

Abstract
Heart failure is one of the largest contributors to disease burden and healthcare outflow in the Western world. Despite significant progress in the treatment of heart failure, disease prognosis remains very poor, with the only curative therapy still being heart transplantation. To counteract the current situation, efforts have been made to better understand the underlying molecular pathways in the progression of cardiac disease towards heart failure, and to link the disease to novel therapeutic targets such as non‐coding RNAs. The non‐coding part of the genome has gained prominence over the last couple of decades, opening a completely new research field and establishing different non‐coding RNAs species as fundamental regulators of cellular functions. Not surprisingly, their dysregulation is increasingly being linked to pathology, including to cardiac disease. Pre‐clinically, non‐coding RNAs have been shown to be of great value as therapeutic targets in pathological cardiac remodelling and also as diagnostic/prognostic biomarkers for heart failure. Therefore, it is to be expected that non‐coding RNA‐based therapeutic strategies will reach the bedside in the future and provide new and more efficient treatments for heart failure. Here, we review recent discoveries linking the function and molecular interactions of non‐coding RNAs with the pathophysiology of cardiac hypertrophy and heart failure.
Keywords: heart failure, hypertrophy, intracellular communication, non‐coding RNA
Abbreviations
- 16K PRL
16‐kDa N‐terminal prolactin fragment
- α‐MHC/β‐MHC
α‐/β‐myosin heavy chain
- ABs
apoptotic bodies
- AKT
protein kinase B
- AngII
angiotensin II
- AR
adrenergic receptor
- ARC
activity‐regulated cytoskeleton‐associated protein
- AT1R
AT2R, angiotensin type 1 receptor, angiotensin type 2 receptor
- Brg1
brahma‐related gene 1
- BVHT
braveheart
- CARL
cardiac‐apoptosis‐related lncRNA
- Cdc42
cell division control protein 42 homologue
- Cdk9
cyclin‐dependent kinase 9
- CDR1
cerebellar degeneration‐related protein 1
- CHAST
cardiac hypertrophy‐associated transcript
- CHRF
cardiac hypertrophy‐related factor
- CircRNAs
circular RNA
- CTGF
connective tissue growth factor
- DYRK1a
dual‐specificity tyrosine‐(Y)‐phosphorylation‐regulated kinase 1a
- ECM
extracellular matrix
- ECs
endothelial cells
- EFNA3
ephrin‐A3 precursor
- ERK1/2
extracellular signal‐regulated kinases
- EVs
extracellular vesicles
- FABP3
fatty acid binding protein 3
- FENDRR
Foxf1 adjacent non‐coding developmental regulatory RNA
- FOXO3
Forkhead Box O3
- GWA
genome‐wide association
- HDAC
histone deacetylase
- HF
heart failure
- HRCR
heart‐related circRNA
- IGF1‐R
insulin‐like growth factor 1 receptor
- IGF1
insulin‐like growth factor 1
- LNA
locked nucleic acid‐modified
- lncRNAs
long non‐coding RNAs
- MAPK
mitogen‐activated protein kinase 1
- MEK1
mitogen‐activated protein kinase kinase
- MHRT
myosin heavy‐chain‐associated RNA transcripts
- miRNAs or miRs
microRNAs
- MVs
microvesicles
- Myd88
myeloid differentiation primary response gene 88
- Myh7
myosin heavy chain 7
- ncRNAs
non‐coding RNAs
- NELFA/Whsc2
negative elongation factor complex member A
- NFAT
nuclear factor of activated T‐cells
- PARP
poly(ADP‐ribose) polymerase family member
- PDLIM5
PDZ and LIM domain 5
- PE
phenylephrine
- PGC‐1α
peroxisome proliferator‐activated receptor gamma coactivator 1‐alpha
- PI3K
phosphatidylinositol‐3‐kinases
- PPAR
peroxisome proliferator‐activated receptor
- PPCM
peripartum cardiomyopathy
- PTEN
phosphatase and tensin homologue
- PTP1b
protein‐tyrosine phosphatase 1b
- Raf1
proto‐oncogene, serine/threonine kinase
- Ras
proto‐oncogene, GTPase
- RasGAP
Ras GTPase activating protein
- Rheb
Ras homologue enriched in brain
- RhoA
Ras homologue gene family, member A
- ROS
reactive oxygen species
- rRNA
ribosomal RNA
- SMAD3
mothers against decapentaplegic homologue 3 also known as SMAD family member 3
- SNPs
single nucleotide polymorphism
- SORBS2
sorbin and SH3 domain containing 2
- TGF‐β
transforming growth factor beta
- tRNA
transfer RNA
- Trx1
thioredoxin1
Introduction
While the existence of non‐coding RNAs (ncRNAs) has been known since the 1960s, it was not until the last decade that their significance and functions began to be understood. Since then there has been a swift extension of our knowledge and understanding of ncRNA biology, in particular regarding small RNAs (Papageorgiou et al. 2015). Although initially less familiar than mRNA, tRNA or rRNA, ncRNAs have received increasing attention due to their crucial roles in cellular mechanisms and regulation. Many ncRNAs discovered so far have roles in DNA replication and chromatin structure, processing of other RNAs, transcriptional and post‐transcriptional control of gene expression, genome integrity, and mRNA stability (Ghildiyal & Zamore, 2009). Accordingly, a growing number of non‐coding transcripts have been assigned roles in gene regulation and RNA processing and cross‐species sequence comparisons have identified conserved functional non‐coding elements (Bejerano et al. 2004; Pennacchio et al. 2006). In fact, SNPs in many non‐coding regions have been linked to disease in genome‐wide association (GWA) studies (Kleinjan & Van Heyningen, 2005).
In particular, microRNAs (miRNAs or miRs) and long non‐coding RNAs (lncRNAs) have emerged as prominent players in human pathophysiological processes. miRs are small RNAs comprising 19–25 base pairs (Bartel, 2004). Over 2000 miRNAs have been identified, and their primary function is to regulate gene expression by either translational inhibition or by promoting degradation of target messenger RNAs (mRNAs) (Bartel, 2004). In turn, lncRNAs are comprised of >200 base pairs and are classified into five subclasses: intergenic, intronic, bidirectional, enhancer, sense and antisense; with each subclass being categorized according to the genomic location of the lncRNA in relation to its neighbouring encoding regions (Archer et al. 2015; Devaux et al. 2015). More unusual species of ncRNA include circular RNAs (Salzman et al. 2013; Wilusz & Sharp, 2013; Cech & Steitz, 2014), which lack 5′ or 3′ ends and are therefore quite stable due to reduced exonuclease susceptibility (Memczak et al. 2013) and able to accumulate to levels that are comparable to mRNAs (Salzman, 2016).
