reconstitution of βAR signalling is particularly prone to cardiac damage but reconstitution of Ca2+ cycling is rather well‐tolerated or even protective are not yet understood. Remarkably, overexpression of adenylyl cyclase 6 (AC6) safely increased left ventricular function beyond standard heart failure therapy in a recently published phase 1/2 trial of AC6 gene transfer in heart failure patients (Pleger et al. 2014; Hammond et al. 2016). Unlike other AC subtypes, AC6 has no effect on basal cAMP levels and is only responsive to βAR stimulation suggesting that selective and non‐constitutive activation of βAR downstream targets may be crucial in distinguishing well‐tolerated from detrimental positive inotropy. AC6 is also thought to improve cardiac performance via cAMP‐independent mechanisms that still need to be elucidated (Gao et al. 2002; Tang et al. 2012).
Recently, RKIP was suggested as a promising strategy to stimulate cardiac contractility and to reconstitute βAR signalling of failing hearts by chronic β1AR activation without triggering adverse effects. Unlike AC6, RKIP enhances adrenergic signalling in cardiomyocytes at a different level. RKIP attenuates GRK2 activity and thereby produces a balanced activation of β1ARs and β2ARs. The following discussion will summarize the potential benefits of this differential activation in failing cardiomyocytes and evaluate RKIP as a therapeutic agent against heart failure. We will further discuss the effects of RKIP on key components of downstream βAR signalling, particularly myocyte Ca2+ kinetics, diastolic Ca2+ leak and myofilament Ca2+ sensitivity since they show characteristic alterations in failing hearts that lead to contractile dysfunction and arrhythmia.
RKIP – a governor of intracellular signalling
RKIP belongs to the evolutionarily conserved phosphatidylethanolamine‐binding protein (PEBP) family, which has been characterized as a modulator of signal transduction cascades in mammalian cells and has been reviewed in detail by Trakul & Rosner (2005), Granovski & Rosner (2008) and Lorenz et al. (2014a). PEBP/RKIP proteins possess a central β‐sheet surrounded by smaller β‐strands and two carboxy‐terminal α‐helices. These structural elements are connected by loops of variable length. Characteristic for this family is a cavity at the surface that consists of dynamically arranged amino acid residues and displays high affinity for small anionic groups such as phosphates, phospholipids and nucleotides (Hengst, 2000; Granovski & Rosner, 2008; Granovski et al. 2009). This cavity is also implicated in the binding of RKIP to the kinase Raf‐1 to the extent that reduced flexibility of the cavity favours Raf binding (Granovski et al. 2009). Raf‐1 is a member of the Raf–mitogen‐activated protein kinase kinase (MEK)–extracellular signal‐regulated protein kinases 1 and 2 (ERK1/2) cascade that is involved in differentiation, proliferation, cell survival and hypertrophy. RKIP has been shown to inhibit Raf‐1 signalling (Yeung et al. 1999), but the mechanism is not yet entirely clarified. While overexpressed RKIP has been postulated to interfere with the interaction of Raf‐1 with its substrate, MEK, endogenous RKIP rather interferes with Raf‐1 activation (Trakul & Rosner, 2005). Phosphorylation of RKIP by protein PKC at serine 153 mediates the release of Raf‐1 from RKIP (Corbit et al. 2003; Lorenz et al. 2003; Deiss et al. 2012). Interestingly, serine 153 phosphorylation triggers an additional mechanistic/structural feature with impact on the control of RKIP interaction partners: it induces RKIP dimerization. A loop structure at the surface of RKIP and in immediate proximity to the PKC phosphorylation site was identified as a part of the dimerization interface (Deiss et al. 2012). RKIP dimerization facilitates the release of Raf‐1 but also participates in the substrate switch of RKIP from Raf‐1 to GRK2 since inhibition of RKIP dimerization prevented RKIP/GRK2 binding and, vice versa, a dimeric RKIP mutant was able to bind GRK2 in the absence of RKIPSer153 phosphorylation (Deiss et al. 2012). As mentioned above, GRK2 is a kinase that phosphorylates activated G‐protein‐coupled receptors (GPCRs), thereby initiating their desensitization and internalization and subsequently blunting receptor signalling (Pierce et al. 2002). In mammalian cells, GRK2 is a major feedback inhibitor of GPCRs and has been implicated in diseases such as immune diseases or heart failure. RKIP does not inhibit the catalytic activity of GRK2 but interferes with the GRK2–receptor interaction via its binding to the N‐terminus of GRK2, a part of GRK2 that is important for GRK–receptor interaction (Lorenz et al. 2003). This inhibitory mechanism of RKIP enables a largely specific interference of RKIP with GRK2 towards receptor substrates while cytosolic substrates of GRK2 are not affected (Schmid et al. 2015). GRK2–RKIP interaction prevents GPCR internalization leading to enhanced GPCR signalling, which, in the heart, enhances contraction and relaxation.
Even though other kinase signalling cascades such as the nuclear factor κ‐light‐chain‐enhancer of activated B‐cells (NFκB) and glycogen synthase kinase‐3β are also known to be regulated by RKIP in cultured cells, thus far only Raf‐1, MEK1 and ERK2 as well as GRK2 have been identified as direct interaction partners of RKIP of which only Raf‐1 and GRK2 have been validated under endogenous conditions (Yeung et al. 2001; Lorenz et al. 2009, 2014b). In line with its influence on several kinase signalling cascades, RKIP impacts on diverse physiological processes including cell transformation, cell cycle, inflammation, metastasis and cardiomyocyte contractility (Granovsky & Rosner, 2008; Lorenz et al. 2014a; Brietz et al. 2016). Deletion or downregulation of RKIP resulted for example in deterioration of metastatic cancer, Alzheimer's disease, pulmonary hypertension and heart failure and increased replication of the Newcastle disease virus (Lorenz et al. 2014a; Schmid et al. 2015; Yin et al. 2016).
RKIP and its function in the heart
Cardiac RKIP expression is up‐regulated in heart failure patients and in mice with pressure overload‐induced heart failure, which implies that RKIP is part of the physiological response to stress in cardiac diseases. Indeed, mice with cardiac overexpression of RKIP are protected from heart failure induced by chronic pressure overload (induced by transverse aortic constriction) while RKIP deficiency exaggerated heart failure under these conditions. AAV9‐mediated gene transfer protected wild‐type and RKIP knockout mice from transverse aortic constriction‐induced heart failure (Schmid et al. 2015). Recent findings strongly suggest that RKIP provides a new and well‐tolerated mode of sustained βAR activation in the heart by differential stimulation of protective vs. detrimental β‐adrenergic signalling (Schmid et al. 2015; Fig. 2). The data indicate that the effects of RKIP in the heart are characterized by the following qualities:
-
(1)
RKIP stimulates β1AR–Gs signalling, which results in enhanced contraction and relaxation via increased PLN and TnI phosphorylation and subsequently increased SERCA2a activity, higher SR Ca2+ load and decreased Ca2+ sensitivity of myofilaments. Cardiac contractility of RKIP‐overexpressing mice was improved compared to control animals up to an age of at least 12–14 months; and lifespan of RKIP‐overexpressing mice under these conditions was at least as long as of non‐transgenic mice (Schmid et al. 2015).
-
(2)
Despite enhanced β1AR–Gs signalling, RKIP‐stimulated hearts are still able to respond adequately to physiological stress situations because the size of the dobutamine response of RKIP‐overexpressing and wild‐type hearts is similar. This moderate or submaximal activation may play an important role for the observed reduction of cardiomyocyte apoptosis, interstitial fibrosis, brain natriuretic peptide and collagen expression in RKIP‐overexpressing mice compared to wild‐type controls and the overall well‐tolerated positive inotropic phenotype of RKIP‐overexpressing mice (Schmid et al. 2015).