Here, we seek to summarize the current knowledge on the role of distinct classes of ncRNAs regulating pathological cardiac remodelling and their potential relevance as diagnostic and/or therapeutic tools in the treatment of heart failure.
Cardiac remodelling towards heart failure
Heart failure (HF) is one of the main causes of mortality and morbidity worldwide (Ziaeian & Fonarow, 2016) and is the common name of cardiovascular diseases such as myocardial infarction and inherited or acquired cardiomyopathies. Whereas the heart is a dynamic organ that is able to react to injuries through cardiomyocyte hypertrophy, extracellular matrix remodelling and reactivation of fetal cardiac genes (Konstam et al. 2011), end‐stage failing hearts besides displaying genetic re‐programming of fetal genes also pose higher vulnerability to necrotic or apoptotic programmes and dysfunctional vascular remodelling (Lips et al. 2003). Cardiac remodelling is a complex pathogenic pathway involving cardiac molecular and cellular adjustments leading to clinical manifestations such as changes in shape, size and function as a response to stress or injury. Cardiac hypertrophy is considered physiological when it develops from intense exercise or during pregnancy in healthy individuals (Shimizu & Minamino, 2016). However, under sustained cardiac stress such as chronic hypertension or myocardial infarction (Kehat & Molkentin, 2010), hypertrophic cardiomyocyte growth may become pathological and lead to increased myocardial mass to compensate for the elevated wall stress. Neurohormonal activation, mechanical stress, haemodynamic load and intracellular reactive cascades originate this clinical response (Lips et al. 2003; Kehat & Molkentin, 2010).
Although the human heart contains an estimated 2–3 billion cardiac muscle cells, they ‘only’ account for a third of the total number of cells in the heart. The remaining cells comprise different cell types, including smooth muscle and endothelial cells of the coronary vasculature and the endocardium, fibroblasts and other connective tissue cells, mast cells, and immune system‐related cells. These distinct cell groups do not function in isolation from one another within the heart but instead interact physically and via a variety of soluble paracrine, autocrine and endocrine factors. Besides hypertrophy of cardiac muscle cells, cardiomyopathies are normally associated with endothelial dysfunction and vascular rarefaction (De Boer et al. 2003; Shiojima et al. 2005). Transition to HF results from decreased angiogenesis, increased fibrosis and extracellular matrix (ECM) deposition (Shiojima et al. 2005). ECM not only provides structural cardiac support, allowing cells to be interconnected, but is also a cytokine and growth factor source. During pathological conditions ECM, influenced by autocrine and paracrine signals, is subjected to remodelling of its structural components, which will induce cardiac fibroblast differentiation into smooth muscle actin‐expressing myofibroblasts and ultimately lead to secretion of a great amount of ECM proteins. Furthermore, in a healthy heart there is a resident population of cardiac macrophages that is established embryonically and persists in adulthood. Macrophages and recruited monocytes may also be involved in cardiac remodelling, a hypothesis supported by the observation that pro‐inflammatory cytokines are released during heart injury; however, the exact role of these inflammatory cells in cardiac hypertrophy is still to be defined (Topkara et al. 2011; Epelman et al. 2014). Because cardiac hypertrophy impairs the relationship between ATP demand and production it is also important to underline the association of cardiac mitochondrial homeostasis with cardiac function, as mitochondrial bioenergetics must keep up with the cardiac hypertrophic phenotype and defective mitochondrial function reduces production of ATP while increasing the generation of reactive oxygen species. All these changes, accompanied by cardiac fibrosis, ventricular stiffening and inflammation, constitute the cardiac response to a chronic insult (Valiente‐Alandi et al. 2016).
Apart from well‐established transcriptional mechanisms, increasing evidence indicates that gene re‐programming in the remodelling/failing heart are under epigenetic control by ncRNAs.
Non‐coding RNAs in cardiomyocyte hypertrophy
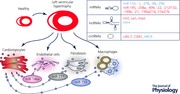
Different ncRNAs have been found to play pivotal regulatory roles in pathophysiological processes, with aberrant expression of miRNAs and lncRNAs in the different cardiac cell types being associated with many diseases, including cardiovascular abnormalities. Only recently, lncRNAs have been identified with important functional contributions to cardiac development and progression towards cardiac disease. Yet, only a limited number of lncRNAs have been identified as regulators of cardiac hypertrophy. Circular RNAs (circRNAs) represent yet another class of prevalent and diverse endogenous RNAs that, based on very few studies, seem to be able to regulate gene expression in eukaryotes. Because only a few circRNAs have been associated with cardiac disease, the regulation and function of human circRNAs in cardiac disease remain largely unknown. Below, we summarize the most distinct ncRNAs that have been associated with cardiac hypertrophy (Fig. 1).
Figure 1. Most prominent non‐coding RNAs in cardiac hypertrophy.
Different non‐coding RNAs species were identified as fundamental regulators of cellular functions. Upon cardiomyocyte hypertrophic growth as a response to chronic stress, microRNAs (miRNAs), long non‐coding RNAs (lncRNAs) and circular RNAs (circRNAs) interplay with their respective targets to regulate specific cellular functions and induce pathological cell growth that will eventually develop into heart failure.
MicroRNAs
MicroRNAs (miRNAs or miRs) are endogenous, single stranded, small non‐coding RNA molecules (∼22 nucleotides) that suppress gene expression at a post‐transcriptional level by targeting specific mRNAs. The mechanisms by which several miRNAs are involved in pathological cardiac remodelling have been extensively studied, resulting in the identification of many functional miRNAs whose importance is gradually being revealed.
In the context of pathological cardiac remodelling, most miRNAs have been studied in relation to their specific expression levels in cardiomyocytes. Several miRNAs were found to be downregulated in cardiac disease and exert, therefore, an anti‐hypertrophic function. In this regard miR‐133, highly abundant in cardiomyocytes, was one of the first miRNAs to be associated with cardiac pathological growth. MiR‐133 is significantly reduced upon severe cardiac hypertrophy (Duisters et al. 2009) where, by acting as a strong repressor of connective tissue growth factor (CTGF), it is able to regulate extracellular matrix deposition. While CTGF is mostly expressed in fibroblasts, during pathological cardiac remodelling it is also secreted by cardiomyocytes (Duisters et al. 2009).
In a different study, the significant role of miR‐133 was confirmed, in combination with miR‐1, as both miRNAs were found to be downregulated in three different murine models of cardiac hypertrophy (Carè et al. 2007). In this setting, miR‐133 exerts its function through direct regulation of Ras homologue gene family, member A (RhoA), cell division control protein 42 homologue (Cdc42) and negative elongation factor complex member A (NELFA/Whsc2), three proteins known to be involved in cardiac hypertrophy (Carè et al. 2007). It is worth noting that downregulation of miR‐133 is also found in human hypertrophic cardiomyopathy, which supports its relevance in the development of the disease. In a miR‐133 cardiac‐specific transgenic mouse model subjected to pressure overload, cardiac performance was maintained and both cardiomyocyte apoptosis and collagen deposition were reduced (Castaldi et al. 2014). These effects are associated with repression of β1‐adrenergic receptor kinase, one of the direct targets of miR‐133 (Castaldi et al. 2014). A different study also demonstrated that overexpression of miR‐133 under cardiac pressure overload, either induced by transverse aortic constriction or stimulation with isoproterenol, did not affect cardiomyocyte hypertrophic growth but did confer protection against cardiac fibrosis (Matkovich et al. 2010). miR‐133 is able to regulate multiple components of the β1AR transduction cascade by exerting a cardioprotective function during heart failure.
miR‐1, on its own, is highly expressed in cardiac tissue and its deletion causes cardiac defects. When following the changes in miRNA expression levels in time, during 14 days of pressure overload, miR‐1 was the only miRNA found to be downregulated as early as day one. Cardiac‐specific overexpression of miR‐1 results in repression of target genes such as Ras GTPase activating protein (RasGAP), cyclin‐dependent kinase 9 (Cdk9), fibronectin and Ras homologue enriched in brain (Rheb) and subsequent attenuation of cardiac hypertrophic growth, reduced fibrosis, lower cardiomyocyte apoptosis and improved calcium signalling (Karakikes et al. 2013). Other targets of miR‐1 with direct effects on cardiac cells are insulin‐like growth factor (IGF1) and IGF1‐receptor (IGF1‐R) (Elia et al. 2009). While IGF1 normally binds to its receptor, inducing activation of different pathways, including PI3K–AKT, and inhibiting the transcription factor FOXO3A, modulation of any of these factors in cardiomyocytes directly affects miR‐1 expression levels. Whereas transfection of AKT reduces miR‐1 expression, overexpression of FOXO3A induces an increase in miR‐1 levels (Elia et al. 2009). In a model of cardiac pressure overload, increased levels of IGF1 due to repression of miR‐1, are also accompanied by upregulation of the circulating fatty acid‐binding protein 3 (FABP3), a small protein implicated in cardiac metabolism that is secreted into the blood stream and could therefore be potentially monitored as a biomarker reflecting myocardial function (Varrone et al. 2013). Altogether, these results underline how miR‐1–IGF1 interplay regulates many processes in the control of cardiac function.
Another miRNA that is downregulated during progression of postnatal cardiac hypertrophy towards failure is miR‐378. A correlation between miR‐378 and insulin receptor (IGF1‐R) expression levels has been established during postnatal remodelling and cardiomyocyte survival, an observation that is in agreement with the fact that cardiac remodelling is, to some extent, characterized by a decrease in both insulin‐like growth factor (IGF1) and IGF1‐R (Knezevic et al. 2012). Moreover, overexpression of miR‐378 in primary cardiomyocytes inhibited phenylephrine (PE)‐induced Ras activity, therefore preventing the activation of two major cell growth signalling pathways, PI3K–AKT and Raf1–MEK1–ERK1/2, acting downstream of Ras signalling (Knezevic et al. 2012).
Although other anti‐hypertrophic miRNAs have been identified, their precise function in regulating cardiac hypertrophic growth has been less extensively described. It is worth noting that cardiac hypertrophy can be repressed by thioredoxin1 (Trx1) via upregulation of miR‐98, a member of the let‐7 family. Accordingly, downregulation of miR‐98 in a rodent model of angiotensin II (AngII)‐induced hypertrophy resulted in exacerbated cardiac growth, most likely due to elevated levels of its target gene, cyclin D2. In this way, Trx1 acts as a negative feedback regulator of Ang II‐induced cardiac hypertrophy (Yang et al. 2011).
One of the first functional miRNAs shown to be upregulated in cardiac pathological remodelling was miR‐195. Overexpression of this miRNA in cardiomyocytes is sufficient to induce hypertrophic cell growth, and in mice increased expression of miR‐195 leads to cardiomyocyte growth disorganization followed by development of severe hypertrophy already at 6 weeks of age (van Rooij et al. 2006). The same research group and others also found miR‐208 to be upregulated in response to pressure overload and in human heart failure (Tatsuguchi et al. 2007; Thum et al. 2007; van Rooij et al. 2007) and to be cardiac‐specific. miR‐208a was shown to be involved in the regulation of the myosin heavy chain isoform switch that occurs during cardiac development and in the adult heart under pathological conditions (Callis et al. 2009). Overexpression of miR‐208 in mice is sufficient to induce arrhythmias and to regulate hypertrophy‐related signalling pathways leading to pathological remodelling and heart failure (Callis et al. 2009). In later studies, this miRNA was identified in the bloodstream of cardiac disease patients and is now also considered as a potential non‐invasive biomarker of myocardial injuries (Corsten et al. 2010; Wang et al. 2010). Another miRNA from the same family, miR‐499 is also found to be upregulated in both human and murine hypertrophic hearts (Matkovich et al. 2012). Upon pressure overload, augmented miR‐499 expression results in cardiac maladaptation and accelerated transition to heart failure, due to targeting of Akt and MAPKs and thus modulation of cardiac kinase and phosphatase pathways (Matkovich et al. 2012).