-
(3)
Besides β1AR–Gs‐signalling that stimulates the activity of both PKA and CaMKII, RKIP activates β2AR–Gi in mouse hearts. The simultaneous activation of β2AR–Gi within the transverse (t)‐tubular region prevents the adverse effects of mere β1AR–Gs, such as diastolic Ca2+ leak and cardiac arrhythmia due to hyperphosphorylation and subsequent activation of the RyR2 or hyperphosphorylation of the LTCC. These RKIP effects appear to be mediated by β2AR–Gi signalling since this protection is absent in RKIP‐overexpressing mice lacking β2ARs and in the presence of the Gi inhibitor pertussis toxin (Communal et al. 1999; Xiao et al. 1999; Lehnart et al. 2009; Eschenhagen, 2010; Nikolaev et al. 2010; Bers, 2014; Schmid et al. 2015). Of note, these experiments show that RKIP predominantly activates β2AR coupled to Gi in mouse hearts, even though RKIP in principle is capable of activating β2AR–Gs as demonstrated in cell cultures (Lorenz et al. 2003). Further evidence for a central role of β2AR on the protective effects of RKIP in the heart is the absence of protection from cardiac remodelling, i.e. apoptosis and interstitial fibrosis, in RKIP transgenic mice lacking the β2AR as well as a reduced overall survival compared to β2KO controls – effects that are reported to result from mere β1AR–Gs signalling (Schmid et al. 2015). The switch of β2AR from Gs to Gi in RKIP transgenic mice seemed to be due to the enduring β1AR–Gs–PKA activation as indicated by the characteristic phosphorylation patterns of βAR downstream targets (Daaka et al. 1997; Xiao et al. 1999). Remarkably, overall β2AR phosphorylation is significantly reduced in RKIP transgenic mice, consistent with GRK2 inhibition and absence of β2AR desensitization in RKIP transgenic mice (Rockmann et al. 2002; Houslay & Baillie, 2005; Schmid et al. 2015).
-
(4)
RKIP was found to inhibit βAR downregulation and thereby secures sustained positive inotropy, which is not achieved by direct agonist‐mediated βAR stimulation (Lorenz et al. 2003; Schmid et al. 2015) (Figs 1 and 2).
-
(5)
RKIP promotes cell survival. RKIP overexpression reduced cardiomyocyte apoptosis, whereas deletion of RKIP (RKIP−/−) dramatically increased it. This effect is β2AR dependent since the protection from apoptosis is absent in RKIP‐transgenic mice lacking the β2AR. Interestingly, β2AR–Gi is known to stimulate the kinase Akt, which in turn mediates anti‐apoptotic effects (Chesley et al. 2000; Talan et al. 2011). In line with β2AR–Gi activation by RKIP, Akt activation was enhanced in RKIP‐overexpressing mice and was dependent on β2AR and pertussis toxin‐sensitive Gi proteins. These findings suggest that RKIP mediates cell survival via Akt.
Figure 2. RKIP induces positive inotropy and protects from cell death and diastolic Ca2+ leak.
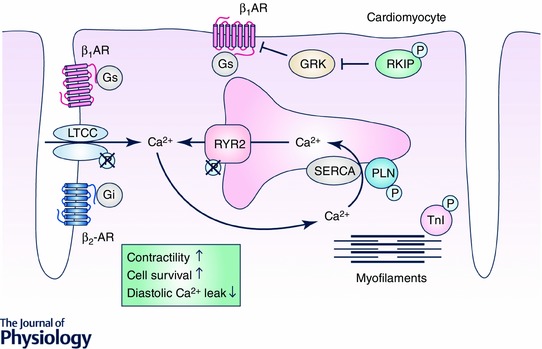
The Raf kinase inhibitor protein (RKIP) binds GRK2 and inhibits G‐protein‐coupled receptor kinase (GRK)‐mediated receptor phosphorylation, which prevents receptor desensitization and internalization and, thus, increases β‐adrenergic receptor signalling. RKIP increases contractility and relaxation of cardiomyocytes via activated β1‐adrenergic receptor (β1AR) coupled to stimulatory G‐proteins (Gs): phosphorylated phospholamban (PLN) dissociates from sarco‐/endoplasmatic reticulum Ca2+‐ATPase (SERCA2a) and thereby increases SERCA2a activity, Ca2+ loading of the sarcoplasmatic reticulum and cardiomyocyte contractility. Phosphorylation of troponin I (TnI) decreases Ca2+ sensivity and thereby increases cardiomyocyte relaxation. RKIP mediates anti‐apoptotic, anti‐fibrotic and anti‐arrhythmic effects via increased β2‐adrenergic receptor (β2AR) signalling. Continuous signalling of β2AR coupled to inhibitory G‐proteins (Gi) prevents β1AR‐stimulated increases in ryanodine receptor 2 (RyR2) and L‐type Ca2+ channel (LTCC) phosphorylation and protects from diastolic Ca2+ leak.
However, as described above, RKIP not only increases GPCR signalling via GRK inhibition, but also inhibits mitogen‐activated protein kinase signalling dependent on its phosphorylation status: RKIP acts as a GRK inhibitor in its PKC phosphorylated form (pRKIPSer153), but in the absence of Ser153 phosphorylation it acts as a Raf‐1 inhibitor. In the heart, Raf–MEK–ERK1/2 signalling promotes cell survival (Punn et al. 2000; Harris et al. 2004; Heineke & Molkentin, 2006; Purcell et al. 2007; Sheikh et al. 2008; Cheng et al. 2011; Van Berlo et al. 2011). Thus, in its unphosphorylated form, RKIP could potentially increase cardiomyocyte death. However, RKIP in the heart mainly exists in its phosphorylated form, so that the Raf‐1 inhibitory effect of RKIP is absent in the heart. Even moderate RKIP overexpression of up to 8‐fold revealed no inhibitory effect of RKIP on Raf‐1/MEK/ERK1/2. Potential side effects of this new cardioprotective strategy may occur at very high overexpression levels of RKIP that exceed the ability of PKC to fully phosphorylate RKIP and may result in RKIP‐mediated Raf/MEK/ERK1/2 inhibition and increased apoptosis associated with signs of heart failure as seen by Fu et al. or by cardiac overexpression of a phosphorylation‐deficient mutant of RKIP, RKIPS153A (Bueno et al. 2000; Lorenz et al. 2003; Fu et al. 2013; Ruppert et al. 2013; Schmid et al. 2015).
Compared to other positive inotropic strategies in heart failure therapy, the biochemical and phenotypic effects of RKIP substantiate the hypothesis that a successful positive inotropic strategy should not induce an unselective activation of βAR downstream targets (as for βAR agonists, PDEIII inhibitors or β1AR‐overexpressing mice) but rather circumvent activation of the RyR2 (as in PLN–/– mice, SERCA2a or βARKct overexpression) or even protect from RyR2 sensitization (as in GRK2–/– mice, S100A1 and RKIP transgenic mice) (Kairouz et al. 2012; Respress et al. 2012; Bers, 2014; Pleger et al. 2014; Ritterhoff et al. 2015). RKIP achieves this well‐tolerated βAR stimulation with positive inotropy and lusitropy in RKIP‐overexpressing mice by concomitant activation of β2ARs (in their Gi‐coupled mode) that counteracts several maladaptive β1AR effects such as RyR2 sensitization, diastolic Ca2+ leaks and arrhythmia as well as apoptosis and fibrosis. Indeed, ‘sole’ β1AR activation as provided in RKIP transgenic mice lacking the β2AR (β2KO) increased cardiac contractility, but also reduced overall survival of ageing RKIP transgenic mice (RKIP/β2KO) compared to β2KO controls (Schmid et al. 2015). Taken together, the consequences of enhanced βAR signalling in the heart appear to be highly dependent on the type and the extent of activated signalling elements.
In sum, RKIP differentially modulates several molecular events downstream of βAR, which appears promising for heart failure therapy. In the following, we will discuss the differential regulation of βAR receptors by RKIP, its effects on myocyte Ca2+ kinetics and distribution and how failing hearts may benefit from these alterations.
Depressed myocyte Ca2+ cycling and heart disease
Ca2+ enters the cardiomyocyte via the LTCCs, which are predominantly located within the t‐tubuli of the sarcolemma in close neighbourhood to the sarcoplasmic Ca2+ release channels, RyR2. These functional dyads facilitate the rapid increase of cytosolic Ca2+ levels upon depolarization of the cell leading to myofilament contraction. Cardiac relaxation is initiated by Ca2+ removal from the cytosol. In human myocytes, 74% of diastolic Ca2+ removal is accomplished by the Ca2+‐ATPase SERCA2a, 24% by the Na+/Ca2+ exchanger (NCX), 1% by the sarcolemmal Ca2+‐ATPase (PMCA) and 1% by the mitochondrial Ca2+ uniporter) (Bers, 2014). Since SERCA2a eliminates the largest share (even 93% of Ca2+ removal in myocytes from mice and rats), the SERCA2a regulator PLN plays a pivotal role in modulating myocyte Ca2+ distribution and kinetics. PLN inhibits SERCA2a, thereby attenuating the rate of Ca2+ transport to the SR. Phosphorylation of PLN by PKA at serine 16 can almost fully relieve this inhibitory effect leading to a pronounced acceleration of SR Ca2+ uptake. As Ca2+ is sequestered by the SR, in the sarcomeres TnI inhibition of actin–myosin interactions is re‐established and myocytes relax (Rockman et al. 2002).
In heart failure, depressed contractility is associated with depressed myocyte Ca2+ cycling. Although the causes of heart failure can vary widely, e.g. myocardial infarction, arterial hypertension, infections or genetic defects, the pattern of abnormal Ca2+ metabolism is relatively uniform (overview in Lehnart et al. 2009). In failing hearts, Ca2+ release is typically reduced, consistent with a decrease in contractility and force generation (van der Velden et al. 2004; Avner et al. 2011; Haghighi et al. 2014). Further, SR Ca2+ reuptake during diastole is slow and diastolic Ca2+ levels are elevated due to a diastolic Ca2+ leak (via RyR2), but primarily due to reduced SERCA2a activity. Depression of the Ca2+ pump results from reduced SERCA2a expression in failing hearts, whereas PLN levels remain stable leading to a reduced SERCA/PLN ratio (Hasenfuss & Pieske, 2002). SR Ca2+ transport is further inhibited by a reduction of PLN phosphorylation, most likely as a result of increased protein phosphatase‐1 activity and downregulation of βAR density (Weber et al. 2016).
The identification of inherited mutations in Ca2+ regulatory proteins that caused alterations of protein function and induced dilated cardiomyopathy and terminal heart failure finally proved the concept that the Ca2+ cycling alterations in failing hearts are not secondary events or bystander in the course of the disease, but play a causative role in myocardial remodelling and the deterioration of cardiac function (Haghighi et al. 2003; Schmitt et al. 2003). Therefore, restoration of SR Ca2+ cycling holds promise for the treatment of heart failure.
RKIP seems to provide a promising approach for the restoration of depressed Ca2+ cycling. It increases myocyte Ca2+ transients at baseline and also upon bolus application of caffeine indicative of increased Ca2+ release. Increased Ca2+ release during systole in RKIP‐overexpressing cardiomyocytes is most likely due to accelerated Ca2+ reuptake into the SR during diastole, which is mainly due to enhanced PLN phosphorylation at serine 16 (PKA site) and threonine 17 (CaMKII site) leading to efficient release of PLN from SERCA2a and subsequent activation of SERCA2a. The RKIP‐induced increase in Ca2+ reuptake during diastole may further be supported by an accelerated Ca2+ release from the myofilaments mediated by enhanced TnI phosphorylation at the serine residues 23 and 24. Further, via GRK inhibition, RKIP prevents βAR desensitization and βAR degradation in a heart under chronic sympathetic stress, which subsequently secures efficient and continuous Ca2+ cycling. In addition, RKIP overexpression is able to prevent a loss of SERCA2a expression in a failing mouse heart, which in turn also ensures effective Ca2+ cycling in cardiomyocytes.
In sum, RKIP improves cardiac performance in healthy hearts and in failing hearts. Since the extent of the contractile response depends on the amount of activating Ca2+, the enhanced Ca2+ load would explain the hypercontractile phenotype of RKIP‐overexpressing hearts and the improved cardiac function in a mouse model of heart failure due to chronic pressure overload (Schmid et al. 2015). RKIP is an elegant example of achieving a stable and physiological (i.e. still regulatable) increase in Ca2+ cycling on several molecular levels via restoring expression and function of both βAR and direct regulators of myocyte Ca2+ cycling.
Increased Ca2+ sensitivity and heart disease
Ca2+ sensitivity of myofilaments affects contraction, relaxation and remodelling of the myocardium as well as cardiac rhythm. Increased Ca2+ sensitivity was observed in end‐stage heart failure and in heart tissue 3–4 days after myocardial infarction (van der Velden et al. 2004; Avner et al. 2011). Further, in hypertrophic cardiomyopathy, the most frequent cause of sudden cardiac death in the young population, an increase of myofilament Ca2+ sensitivity has been proposed as a central disease mechanism (Landstrom & Ackerman 2012; Deftereos et al. 2016). Increased Ca2+ sensitivity is often associated with high susceptibility for ventricular tachycardia and sudden cardiac death. Desensitization of myofilaments to Ca2+ was suggested to reduce the risk of arrhythmias by stabilizing action potential generation and propagation, because high Ca2+ sensitivity would prolong Ca2+ transients and slow down the propagation of action potentials, thereby fostering the generation of electrical re‐entry (Huke & Knollmann, 2010; Tardiff et al. 2015). The causative relation to arrhythmia was underlined by human and animal studies that found increased episodes of ventricular tachycardia after myocardial infarction and in heart failure upon treatment with the Ca2+ sensitizer levosimendan (Flevari et al. 2006). In contrast, the myosin inhibitor blebbistatin reduced myofilament Ca2+ sensitivity and prevented ventricular tachycardia in troponin T mutant mice (Baudenbacher et al. 2008). These examples demonstrate the broad therapeutic possibilities of Ca2+ desensitizing agents to fight arrhythmias and myocardial remodelling as well as contractile dysfunction.
Schmid et al. (2015) found RKIP to increase TnI phosphorylation at S23/S24. This PKA‐dependent phosphorylation decreases Ca2+ sensitivity of the Tn complex because phosphorylation reduces its Ca2+ affinity (Cheng et al. 2015). The strong therapeutic potential of decreasing Ca2+ sensitivity by TnI phosphorylation was demonstrated by the expression of a pseudo‐phosphorylated TnI mutant that rescued the morphological and functional changes of the heart in an animal model of hypertrophic cardiomyopathy caused by an E180G α‐Tm mutant with increased myofilament Ca2+ sensitivity (Alves et al. 2014). RKIP appears as a particularly attractive tool for Ca2+ desensitization of myofilaments because it also exhibits positive inotropy via enhanced β1AR signalling and antiarrhythmic effects via reduced RyR2 and LTCC phosphorylation, and it prevents apoptosis and maladaptive remodelling by Akt stimulation.
Diastolic Ca2+ leak and heart disease
Besides desensitization of myofilaments by increasing TnI phosphorylation, Schmid et al. (2015) also showed RKIP to reduce the frequency of Ca2+ sparks and Ca2+ waves. Ca2+ sparks occur if a cluster of RyR2 produces a local Ca2+ release from the SR. With every heartbeat, the action potential synchronizes the almost simultaneous opening of thousands of RyR2 clusters within a myocyte leading to a Ca2+ transient that initiates contraction. In contrast, the local Ca2+ increase caused by a spontaneous Ca2+ spark can trigger Ca2+ release only from neighbouring RyR2 clusters via a Ca2+‐induced Ca2+ release. The resulting propagating wave may induce Ca2+ elimination by the NCX causing an inward current (1 Ca2+ out–3 Na+ in) and both early and delayed afterdepolarizations that trigger aberrant electrical activity and arrhythmias of the heart (reviewed in Bers, 2014). The clinical relevance of this pathomechanism was demonstrated by the finding of disease‐causing RyR2 mutations in patients with catecholaminergic polymorphic ventricular tachycardia, because the genetic defects put affected individuals at risk for stress‐induced ventricular tachycardia (Priori & Chen, 2011). Not only do Ca2+ sparks trigger arrhythmia, but the diastolic loss of Ca2+ also reduces SR Ca2+ content. As a consequence of this loss, the systolic Ca2+‐induced Ca2+ release is smaller leading to reduced contraction of the heart. The Ca2+ leak also impairs myocardial relaxation, because it slows down cytosolic Ca2+ clearance during diastole of the heart and it may cause diastolic activation of contractile proteins.