Several independent in vivo studies in mice suggest that miR‐22, another heart‐enriched miRNA, is involved in the pathogenesis of HF, despite some controversial observations. While forced miR‐22 expression in cardiomyocytes is sufficient to induce hypertrophy as reflected by increased cell surface areas and increased expression of stress markers (Jentzsch et al. 2012; Huang et al. 2013), loss‐of‐function of miR‐22 also seems to cause premature cardiac dilatation in stressed mouse hearts (Gurha et al. 2012; Huang et al. 2013). In contrast, inhibition of miR‐22 in mice and rats that were subjected to cardiac stress, by using an antagomir, resulted in repressed cardiac hypertrophy without any signs of cardiac dilatation or dysfunction (Friese et al. 2013; Tu et al. 2013). While these results suggest the therapeutic value of modulating miR‐22 to treat cardiomyopathies, one should take into account the fact that miR‐22 was also shown to target PTEN (Xu et al. 2012), a known tumour suppressor. Therefore, reduced expression of miRNA‐22 in other organs, rather than the heart, may increase the risk of promoting cell proliferation and tumour formation.
Several miRNAs have been associated with regulation of calcineurin–NFAT signalling. Increased expression levels of the miR‐212/132 family lead to cardiac hypertrophic growth, heart failure and death (Ucar et al. 2012). These severe effects are due to direct regulation of the transcription factor FOXO3, and subsequent alterations in calcineurin–NFAT signalling. Hence, miR‐212/132 depletion either in genetic animal models or through treatment of animals with an antagomir confers protection from cardiac hypertrophic growth and from eventual development into heart failure (Ucar et al. 2012). miR‐199b, in turn, not only targets calcineurin–NFAT signalling but is also regulated by NFAT itself (da Costa Martins et al. 2010). Upregulation of this miRNA in diseased hearts is responsible for inhibition of NFAT (re)translocation to the cytoplasm by direct targeting of the nuclear NFAT kinase dual‐specificity tyrosine‐(Y)‐phosphorylation regulated kinase 1a (Dyrk1a) (da Costa Martins et al. 2010). In a mouse model of pressure overload, inhibition of miR‐199 restored Dyrka1a expression and this led to reduction of nuclear NFAT activity, preventing or even, more importantly, reversing hypertrophy, fibrosis and cardiac dysfunction (da Costa Martins et al. 2010). Altogether, these findings may contribute to future clinical applications in the treatment of hypertrophic cardiac disease.
Cardiac hypertrophy and heart failure are accompanied by unphysiological accumulation of collagens and other extracellular matrix proteins, leading to cardiac fibrotic lesions. In agreement with these observations, several fibroblast‐enriched miRNAs have been implicated in the development of cardiac hypertrophic growth. Despite several controversial reports on the function and mechanisms of action of miR‐21, this is currently a well‐established regulator of cardiac fibrosis and hypertrophy. miR‐21 was shown to be specifically upregulated in fibroblasts of failing murine hearts and to target intracellular signalling in cardiomyocytes (Thum et al. 2008). In vivo silencing of miR‐21, by means of a cholesterol‐conjugated antagomir, was followed by reduced cardiac fibrosis and hypertrophy after pressure overload, mostly related to lower ERK–MAPK activity in fibroblasts (Thum et al. 2008). Curiously, a different group reported contrasting data showing that in vivo inhibition of miR‐21, with locked nucleic acid‐modified (LNA) nucleotides, did not prevent cardiac remodelling in a mouse model of pressure overload (Patrick et al. 2010). This discrepancy could be due to technical differences such as the chemistry used to inhibit miRNA‐21 or different administration routes. miR‐21 seems to exert its detrimental effect during pathological cardiac remodelling by inducing both proliferation and survival of cardiac fibroblasts (Thum et al. 2008).
Another miRNA displaying dysregulated expression in situations of excessive cardiac fibrosis is miR‐29. This miRNA, simultaneously targets multiple genes that regulate proteins such as elastin, collagens and fibrillins, all associated with the fibrotic response (Abonnenc et al. 2013). Transforming growth factor beta (TGFβ), a known agonist of collagen production and deposition in the heart, was shown to be an upstream regulator of miR‐29, as activation of TGFβ signalling in fibroblasts leads to decreased expression of miR‐29 (van Rooij et al. 2008; Zhang et al. 2014). In a mouse model of AngII‐induced cardiac remodelling, overexpression of miR‐29b is sufficient to inhibit cardiac fibrosis by arresting TGF‐β–Smad3‐dependent signalling (Zhang et al. 2014). Moreover, miR‐29 is significantly more abundant in plasma of patients affected by hypertrophic cardiomyopathy, compared to plasma of healthy controls, underlining the potential of this miRNA as a novel clinical biomarker for the disease (Roncarati et al. 2014; Derda et al. 2015).
Overall, these findings indicate that both miR‐21 and miR‐29 can contribute to myocardial remodelling by primarily acting within cardiac fibroblasts, and suggest that their downregulation could be beneficial by inhibiting fibroblast proliferation and, subsequently, moderating cardiac pathological remodelling.