RKIP reduces the frequency of Ca2+ sparks and Ca2+ waves by reducing phosphorylation of RyR2 at SS2808/2814 (Schmid et al. 2015). Hyperphosphorylation of RyR2 is known to induce diastolic SR Ca2+ leakage, which predisposes for arrhythmias (Bers, 2014). The respective role of S2808 and S2814 phosphorylation by PKA or CaMKII in this scenario, however, is controversial in the field (Eschenhagen, 2010; Bers, 2014). The reduction of RyR2 phosphorylation by RKIP despite enhancing β‐adrenergic signalling is explained by β2AR–Gi‐coupled signalling induced by RKIP (Schmid et al. 2015). Since β2ARs are believed to be concentrated within the t‐tubular region of the cardiomyocyte (Kuschel et al. 1999; Orchard & Brette, 2007; Nikolaev et al. 2010), β2AR–Gi signalling within the t‐tubular region seems to protect βAR targets in close proximity to β2ARs such as the RyR2 and LTCC from hypersensitization. It is noteworthy that the resulting stabilization of RyR2 is strong enough to reduce the incidence of Ca2+ sparks despite increased SR Ca2+ contents, a condition that can trigger spontaneous Ca2+ release.
Pharmacological and gene therapeutic studies are aimed at fixing the Ca2+ leak, increasing the Ca2+ transient and enhancing cytosolic Ca2+ clearance (reviewed in Marks, 2013). RKIP is a positive inotropic strategy that circumvents RyR2 sensitization and seems to fulfil all of these requirements. RKIP (1) reduces the occurrence of Ca2+ sparks by β2AR‐mediated stabilization of RyR2, (2) accelerates cytosolic Ca2+ elimination by PKA‐dependent phosphorylation of PLN and (3) increases SR Ca2+ filling and therefore the size of the Ca2+ transients. Finally, RKIP overexpression prevented morphological and functional maladaptation of the heart in mice subjected to long‐term pressure overload. Future studies will show if these beneficial effects hold true for the rescue of independent models of heart failure.
Conclusion
RKIP is an endogenous protein that exhibits a combination of favourable effects for heart failure patients: (1) the gain/increase of cardiac contractile efficiency by the activation of Gs signalling/β1AR leading to functional recovery of the heart and (2) the protection of the heart under sympathetic stress from exaggerated β1AR downstream signalling including protection from apoptosis and pro‐arrhythmic adverse effects via β2AR activation. This approach promises a new therapeutic strategy to achieve well‐tolerated long‐term increases in cardiac contractility. RKIP comprises several favourable characteristic effects on calcium cycling, calcium sensitivity, G‐protein recruitment to βARs and a physiological extent or range of βAR activation and has proven protective in murine heart failure. Future studies will further unravel the signalling network induced by RKIP that is responsible for the well‐tolerated mode of βAR activation and evaluate its therapeutic efficacy in various disease entities.
Additional information
Competing interests
The authors have no competing interests.
Author contributions
All authors have approved the final version of the manuscript and agreed to be accountable for all aspects of the work. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
This work was supported by Deutsche Forschungsgemeinschaft (DFG; Sonderforschungsbereich SFB688 (TPA17 to K.L., Fonds 881067) and SFB 1116 (TPA02 to J.P.S.)), by Bundesministerium für Bildung und Forschung (BMBF; Comprehensive Heart Failure Centre Würzburg; project MY1 to K.L., 98519102 (8502/0)) and by the Ministry for Innovation, Science and Research of the Federal State of North Rhine‐Westphalia (K.L.). A PhD position was awarded to T.B. by the Elite Network of Bavaria within the IDK ‘receptor dynamics’.
Biographies
Kristina Lorenz is the Director of the Biomedical Research Department at the Leibniz‐Institute of Analytical Sciences ‐ISAS‐ e.V and Professor for mechanisms in cardiovascular diseases at the University of Duisburg‐Essen. Her group investigates signaling pathways involved in heart failure and cardiac hypertrophy with major focus on mitogen‐activated protein kinase (MAPK) and G protein coupled receptor (GPCR) signaling.

Joachim Schmitt is a professor of cardiovascular pharmacology at the Heinrich‐Heine‐University in Düsseldorf, Germany. His group investigates pathomechanisms of heart diseases with a focus on myocyte calcium cycling and sarcomere function.
References
- Ahmet I, Krawczyk M, Zhu W, Woo AYH, Morrell C, Poosala S, Xiao RP, Lakatta EG & Talan MI (2008). Cardioprotective and survival benefits of long‐term combined therapy with β2 adrenoreceptor (AR) agonist and β1AR blocker in dilated cardiomyopathy postmyocardial infarction. J Pharmacol Exp Ther 325, 491–499. [DOI] [PubMed] [Google Scholar]
- Alves ML, Dias FA, Gaffin RD, Simon JN, Montminy EM, Biesiadecki BJ, Hinken AC, Warren CM, Utter MS, Davis RT, Sadayappan S, Robbins J, Wieczorek DF, Solaro RJ & Wolska BM. (2014) Desensitization of myofilaments to Ca2+ as a therapeutic target for hypertrophic cardiomyopathy with mutations in thin filament proteins. Circ Cardiovasc Genet 2, 123–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avner BS, Shioura KM, Scruggs SB, Grachoff M, Geenen DL, Helseth DL, Farjah M, Goldspink PH & Solaro JR (2011). Myocardial infarction in mice alters sarcomeric function via post‐translational protein modification. Mol Cell Biochem 363, 203–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker AJ (2014). Adrenergic signaling in heart failure: a balance of toxic and protective effects. Pflugers Arch 466, 1139–1150. [DOI] [PubMed] [Google Scholar]
- Balligand JL (2013). Beta3‐adrenoreceptors in cardiovasular diseases: new roles for an "old" receptor. Curr Drug Deliv 10, 64–66. [DOI] [PubMed] [Google Scholar]
- Baudenbacher F, Schober T, Pinto JR, Sidorov VY, Hilliard F, Solaro RJ, Potter JD & Knollmann BC (2008). Myofilament Ca2+ sensitization causes susceptibility to cardiac arrhythmia in mice. J Clin Invest 118, 3893–3903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belge C, Hammond J, Dubois‐Deruy E, Manoury B, Hamelet J, Beauloye C, Markl A, Pouleur AC, Bertrand L, Esfahani H, Jnaoui K, Götz KR, Nikolaev VO, Vanderper A, Herijgers P, Lobysheva I, Iaccarino G, Hilfiker‐Kleiner D, Tavernier G, Langin D, Dessy C & Balligand JL (2014). Enhanced expression of β3‐adrenoceptors in cardiac myocytes attenuates neurohormone‐induced hypertrophic remodeling through nitric oxide synthase. Circulation 129, 451‐462. [DOI] [PubMed] [Google Scholar]
- Bers DM (2014). Cardiac sarcoplasmic reticulum calcium leak: basis and roles in cardiac dysfunction. Annu Rev Physiol 76, 107–127. [DOI] [PubMed] [Google Scholar]
- Brietz A, Schuch KV, Wangorsch G, Lorenz K & Dandekar T (2016). Analyzing ERK 1/2 signalling and targets. Mol Biosyst 12, 2436–2446. [DOI] [PubMed] [Google Scholar]
- Bristow MR, Anderson FL, Port JD, Skerl L, Hershberger RE, Larrabee P, O'Connell JB, Renlund DG, Volkman K, Murray J & Feldman AM (1991). Differences in beta‐adrenergic neuroeffector mechanisms in ischemic versus idiopathic dilated cardiomyopathy. Circulation 84, 1024–1039. [DOI] [PubMed] [Google Scholar]
- Bristow MR, Ginsburg R, Umans V, Fowler M, Minobe W, Rasmussen R, Zera P, Menlove R, Shah P & Jamieson S (1986). β1‐ and β2‐Adrenergic‐receptor subpopulations in nonfailing and failing human ventricular myocardium: coupling of both receptor subtypes to muscle contraction and selective beta 1‐receptor down‐regulation in heart failure. Circ Res 59, 297–309. [DOI] [PubMed] [Google Scholar]
- Brodde OE (1991). β1‐ and β2‐Adrenoceptors in the human heart: properties, function, and alterations in chronic heart failure. Pharmacol Rev 43, 203–242. [PubMed] [Google Scholar]
- Brodde OE, Schüler S, Kretsch R, Brinkmann M, Borst HG, Hetzer R, Reidemeister JC, Warnecke H & Zerkowski HR (1986). Regional distribution of beta‐adrenoceptors in the human heart: coexistence of functional β1‐ and β2‐adrenoceptors in both atria and ventricles in severe congestive cardiomyopathy. J Cardiovasc Pharmacol 8, 1235–1242. [DOI] [PubMed] [Google Scholar]
- Bueno OF, De Windt LJ, Tymitz KM, Witt SA, Kimball TR, Klevitsky R, Hewett TE, Jones SP, Lefer DJ, Peng CF, Kitsis RN & Molkentin JD (2000). The MEK1‐ERK1/2 signaling pathway promotes compensated cardiac hypertrophy in transgenic mice. EMBO J 19, 6341–6350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng H, Kari G, Dicker AP, Rodeck U, Koch WJ & Force T (2011). A novel preclinical strategy for identifying cardiotoxic kinase inhibitors and mechanisms of cardiotoxicity. Circ Res 109, 1401–1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Y, Rao V, Tu A‐Y, Lindert S, Wang D, Oxenford L, McCulloch AD, McCammon JA & Regnier M (2015). Troponin I mutations R146G and R21C alter cardiac troponin function, contractile properties and modulation by PKA‐mediated phosphorylation. J Biol Chem 68, 3045–3119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chesley A, Lundberg MS, Asai T, Xiao RP, Ohtani S, Lakatta EG & Crow MT (2000) The β2‐adrenergic receptor delivers an antiapoptotic signal to cardiac myocytes through Gi‐dependent coupling to phosphatidylinositol 3′‐kinase. Circ Res 87, 1172–1179. [DOI] [PubMed] [Google Scholar]
- Cho GW, Altamirano F & Hill JA (2016). Chronic heart failure: Ca2+, catabolism, and catastrophic cell death. Biochim Biophys Acta 1862, 763–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohn JN, Johnson GR, Shabetai R, Loeb H, Tristani F, Rector T, Smith R & Fletcher R (1993). Ejection fraction, peak exercise oxygen consumption, cardiothoracic ratio, ventricular arrhythmias, and plasma norepinephrine as determinants of prognosis in heart failure. The V‐HeFT VA Cooperative Studies Group. Circulation 87, VI5–VI16. [PubMed] [Google Scholar]
- Communal C, Singh K, Sawyer DB & Colucci WS (1999). Opposing effects of β1‐ and β2‐adrenergic receptors on cardiac myocyte apoptosis: role of a pertussis toxin‐sensitive G protein. Circulation 100, 2210–2212. [DOI] [PubMed] [Google Scholar]
- Corbit KC, Trakul N, Eves EM, Diaz B, Marshall M & Rosner MR (2003). Activation of Raf‐1 signaling by protein kinase C through a mechanism involving Raf kinase inhibitory protein. J Biol Chem 278, 13061–13068. [DOI] [PubMed] [Google Scholar]
- Daaka Y, Luttrell LM & Lefkowitz RJ (1997). Switching of the coupling of the β2‐adrenergic receptor to different G proteins by protein kinase A. Nature 390, 88–91. [DOI] [PubMed] [Google Scholar]
- Deftereos S, Papoutsidakis N, Giannopoulos G, Angelidis C, Raisakis K, Bouras G, Davlouros P, Panagopoulou V, Goudevenos J, Cleman MW & Lekakis J. (2016) Calcium ions in inherited cardiomyopathies. J Med Chem 2, 139–150. [DOI] [PubMed] [Google Scholar]
- Deiss K, Kisker C, Lohse MJ & Lorenz K (2012). Raf kinase inhibitor protein (RKIP) dimer formation controls its target switch from Raf1 to G protein‐coupled receptor kinase (GRK) 2. J Biol Chem 287, 23407–23417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorn GW, Tepe NM, Lorenz JN, Koch WJ & Liggett SB (1999). Low‐ and high‐level transgenic expression of β2‐adrenergic receptors differentially affect cardiac hypertrophy and function in Galphaq‐overexpressing mice. Proc Natl Acad Sci USA 96, 6400–6405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El‐Armouche A, Wittkopper K, Degenhardt F, Weinberger F, Didie M, Melnychenko I, Grimm M, Peeck M, Zimmermann WH, Unsold B, Hasenfuss G, Dobrev D & Eschenhagen T (2008). Phosphatase inhibitor‐1‐deficient mice are protected from catecholamine‐induced arrhythmias and myocardial hypertrophy. Cardiovasc Res 80, 396–406. [DOI] [PubMed] [Google Scholar]
- Engelhardt S, Hein L, Wiesmann F & Lohse MJ (1999). Progressive hypertrophy and heart failure in β1‐adrenergic receptor transgenic mice. Proc Natl Acad Sci USA 96, 7059–7064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eschenhagen T (2010). Is ryanodine receptor phosphorylation key to the fight or flight response and heart failure? J Clin Invest 120, 4197–4203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fajardo G, Zhao M, Urashima T, Farahani S, Hu DQ, Reddy S & Bernstein D. (2013) Deletion of the β2‐adrenergic receptor prevents the development of cardiomyopathy in mice. J Mol Cell Cardiol 63, 155–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felker G (2001). Inotropic therapy for heart failure: An evidence‐based approach. Am Heart J 142, 393–401. [DOI] [PubMed] [Google Scholar]
- Flevari P, Parissis JT, Leftheriotis D, Panou F, Kourea K & Kremastinos DT (2006). Effect of levosimendan on ventricular arrhythmias and prognostic autonomic indexes in patients with decompensated advanced heart failure secondary to ischemic or dilated cardiomyopathy. Am J Cardiol 98, 1641–1645. [DOI] [PubMed] [Google Scholar]
- Francis GS, Cohn JN, Johnson G, Rector TS, Goldman S & Simon A (1993). Plasma norepinephrine, plasma renin activity, and congestive heart failure. Relations to survival and the effects of therapy in V‐HeFT II. The V‐HeFT VA Cooperative Studies Group. Circulation 87, 40–48. [PubMed] [Google Scholar]
- Fu X, Koller S, Abd Alla J & Quitterer U (2013). Inhibition of G protein‐coupled receptor kinase 2 (GRK2) triggers the growth‐promoting mitogen‐activated protein kinase (MAPK) pathway. J Biol Chem 288, 7738–7755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao MH, Bayat H, Roth DM, Yao Zhou J, Drumm J, Burhan J & Hammond HK (2002). Controlled expression of cardiac‐directed adenylylcyclase type VI provides increased contractile function. Cardiovasc Res 56, 197–204. [DOI] [PubMed] [Google Scholar]
- Gorelik J, Wright PT, Lyon AR & Harding SE (2013). Spatial control of the AR system in heart failure: the transverse tubule and beyond. Cardiovasc Res 98, 216–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granovsky AE & Rosner MR (2008). Raf kinase inhibitory protein: a signal transduction modulator and metastasis suppressor. Cell Res 18, 452–457. [DOI] [PubMed] [Google Scholar]