Hypoxia is the driving force in the regulation of the metabolic switch in the failing heart from main healthy fatty acid oxidation to pathological glucose utilization. While it is well accepted that a cardiac hypoxic environment contributes to the development of heart failure, the molecular mechanisms behind this response remain unclear, and more and more it is becoming clear that it involves not only hypoxia‐inducible factors but also several miRNAs (Azzouzi et al. 2015). Early in vitro studies demonstrated upregulated levels of certain miRNAs, such as miR‐21, in cardiac cells exposed to hypoxia and with increased generation of ROS (Cheng et al. 2009; Simone et al. 2009). In this regard, the expression of the peroxisome proliferator‐activated receptor (PPAR) family of proteins, transcription factors that regulate the expression of fatty acid anabolic and catabolic genetic programmes, is tightly regulated by miRNAs. While upregulation of miR‐21 in heart failure is associated with suppression of PPAR‐a (Matkovich et al. 2009; Leptidis et al. 2013; Yang et al. 2015), a different mechanism for the regulation of PPAR‐d involving miR‐199a and miR‐214 has been proposed (El Azzouzi et al. 2013). PPAR‐g, in turn, seems to be regulated by miR‐27a and ‐27b, both upregulated in hypertrophied hearts (Jentzsch et al. 2012; Wang et al. 2012). Furthermore, regulation of the peroxisome proliferator‐activated receptor‐gamma coactivator‐1α (PGC‐1α), an additional protein with reduced expression in the failing heart, by miR‐696 results not only in less mitochondria but also reduced mitochondrial activity (Aoi et al. 2010). Under cardiac ischaemic conditions, increased levels of the ‘master hypoxamiR’, miR‐210, in mouse hearts result in cardioprotective effects such as preserved cardiac function, improved neovascularization, and inhibition of apoptosis, effects which were mechanistically explained by direct targeting of ephrin A3 (EFNA3) and protein tyrosine phosphatase, non‐receptor type 1 (PTP1b) (Hu et al. 2010; Chan et al. 2012). Many other miRNAs have been associated with cardiac hypoxia and subsequent mitochondrial dysfunction (Azzouzi et al. 2015). It is noteworthy that miR‐181c, being encoded in the nucleus and processed to the mature form in the cytosol, can be translocated into the mitochondria, and ultimately regulate mitochondrial gene expression. Increased mitochondrial miR‐181c expression levels lead to decreased exercise capacity in rats and development of heart failure as a result of altered O2 consumption, ROS production, and mitochondrial membrane potential (Das et al. 2014). The above examples indicate a strong link between the hypoxic response and alterations in cardiac mitochondrial integrity and energy metabolism, both accompanied by direct or indirect miRNA regulation to fine‐tune the balance between fatty acid oxidation versus glucose oxidation and glycolysis.
Long non‐coding RNAS
Long non‐coding RNAs (lncRNA) are a class of pseudogenes (∼200 nucleotides in length) that have lost their protein‐coding function, despite looking similar to protein‐coding genes, as they can also incorporate exons, display a 3′ poli‐A tail and CpG islands in their promoter region and be subject of alternative splicing. Moreover, lncRNAs are poorly conserved throughout evolution, do not display a common sequence or structural motifs and can be intergenic, intronic, antisense (opposite strand relative to annotated transcripts), adjacent to protein‐coding loci or extragenic (Devaux et al. 2015). By targeting promoters, enhancers and insulators as functional cis‐ or trans‐regulatory elements, lncRNAs are a central component in the regulation of epigenetic modifications, allelic modulation (genomic imprinting) and transcriptional/posttranscriptional gene regulation (Nakagawa, 2016). Their ability to switch conformation can either activate or inhibit interactions with other molecules within the cell as lncRNAs can be found in both the cytoplasm and the nucleus. Such changes in lncRNA primary/secondary structure or in expression levels are found to contribute to numerous human diseases by interfering with gene expression and signalling pathways. In the cardiovascular field, several studies have detected and characterized the expression of lncRNAs under normal physiological conditions and in disease states, linking several lncRNAs to different heart failure aetiologies.
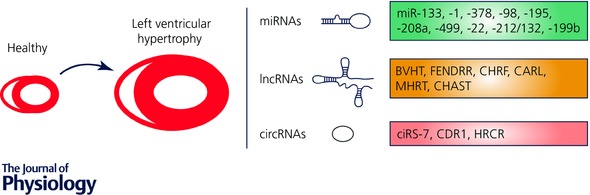
lncRNAs such as Braveheart (BVHT) and Foxf1 adjacent non‐coding developmental regulatory RNA (FENDRR) have been recently proposed to be involved in cardiac development (Grote et al. 2013; Klattenhoff et al. 2013). While BVHT seems to play a pivotal role in regulating cardiomyocyte differentiation, this lncRNA is mouse specific (Klattenhoff et al. 2013). In turn, FENDRR, being more conserved, regulates central transcription factors for cardiac development and its silencing in mice results in severe cardiac developmental defects (Grote et al. 2013). To date, only few studies were set at unveiling the role of lncRNAs in cardiac pathophysiology, with deep sequencing‐based studies having recently characterized cardiac‐enriched lncRNAs and shown altered lncRNA expression patterns in heart failure (Matkovich et al. 2014; Yang et al. 2014). While only 17 lncRNAs were shown to be regulated after early and late (1 and 4 weeks, respectively) pressure overload with mild decline in heart function (Matkovich et al. 2014), more than 100 lncRNAs are differentially regulated in later remodelling stages and heart failure (Lee et al. 2011), suggesting that severe disease states are more intensely regulated by lncRNAs.
Despite the restricted number of lncRNAs found to be highly regulated in cardiac hypertrophy, recent studies have better characterized few of them. Wang and colleagues have shown that lncRNAs can act in concert with miRNAs to modulate cardiac hypertrophic growth. While trying to better understand the mechanism by which miRNA‐489 is regulated, the authors identified a lncRNA, cardiac hypertrophy related factor (CHRF), that, besides being upregulated in murine hearts after transverse aortic constriction and in human heart failure samples, is also widely expressed in cardiovascular cells with a specific function in cardiomyocytes. CHRF induces cardiomyocyte hypertrophy in vitro and apoptosis in vivo by acting as a sponge for miRNA‐489 (Wang et al. 2014a). By sequestering miR‐489, CHFR leads to de‐repression of the target gene myeloid differentiation primary response gene 88 (Myd88), and induces a cardiac hypertrophic phenotype (Wang et al. 2014a). The same group also identified cardiac‐apoptosis‐related lncRNA (CARL) that interacts with miR‐539 like a sponge to inhibit miR‐539 functions in mitochondria fission (Wang et al. 2014b). CARL plays an important role in anoxic conditions such as injury provoked by ischaemia–reperfusion (Wang et al. 2014b). The lncRNA myosin heavy‐chain‐associated RNA transcripts (MHRT), a cluster of antisense transcripts from the Myh7 loci, was recently shown to affect cardiac hypertrophic remodelling and progression to heart failure (Han et al. 2014). Cardiac stress activates the chromatin‐remodelling factor Brg1 (part of the Brg1—HDAC–PARP complex 3) that by repressing α‐MHC and activating β‐MHC maintains a fetal state of MHC expression (Han et al. 2011). MHRT, highly abundant in the nuclear fraction of cardiomyocytes, is downregulated in cardiac pressure overload (Han et al. 2014). Induced expression of MHRT, by antagonizing Brg1 functions, inhibits the pathological switching of α‐MHC to β‐MHC and protects the heart from failing (Han et al. 2014). Interestingly, MHRT and miR‐208a/b are encoded by the same locus but exhibit divergent functions on heart function after pressure overload by conferring protective (MHRT) versus detrimental (miR‐208) effects (van Rooij et al. 2007; Han et al. 2014). These opposing biological functions seem to depend on inverse transcriptional regulation of the RNAs encoded by the locus since cardiac stress induces both transcriptional downregulation of MHRT and β‐MHC–miR‐208 upregulation, suggesting the action of independent promoters in the same genomic region.
It is important to underline that human MHRT also originates from Myh7 (β‐MHC) loci, indicating that this lncRNA is conserved through species and suggesting that this mechanism of feedback between MHRT and Brg1 is important in the onset of human hypertrophic cardiomyopathy.
The most recent lncRNA to be associated with cardiac hypertrophic remodelling and failure was cardiac hypertrophy‐associated transcript (CHAST). Pressure overload‐induced cardiac stress leads to specific CHAST upregulation in murine cardiomyocytes (Viereck et al. 2016). In humans the expression levels of this lncRNA are increased in cardiac tissue of aortic stenosis patients and in human embryonic stem cell‐derived cardiomyocytes after hypertrophic stimulation (Viereck et al. 2016). Overexpression of CHAST in mice is sufficient to induce cardiomyocyte hypertrophic growth, while its silencing allows for both prevention and reversal of pressure overload‐induced pathological cardiac remodelling (Viereck et al. 2016). Mechanistically, CHAST inhibits cardiomyocyte autophagy by targeting pleckstrin homology domain‐containing protein family M member 1, expressed on the opposite strand of CHAST.
By using diverse mechanisms to regulate pivotal signalling pathways in the development of cardiac disease such as chromatin modification and transcriptional regulation, lncRNAs are now emerging as potential targets to prevent pathological cardiac remodelling.
Circular RNAS
Deep sequencing and advanced data analysis identified a diversity of circular RNA isoforms (circRNAs) in different biological systems. It is assumed that circRNAs do not code for proteins due to their circular structure and, in contrast to other non‐coding RNAs, the lack of both 5′ and 3′ polarity and a poli‐A tail (Qu et al. 2015; Chen, 2016). Regarding their genomic location, circRNAs can be classified as being exonic, intronic, antisense, intragenic or intergenic, depending on their genomic adjacency to a close gene (Qu et al. 2015), and display distinctive properties, including the potential for circle amplification of RNA, the ability to reorganize the order of genomic information, protection from exonucleases, and constraints on RNA folding. Furthermore, the production of circRNAs is mostly certain to influence mRNA processing by playing a role in alternative splicing, since splicing patterns of any pre‐mRNA are dictated by direct competition between alternative pairing of 5′ and 3′ splice sites (Ashwal‐Fluss et al. 2014). It is worth noting that circRNAs are ancient non‐coding RNAs, but only lately have they started being newly appreciated for their essential role in many processes such as ageing, cancer, cardiovascular diseases and tissue development (Qu et al. 2015). In fact, it has been reported for several circRNAs that they can function as endogenous sponges to interact with miRNAs and influence the expression of their target genes. This was shown for miR‐7 as circRNAs ciRS‐7 (Hansen et al. 2013) and CDR1 (Memczak et al. 2013) are able to sequester miR‐7 away from its target genes in different biological settings. More recently, a new circRNA, heart‐related circRNA (HRCR) was identified as being anti‐hypertrophic by targeting the positive regulator of hypertrophy, miR‐223. miR‐223 inhibition through HRCR leads to increased levels of the target gene activity‐regulated cytoskeleton‐associated protein, ARC, associated with reduced pressure overload‐induced cardiac hypertrophy in mice. Whether this is a general mechanism of action for the many existent circRNAs or just a gene‐specific function remains to be clarified.
An inventory of circRNA expression and respective alteration in the hearts of different species was recently generated after comparing rat neonatal cardiomyocytes with adult ones, mouse hearts subjected to sham surgery with hearts subjected to pressure overload by transverse aortic constriction and, finally, human non‐failing with failing hearts. This set of data, besides providing information on the conservation of the different circRNAs between the three studied species, also demonstrates that overall changes in circRNA regulation are higher in rat cardiomyocytes than in failing murine and human hearts. As mentioned earlier, the formation of circRNAs may influence mRNA processing by competing with splice events (Ashwal‐Fluss et al. 2014). This has been suggested to explain the diversity of titin splicing, which is linked to hereditary cardiomyopathy in rats and humans (Khan et al. 2016). Altogether, it seems reasonable to speculate that altered circRNA levels may contribute to the disease phenotypes in different species.
Cell‐to‐cell transfer of non‐coding RNAs
Cells communicate with one another to create specific microenvironments and share resources that are essential to maintain homeostasis and respond to external stimuli. To fully understand the biology and pathobiology of the heart and to gain new insights into the molecular regulation of a myocardial hypertrophic response, the contribution of cell–cell crosstalk in the heart to this process must be considered.
In recent years, more interest has been concentrated on extracellular vesicles (EVs) that allow long‐range cellular communication (EL Andaloussi et al. 2013). EVs are secreted by cells and act as transport vehicles for a range of small molecules like mRNA, miRNAs, lncRNAs, small amounts of DNA and low molecular weight lipids and proteins (including transcription factors and cytokines) (Valadi et al. 2007; EL Andaloussi et al. 2013).
EVs can be classified in three different subgroups according to their subcellular origin, content and size: microvesicles (MVs) (0.1–1 μm), exosomes (20–100 nm) and apoptotic bodies (ABs) (0.5–2 μm) (EL Andaloussi et al. 2013). Cardiac cells are able to secrete MVs and exosomes under physiological and pathological conditions. All major cardiac cell types, including cardiomyocytes, endothelial cells and fibroblasts, can release exosomes that modulate recipient cellular functions and may therefore be involved in many cardiovascular disorders. Nevertheless, only a relatively small number of studies have been dedicated to investigating the potential role of exosomes during cardiomyocyte hypertrophy, specifically, and cardiac disease in general (Fig. 2).