- Granovsky AE, Clark MC, McElheny D, Heil G, Hong J, Liu X, Kim Y, Joachimiak G, Joachimiak A, Koide S & Rosner MR (2009). Raf kinase inhibitory protein function is regulated via a flexible pocket and novel phosphorylation‐dependent mechanism. Mol Cell Biol 29, 1306–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenberg B (2015). Gene therapy for heart failure. J Cardiol 66, 195–200. [DOI] [PubMed] [Google Scholar]
- Greenberg B, Butler J, Felker GM, Ponikowski P, Voors AA, Pogoda JM, Provost R, Guerrero J, Hajjar RJ, Zsebo KM (2016). Prevalence of AAV1 neutralizing antibodies and consequences for a clinical trial of gene transfer for advanced heart failure. Gene Ther 23, 313–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenberg B, Yaroshinsky A, Zsebo KM, Butler J, Felker GM, Voors AA, Rudy JJ, Wagner K & Hajjar RJ (2014). Design of a phase 2b trial of intracoronary administration of AAV1/SERCA2a in patients with advanced heart failure: the CUPID 2 trial (calcium up‐regulation by percutaneous administration of gene therapy in cardiac disease phase 2b). JACC Heart Fail 2, 84–92.24622121 [Google Scholar]
- Haghighi K, Kolokathis F, Pater L, Lynch RA, Asahi M, Gramolini AO, Fan G‐C, Tsiapras D, Hahn HS, Adamopoulos S, Liggett SB, Dorn GW, MacLennan DH, Kremastinos DT & Kranias EG (2003). Human phospholamban null results in lethal dilated cardiomyopathy revealing a critical difference between mouse and human. J Clin Invest 111, 869–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haghighi K, Bidwell P & Kranias EG (2014). Phospholamban interactome in cardiac contractility and survival: A new vision of an old friend. J Mol Cell Cardiol 77, 160–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond HK, Penny WF, Traverse JH, Henry TD, Watkins MW, Yancy CW, Sweis RN, Adler ED, Patel AN, Murray DR, Ross RS, Bhargava V, Maisel A, Barnard DD, Lai NC, Dalton ND, Lee ML, Narayan SM, Blanchard DG & Gao MH (2016). Intracoronary gene transfer of adenylyl cyclase 6 in patients with heart failure: a randomized clinical trial. JAMA Cardiol 1, 163–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris IS, Zhang S, Treskov I, Kovacs A, Weinheimer C & Muslin AJ (2004). Raf‐1 kinase is required for cardiac hypertrophy and cardiomyocyte survival in response to pressure overload. Circulation 110, 718–723. [DOI] [PubMed] [Google Scholar]
- Hasenfuss G & Pieske B (2002). Calcium cycling in congestive heart failure. J Mol Cell Cardiol 34, 951–969. [DOI] [PubMed] [Google Scholar]
- Heineke J & Molkentin JD (2006). Regulation of cardiac hypertrophy by intracellular signalling pathways. Nat Rev Mol Cell Biol 7, 589–600. [DOI] [PubMed] [Google Scholar]
- Hengst U (2000). The phosphatidylethanolamine‐binding protein is the prototype of a novel family of serine protease inhibitors. J Biol Chem 276, 535–540. [DOI] [PubMed] [Google Scholar]
- Houslay MD & Baillie GS (2005). β‐Arrestin‐recruited phosphodiesterase‐4 desensitizes the AKAP79/PKA‐mediated switching of β2 adrenoceptor signalling to activation of ERK. Biochem Soc Trans 33, 1333–1336. [DOI] [PubMed] [Google Scholar]
- Huke S & Knollmann BC (2010). Increased myofilament Ca2+‐sensitivity and arrhythmia susceptibility. J Mol Cell Cardiol 48, 824–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen BC, Swigart PM, De Marco T, Hoopes C & Simpson PC (2009). α1‐Adrenergic receptor subtypes in nonfailing and failing human myocardium. Circ Heart Fail 2, 654–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kairouz V, Lipskaia L, Hajjar RJ & Chemaly ER (2012). Molecular targets in heart failure gene therapy: current controversies and translational perspectives. Ann N Y Acad Sci 1254, 42–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krüger M & Linke WA (2006). Protein kinase‐A phosphorylates titin in human heart muscle and reduces myofibrillar passive tension. J Muscle Res Cell Motil 27, 435–444. [DOI] [PubMed] [Google Scholar]
- Kuschel M, Zhou YY, Cheng H, Zhang SJ, Chen Y, Lakatta EG & Xiao RP (1999). Gi protein‐mediated functional compartmentalization of cardiac β2‐adrenergic signaling. J Biol Chem 274, 22048–22052. [DOI] [PubMed] [Google Scholar]
- Landstrom AP, Ackerman MJ. (2012). Beyond the cardiac myofilament: hypertrophic cardiomyopathy‐associated mutations in genes that encode calcium‐handling proteins. Curr Mol Med 5, 507–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lechat P, Packer M, Chalon S, Cucherat M, Arab T & Boissel JP (1998). Clinical effects of β‐adrenergic blockade in chronic heart failure: a meta‐analysis of double‐blind, placebo‐controlled, randomized trials. Circulation 98, 1184–1191. [DOI] [PubMed] [Google Scholar]
- Lefkowitz RJ, Pierce KL & Luttrell LM (2002). Dancing with different partners: protein kinase a phosphorylation of seven membrane‐spanning receptors regulates their G protein‐coupling specificity. Mol Pharmacol 62, 971–974. [DOI] [PubMed] [Google Scholar]
- Lehnart SE, Maier LS & Hasenfuss G (2009). Abnormalities of calcium metabolism and myocardial contractility depression in the failing heart. Heart Fail Rev 14, 213–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liggett SB, Tepe NM, Lorenz JN, Canning AM, Jantz TD, Mitarai S, Yatani A & Dorn GW (2000). Early and delayed consequences of β2‐adrenergic receptor overexpression in mouse hearts: critical role for expression level. Circulation 101, 1707–1714. [DOI] [PubMed] [Google Scholar]
- Liggett SB, Wagoner LE, Craft LL, Hornung RW, Hoit BD, McIntosh TC & Walsh RA (1998). The Ile164 β2‐adrenergic receptor polymorphism adversely affects the outcome of congestive heart failure. J Clin Invest 102, 1534–1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lompré A‐M, Hajjar RJ, Harding SE, Kranias EG, Lohse MJ & Marks AR (2010). Ca2+ cycling and new therapeutic approaches for heart failure. Circulation 121, 822–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenz K, Lohse MJ & Quitterer U (2003). Protein kinase C switches the Raf kinase inhibitor from Raf‐1 to GRK‐2. Nature 426, 574–579. [DOI] [PubMed] [Google Scholar]
- Lorenz K, Schmid E & Deiss K (2014a). RKIP: a governor of intracellular signaling. Crit Rev in Oncog 19, 489‐496. [DOI] [PubMed] [Google Scholar]
- Lorenz K, Schmitt JP, Vidal M & Lohse MJ (2009). Cardiac hypertrophy: targeting Raf/MEK/ERK1/2‐signaling. Int J Biochem Cell Biol 41, 2351–2355. [DOI] [PubMed] [Google Scholar]
- Lorenz K, Stathopoulou K, Schmid E, Eder P & Cuello F (2014b). Heart failure‐specific changes in protein kinase signalling. Pflugers Arch 466, 1151–1162. [DOI] [PubMed] [Google Scholar]
- Lother A & Hein L (2016). Pharmacology of heart failure: From basic science to novel therapies. Pharmacol Ther 166, 136–149. [DOI] [PubMed] [Google Scholar]
- Maier LS & Bers DM (2007). Role of Ca2+/calmodulin‐dependent protein kinase (CaMK) in excitation‐contraction coupling in the heart. Cardiovasc Res 73, 631–640. [DOI] [PubMed] [Google Scholar]
- Marks AR (2013). Calcium cycling proteins and heart failure: mechanisms and therapeutics. J Clin Invest 123, 46–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Métrich M, Berthouze M, Morel E, Crozatier B, Gomez AM & Lezoualc'h F (2009). Role of the cAMP‐binding protein Epac in cardiovascular physiology and pathophysiology. Pflugers Arch 459, 535–546. [DOI] [PubMed] [Google Scholar]