Figure 2. Cardiac cell‐to‐cell transfer of non‐coding RNAs.
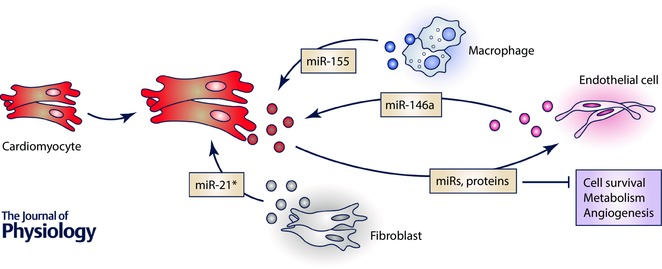
In general cells communicate with each other to create distinct microenvironments and share resources. Cardiac cells, including cardiomyocytes, fibroblasts, endothelial and immune cells, also communicate with each other by means of extracellular vesicles, namely exosomes (represented by dots in different colours, depending on cell source). Cardiomyocytes are known to directly transfer exosome cargo to endothelial cells and thereby alter several basic cellular functions on the recipient cells such as cell cycle, metabolism and angiogenesis. To date, a very limited number of studies have mainly focused on identifying the content of exosomes that are transferred from other cell types (endothelial cells, fibroblasts and macrophages) into cardiomyocytes and how this cargo interferes with cardiomyocyte behaviour and, subsequently, cardiac remodelling.
It has been proposed that exosomal signalling during hypoxia mediates microvascular endothelial cell migration and, subsequently, vasculogenesis (Salomon et al. 2013). In the heart, exosomes released by cardiomyocytes can induce different metabolic functions in the recipient cells (Waldenström et al. 2012). Cardiomyocytes and endothelial cells (ECs) have a close anatomical relationship that is essential for maintaining normal cardiac development and function. However, little is known about the mechanisms by which both cell types crosstalk not only under physiological conditions but also upon cardiac stress. Under conditions of glucose deficiency, cardiomyocytes secrete not only larger numbers of exosomes but their protein content also significantly differs from those produced in the presence of glucose (Garcia et al. 2015). In the presence of glucose, the exosomal proteins were mainly associated with cardiovascular processes, while in deprived cardiomyocyte‐released exosomes the protein content was related to vesicle trafficking and metabolic processes (Garcia et al. 2015). Although these findings were only supported by in vitro data, cardiomyocytes were able to exchange their exosomal content with the surrounding endothelium, altering the gene transcription in the recipient cells and affecting their angiogenesis‐related capacities such as proliferation and tube formation. Furthermore, exosomes in deprivation conditions were enriched in a set of miRNAs known to be pro‐angiogenic when internalized in ECs. In turn, ECs also released exosomes that could be taken up by cardiomyocytes and influence their physiological functions and responsive mechanisms to stress. This has been shown in women affected with peripartum cardiomyopathy (PPCM), where an anti‐angiogenic 16‐kDa N‐terminal prolactin fragment (16K PRL) acts on ECs to inhibit their migratory capacity, block cell cycle progression, induce apoptosis and, subsequently, induce the release of miR‐146a‐enriched exosomes (Halkein et al. 2013). These exosomes can be taken up by cardiomyocytes where miR‐146a will interfere with the physiological metabolism and contraction capacity of the cell, leading to the development of hypertrophy (Halkein et al. 2013). Pharmacological inhibition of miR‐146a in a mouse model of PPCM induced metabolic activity in improved cardiac function by increasing the expression levels of miR‐146a target genes in cardiomyocytes (Halkein et al. 2013). The fact that the cardiac tissue of PPCM patients exhibits increased expression of miR‐146a and decreased respective target levels when compared to non‐failing tissue suggests that the proposed mechanism is also conserved in humans. In addition, this study also established miR‐146a as a potential highly specific blood biomarker in the diagnosis and risk stratification of patients with PPCM (Halkein et al. 2013).
A recent study by Climent and colleagues (Climent et al. 2015) identified the transfer of miR‐143/‐145 cluster from smooth muscle cells towards ECs via a different form of cell‐to‐cell communication that involves membrane protrusions named tunnelling nanotubes and a feedback regulatory mechanism between the two types of cells (Climent et al. 2015). Endothelial cells receiving the miRNA cluster display reduced angiogenic capacity and will release transforming growth factor b (TGFb), which in turn stimulates the differentiation of smooth muscle cells and upregulation of miR‐143/145 cluster in these cells. Once transferred to ECs the miRNA cluster will reduce endothelial capacity to form capillary structures (Climent et al. 2015).
Cardiac fibroblasts can also respond to pathological situations by releasing exosomes. When treated with angiotensin II, fibroblasts secrete exosomes that, in turn, stimulate more angiotensin production and respective receptor expression (AT1R and AT2R) on cardiomyocytes, leading to hypertrophic cell growth. This exosome‐induced effect could be blocked either by AT1R and AT2R antagonists or with exosome inhibitors. Moreover, fibroblasts can exert paracrine effects through the release of exosomes containing the passenger strand of miR‐21 (miR21*), which will be taken up by cardiomyocytes and induce their hypertrophic growth (Bang et al. 2014). miR‐21* induces cardiac hypertrophy by downregulating sorbin and SH3 domain containing 2 (SORBS2) or PDZ and LIM domain 5 (PDLIM5), both involved in regulation of cardiac muscle structure and function (Bang et al. 2014). By using cardiac allografts in mice it was demonstrated that inhibition of miR‐21* prevents fibrosis and decreases the infiltration of fibroblasts in the allograft (Bang et al. 2014). This is a striking example of how miRNAs* are not necessarily degraded, but can have functional roles and great potential as diagnostic and therapeutic tools in cardiovascular diseases, similar to mature strands. Altogether, such findings suggest that targeting exosome release from cardiac fibroblasts may serve as a novel therapeutic strategy to treat cardiac pathological hypertrophy and heart failure.