- Métrich M, Lucas A, Gastineau M, Samuel J‐L, Heymes C, Morel E & Lezoualc'h F (2008). Epac mediates β‐adrenergic receptor‐induced cardiomyocyte hypertrophy. Circ Res 102, 959–965. [DOI] [PubMed] [Google Scholar]
- Najafi A, Sequeira V, Kuster DWD & van der Velden J (2016). β‐Adrenergic receptor signalling and its functional consequences in the diseased heart. Eur J Clin Invest 46, 362–374. [DOI] [PubMed] [Google Scholar]
- Nikolaev VO, Moshkov A, Lyon AR, Miragoli M, Novak P, Paur H, Lohse MJ, Korchev YE, Harding SE & Gorelik J (2010). β2‐Adrenergic receptor redistribution in heart failure changes cAMP compartmentation. Science 327, 1653–1657. [DOI] [PubMed] [Google Scholar]
- O'Connor CM, Gattis WA, Uretsky BF, Adams KF Jr, McNulty SE, Grossman SH, McKenna WJ, Zannad F, Swedberg K, Gheorghiade M & Califf RM (1999). Continuous intravenous dobutamine is associated with an increased risk of death in patients with advanced heart failure: insights from the Flolan International Randomized Survival Trial (FIRST). Am Heart J 138, 78–86. [DOI] [PubMed] [Google Scholar]
- Ohsuzu F, Katsushika S, Akanuma M, Hakamada N, Hamada H, Maie S, Muneoka K, Okumori M & Nakamura H (1994). Reduction of β‐adrenergic receptors in atrial cell membranes of patients following mild to moderate heart failure. Jpn Heart J 35, 27–34. [DOI] [PubMed] [Google Scholar]
- Orchard C & Brette F (2007). t‐Tubules and sarcoplasmic reticulum function in cardiac ventricular myocytes. Cardiovasc Res 77, 237–244. [DOI] [PubMed] [Google Scholar]
- Packer M, Bristow MR, Cohn JN, Colucci WS, Fowler MB, Gilbert EM & Shusterman NH (1996a). The effect of carvedilol on morbidity and mortality in patients with chronic heart failure. U.S. carvedilol heart failure study group. N Engl J Med 334, 1349–1355. [DOI] [PubMed] [Google Scholar]
- Packer M, Colucci WS, Sackner‐Bernstein JD, Liang CS, Goldscher DA, Freeman I, Kukin ML, Kinhal V, Udelson JE, Klapholz M, Gottlieb SS, Pearle D, Cody RJ, Gregory JJ, Kantrowitz NE, LeJemtel TH, Young ST, Lukas MA & Shusterman NH (1996b). Double‐blind, placebo‐controlled study of the effects of carvedilol in patients with moderate to severe heart failure. The PRECISE trial. Prospective randomized evaluation of carvedilol on symptoms and exercise. Circulation 94, 2793–2799. [DOI] [PubMed] [Google Scholar]
- Patterson AJ, Zhu W, Chow A, Agrawal R, Xiao RP & Kobilka B (2004). Protecting the myocardium: a role for the beta2 adrenergic receptor in the heart. Crit Care Med 32, 1041–1048. [DOI] [PubMed] [Google Scholar]
- Paur H, Wright PT, Sikkel MB, Tranter MH, Mansfield C, O'Gara P, Stuckey DJ, Nikolaev VO, Diakonov I, Pannell L, Gong H, Sun H, Peters NS, Petrou M, Zheng Z, Gorelik J, Lyon AR & Harding SE (2012). High levels of circulating epinephrine trigger apical cardiodepression in a β2‐adrenergic receptor/Gi‐dependent manner: a new model of Takotsubo cardiomyopathy. Circulation 126, 697–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira L, Cheng H, Lao D‐H, Na L, van Oort RJ, Brown JH, Wehrens XHT, Chen J & Bers DM (2013). Epac2 mediates cardiac β1‐adrenergic‐dependent sarcoplasmic reticulum Ca2+ leak and arrhythmia. Circulation 127, 913–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce KL, Premont RT & Lefkowitz RJ (2002). Signalling: Seven‐transmembrane receptors. Nat Rev Mol Cell Biol 3, 639–650. [DOI] [PubMed] [Google Scholar]
- Pleger ST, Raake P, Katus HA & Most P (2014). Cardiac calcium handling on trial: targeting the failing cardiomyocyte signalosome. Circ Res 114, 12–14. [DOI] [PubMed] [Google Scholar]
- Pleger ST, Shan C, Ksienzyk J, Bekeredjian R, Boekstegers P, Hinkel R, Schinkel S, Leuchs B, Ludwig J, Qiu G, Weber C, Raake P, Koch WJ, Katus HA, Muller OJ & Most P (2011). Cardiac AAV9‐S100A1 gene therapy rescues post‐ischemic heart failure in a preclinical large animal model. Sci Transl Med 3, 92ra64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponikowski P, Voors AA, Anker SD, Bueno H, Cleland JG, Coats AJ, Falk V, González‐Juanatey JR, Harjola VP, Jankowska EA, Jessup M, Linde C, Nihoyannopoulos P, Parissis JT, Pieske B, Riley JP, Rosano GM, Ruilope LM, Ruschitzka F, Rutten FH & van der Meer P; Authors/Task Force Members; Document Reviewers (2016). 2016 ESC guidelines for the diagnosis and treatment of acute and chronic heart failure. Eur J Heart Fail 18, 891–975. [DOI] [PubMed] [Google Scholar]
- Priori SG & Chen SR (2011). Inherited dysfunction of sarcoplasmic reticulum Ca2+ handling and arrhythmogenesis. Circ Res 7, 871–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Punn A, Mockridge JW, Farooqui S, Marber MS & Heads RJ (2000). Sustained activation of p42/p44 mitogen‐activated protein kinase during recovery from simulated ischaemia mediates adaptive cytoprotection in cardiomyocytes. Biochem J 350, 891–899. [PMC free article] [PubMed] [Google Scholar]
- Purcell NH, Wilkins BJ, York A, Saba‐El‐Leil MK, Meloche S, Robbins J & Molkentin JD (2007). Genetic inhibition of cardiac ERK1/2 promotes stress‐induced apoptosis and heart failure but has no effect on hypertrophy in vivo. Proc Natl Acad Sci USA 104, 14074–14079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Respress JL, van Oort RJ, Li N, Rolim N, Dixit SS, deAlmeida A, Voigt N, Lawrence WS, Skapura DG, Skardal K, Wisloff U, Wieland T, Ai X, Pogwizd SM, Dobrev D & Wehrens XHT (2012). Role of RyR2 phosphorylation at S2814 during heart failure progression. Circ Res 110, 1474–1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritterhoff J, Völkers M, Seitz A, Spaich K, Gao E, Peppel K, Pleger ST, Zimmermann WH, Friedrich O, Fink RHA, Koch WJ, Katus HA & Most P (2015). S100A1 DNA‐based inotropic therapy protects against proarrhythmogenic ryanodine receptor 2 dysfunction. Mol Ther 23, 1320–1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rockman HA, Koch WJ & Lefkowitz RJ (2002). Seven‐transmembrane‐spanning receptors and heart function. Nature 415, 206–212. [DOI] [PubMed] [Google Scholar]
- Ruppert C, Deiss K, Herrmann S, Vidal M, Oezkur M, Gorski A, Weidemann F, Lohse MJ & Lorenz K (2013). Interference with ERK(Thr188) phosphorylation impairs pathological but not physiological cardiac hypertrophy. Proc Natl Acad of Sci USA 110, 7440–7445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadayappan S, Gulick J, Osinska H, Barefield D, Cuello F, Avkiran M, Lasko VM, Lorenz JN, Maillet M, Martin JL, Brown JH, Bers DM, Molkentin JD, James J & Robbins J (2011). A critical function for Ser‐282 in cardiac Myosin binding protein‐C phosphorylation and cardiac function. Circ Res 109, 141–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid E, Neef S, Berlin C, Tomasovic A, Kahlert K, Nordbeck P, Deiss K, Denzinger S, Herrmann S, Wettwer E, Weidendorfer M, Becker D, Schäfer F, Wagner N, Ergün S, Schmitt JP, Katus HA, Weidemann F, Ravens U, Maack C, Hein L, Ertl G, Müller OJ, Maier LS, Lohse MJ & Lorenz K (2015). Cardiac RKIP induces a beneficial beta‐adrenoceptor‐dependent positive inotropy. Nat Med 21, 1298–1306. [DOI] [PubMed] [Google Scholar]
- Schmitt JP, Ahmad F, Lorenz K, Hein L, Schulz S, Asahi M, MacLennan DH, Seidman CE, Seidman JG & Lohse MJ (2009). Alterations of phospholamban function can exhibit cardiotoxic effects independent of excessive sarcoplasmic reticulum Ca2+‐ATPase inhibition. Circulation 119, 436–444. [DOI] [PubMed] [Google Scholar]
- Schmitt JP, Kamisago M, Asahi M, Li GH, Ahmad F, Mende U, Kranias EG, MacLennan DH, Seidman JG & Seidman CE (2003). Dilated cardiomyopathy and heart failure caused by a mutation in phospholamban. Science 299, 1410–1413. [DOI] [PubMed] [Google Scholar]
- Sheikh F, Raskin A, Chu P‐H, Lange S, Domenighetti AA, Zheng M, Liang X, Zhang T, Yajima T, Gu Y, Dalton ND, Mahata SK, Dorn GW II, Heller Brown J, Peterson KL, Omens JH, McCulloch AD & Chen J (2008). An FHL1‐containing complex within the cardiomyocyte sarcomere mediates hypertrophic biomechanical stress responses in mice. J Clin Invest 118, 3870–3880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shizukuda Y & Buttrick PM (2002). Subtype specific roles of β‐adrenergic receptors in apoptosis of adult rat ventricular myocytes. J Mol Cell Cardiol. 34, 823–831. [DOI] [PubMed] [Google Scholar]
- Siedlecka U, Arora M, Kolettis T, Soppa GKR, Lee J, Stagg MA, Harding SE, Yacoub MH & Terracciano CMN (2008). Effects of clenbuterol on contractility and Ca2+ homeostasis of isolated rat ventricular myocytes. Am J Physiol Heart Circ Physiol 295, H1917–H1926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slack J, Grupp IL, Dash R, Holder D, Schmidt A, Gerst MJ, Tamura T, Tilgmann C, James PF, Johnson R, Gerdes AM & Kranias EG (2001). The enhanced contractility of the phospholamban‐deficient mouse heart persists with aging. J Mol Cell Cardiol 33, 1031–1040. [DOI] [PubMed] [Google Scholar]
- Steinfath M, Geertz B, Schmitz W, Scholz H, Haverich A, Breil I, Hanrath P, Reupcke C, Sigmund M & Lo HB (1991). Distinct down‐regulation of cardiac β1‐ and β2‐adrenoceptors in different human heart diseases. Naunyn Schmiedebergs Arch Pharmacol 343, 217–220. [DOI] [PubMed] [Google Scholar]
- Steinfath M, Lavicky J, Schmitz W, Scholz H, Döring V & Kalmár P (1992). Regional distribution of β1‐ and β2‐adrenoceptors in the failing and nonfailing human heart. Eur J Clin Pharmacol 42, 607–611. [DOI] [PubMed] [Google Scholar]
- Tacon CL, McCaffrey J & Delaney A (2012). Dobutamine for patients with severe heart failure: a systematic review and meta‐analysis of randomised controlled trials. Intensive Care Med 38, 359–367. [DOI] [PubMed] [Google Scholar]
- Talan MI, Ahmet I, Xiao R‐P & Lakatta EG (2011). β2 AR agonists in treatment of chronic heart failure: long path to translation. J Mol Cell Cardiol 51, 529–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang T, Gao MH & Hammond HK (2012). Prospects for gene transfer for clinical heart failure. Gene Ther 19, 606–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tardiff JC, Carrier L, Bers DM, Poggesi C, Ferrantini C, Coppini R, Maier LS, Ashrafian H, Huke S & van der Velden J (2015). Targets for therapy in sarcomeric cardiomyopathies. Cardiovasc Res 105, 457–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas JA & Marks BH (1987). Plasma norepinephrine in congestive heart failure. Am J Cardiol 41, 233–243. [DOI] [PubMed] [Google Scholar]
- Trakul N & Rosner MR (2005). Modulation of the MAP kinase signaling cascade by Raf kinase inhibitory protein. Cell Res 15, 19–23. [DOI] [PubMed] [Google Scholar]
- Uchinoumi H, Yang Y, Oda T, Li N, Alsina KM, Puglisi JL, Chen‐Izu Y, Cornea RL, Wehrens XHT & Bers DM (2016). CaMKII‐dependent phosphorylation of RyR2 promotes targetable pathological RyR2 conformational shift. J Mol Cell Cardiol 98, 62–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Berlo JH, Elrod JW, Aronow BJ, Pu WT & Molkentin JD (2011). Serine 105 phosphorylation of transcription factor GATA4 is necessary for stress‐induced cardiac hypertrophy in vivo. Proc Natl Acad Sci USA 108, 12331–12336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Velden J, Merkus D, Klarenbeek BR, James AT, Boontje NM, Dekkers DH, Stienen GJ, Lamers JM & Duncker DJ (2004). Alterations in myofilament function contribute to left ventricular dysfunction in pigs early after myocardial infarction. Circ Res 11, 85–95. [DOI] [PubMed] [Google Scholar]
- Vidal M, Wieland T, Lohse MJ & Lorenz K (2012). β‐Adrenergic receptor stimulation causes cardiac hypertrophy via a Gβγ/Erk‐dependent pathway. Cardiovasc Res 96, 255–264. [DOI] [PubMed] [Google Scholar]
- Völkers M, Weidenhammer C, Herzog N, Qiu G, Spaich K, Wegner von F, Peppel K, Muller OJ, Schinkel S, Rabinowitz JE, Hippe HJ, Brinks H, Katus HA, Koch WJ, Eckhart AD, Friedrich O & Most P (2011). The inotropic peptide ARKct improves AR responsiveness in normal and failing cardiomyocytes through Gβγ‐Mediated L‐Type calcium current disinhibition. Circ Res 108, 27–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber S, Meyer‐Roxlau S, El‐Armouche A (2016). Role of protein phosphatase inhibitor‐1 in cardiac beta adrenergic pathway. J Mol Cell Cardiol 101, 116–126. [DOI] [PubMed] [Google Scholar]
- Woo AYH & Xiao R‐P (2012). β‐Adrenergic receptor subtype signaling in heart: From bench to bedside. Acta Pharmacol Sin 33, 335–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao RP, Avdonin P, Zhou YY, Cheng H, Akhter SA, Eschenhagen T, Lefkowitz RJ, Koch WJ & Lakatta EG (1999). Coupling of β2‐adrenoceptor to Gi proteins and its physiological relevance in murine cardiac myocytes. Circ Res 84, 43–52. [DOI] [PubMed] [Google Scholar]
- Yeung K, Seitz T, Li S, Janosch P, McFerran B, Kaiser C, Fee F, Katsanakis KD, Rose DW, Mischak H, Sedivy JM & Kolch W (1999). Suppression of Raf‐1 kinase activity and MAP kinase signalling by RKIP. Nature 401, 173–177. [DOI] [PubMed] [Google Scholar]
- Yeung KC, Rose DW, Dhillon AS, Yaros D, Gustafsson M, Chatterjee D, McFerran B, Wyche J, Kolch W & Sedivy JM (2001). Raf kinase inhibitor protein interacts with NF‐κB‐inducing kinase and TAK1 and inhibits NF‐kB activation. Mol Cell Biol 21, 7207–7217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin R, Liu X, Zhang P, Chen Y, Xie G, Ai L, Xue C, Qian J, Bi Y, Chen J, Sun Y, Stoeger T & Ding Z (2016). A Raf kinase inhibitor demonstrates antiviral activities both in vitro and in vivo against different genotypes of virulent Newcastle disease virus. Antiviral Res 133, 140–144. [DOI] [PubMed] [Google Scholar]
- Zaugg M, Xu W, Lucchinetti E, Shafiq SA, Jamali NZ & Siddiqui MA (2000). β‐Adrenergic receptor subtypes differentially affect apoptosis in adult rat ventricular myocytes. Circulation 102, 344–350. [DOI] [PubMed] [Google Scholar]
- Zhang X, Szeto C, Gao E, Tang M, Jin J, Fu Q, Makarewich C, Ai X, Li Y, Tang A, Wang J, Gao H, Wang F, Ge XJ, Kunapuli SP, Zhou L, Zeng C, Xiang KY & Chen X (2013). Cardiotoxic and cardioprotective features of chronic β‐adrenergic signaling. Circ Res 112, 498–509. [DOI] [PMC free article] [PubMed] [Google Scholar]