No less importantly, immune cells may also contribute to the cardiac response to stress. miR‐155 was shown to regulate cardiac hypertrophy in different mouse models of cardiac failure. miR‐155 null mice are protected from developing cardiac hypertrophy, an effect attributed to reduced miR‐155 expression in macrophages rather than in cardiomyocytes (Heymans et al. 2013). Supporting this hypothesis, miR‐155‐deficient macrophages were able to inhibit the hypertrophic phenotype in stimulated cardiomyocytes through a paracrine effect (Heymans et al. 2013). This is again a very neat example of crosstalk between different myocardial cell types and how they respond to different sources of cardiac stress.
Although not within the scope of this review, it is important to highlight that circulating exosomes found in body fluids such as plasma and serum are becoming an attractive tool for analytical studies and subsequent disease diagnosis, underpinning the prospect that differential exosomal content can lead to new cardiac‐specific diagnostic markers.
Conclusion and future perspectives
In the past decade, the fundamental role of ncRNAs in cardiac pathophysiology has become more evident. While, using different animal models, scientists could use loss‐ or gain‐of‐function strategies to treat, and in certain cases, reverse cardiac hypertrophy, the prime challenge lies in modulating these ncRNAs and their targets in an organ/cell‐specific manner as a potential therapeutic for heart failure. Nevertheless, promising results have been generated that open a window to patients’ treatment and/or improvement of lifespan and quality of life.
Most of the ncRNAs described above were unknown until the 1980s, with most having been discovered only in the 1990s. What appeared to be a relatively simple picture of genetic control in cells has now gained many, previously hidden, layers of complexity involving many RNA species, ranging from a mere 20 nucleotides in length up to 100,000. And, as many believe, this might be just a glimpse of the tip of the iceberg that still needs to be discovered to fully understand the importance of non‐coding RNAs. Genomics, the study of the DNA sequence of genomes, has been a sizzling field for some time now, and is more and more focused on the discovery of ncRNAs. Because the field of ncRNAs is in its infancy and the functions of many of the ncRNAs are just barely understood, it may be premature to predict specific medical applications, but certainly the potential is there. The population of ncRNAs in a cell, in some sense, resembles a complex set of switches that turn genes on and off before they are transcribed, while they are being transcribed, or even once translation has begun. Once these switches are better understood, researchers may be able to better exploit the system with artificially produced RNAs, enabling clarification of a number of diseases, including cardiac diseases, that currently appear to be associated with unexplained genetic behaviours. Moreover, the connection between ncRNAs and epigenetic modifications is another important aspect that is still poorly understood in the heart, adding to the regulatory complexity of pathological processes.
Additional information
Competing interests
L.O. has no conflicts of interest with regard to this manuscript. P.C.M. is co‐founder of Mirabilis Therapeutics, a spin‐off company of the University of Maastricht.
Author contributions
L.O. collected relevant literature to compile this review and wrote a first draft of the manuscript; P.C.M. structured the manuscript, collected relevant literature, prepared the figures and wrote the paper as submitted.
Funding
This work was funded by a Dr Dekker Established Investigator grant of the Dutch Heart Foundation (2015T066) to P.C.M.
Biographies
Lara Ottaviani completed her MSc in Medical Biotechnology at the Catholic University in Rome, Italy. Currently, she is following her PhD programme at the University of Maastricht by focusing her research on exosomal transfer of microRNAs between distinct cardiac cell types.

Paula da Costa Martins is an Associate Professor in the Faculty of Health, Medicine and at the University of Maastricht. Her major research interests revolve around the regulation of cellular processes related to pathological cardiac remodelling towards heart failure by non‐coding RNAs. She focuses her research in understanding how contracting cardiac muscle cells communicate their needs to the surrounding cells and how these cells respond in synchrony to meet such needs.
References
- Abonnenc M, Nabeebaccus AA, Mayr U, Barallobre‐Barreiro J, Dong X, Cuello F, Sur S, Drozdov I, Langley SR, Lu R, Stathopoulou K, Didangelos A, Yin X, Zimmermann WH, Shah AM, Zampetaki A & Mayr M (2013). Extracellular matrix secretion by cardiac fibroblasts: Role of MicroRNA‐29b and MicroRNA‐30c. Circ Res 113, 1138–1147. [DOI] [PubMed] [Google Scholar]
- Aoi W, Naito Y, Mizushima K, Takanami Y, Kawai Y, Ichikawa H & Yoshikawa T (2010). The microRNA miR‐696 regulates PGC‐1α in mouse skeletal muscle in response to physical activity. Am J Physiol Endocrinol Metab 298, E799–E806. [DOI] [PubMed] [Google Scholar]
- Archer K, Broskova Z, Bayoumi AS, Teoh JP, Davila A, Tang Y, Su H & Kim IM (2015). Long non‐coding RNAs as master regulators in cardiovascular diseases. Int J Mol Sci 16, 23651–23667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashwal‐Fluss R, Meyer M, Pamudurti NR, Ivanov A, Bartok O, Hanan M, Evantal N, Memczak S, Rajewsky N & Kadener S (2014). CircRNA Biogenesis competes with Pre‐mRNA splicing. Mol Cell 56, 55–66. [DOI] [PubMed] [Google Scholar]
- Azzouzi H el, Leptidis S, Doevendans PA & De Windt LJ (2015). HypoxamiRs: regulators of cardiac hypoxia and energy metabolism. Trends Endocrinol Metab 26, 502–508. [DOI] [PubMed] [Google Scholar]
- Bang C, Batkai S, Dangwal S, Gupta SK, Foinquinos A, Holzmann A, Just A, Remke J, Zimmer K, Zeug A, Ponimaskin E, Schmiedl A, Yin X, Mayr M, Halder R, Fischer A, Engelhardt S, Wei Y, Schober A, Fiedler J & Thum T (2014). Cardiac fibroblast‐derived microRNA passenger strand‐enriched exosomes mediate cardiomyocyte hypertrophy. J Clin Invest 124, 2136–2146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartel DP (2004). MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 116, 281–297. [DOI] [PubMed] [Google Scholar]
- Bejerano G, Pheasant M, Makunin I, Stephen S, Kent WJ, Mattick JS & Haussler D (2004). Ultraconserved elements in the human genome. Science 304, 1321–1325. [DOI] [PubMed] [Google Scholar]
- De Boer RA, Pinto YM & Van Veldhuisen DJ (2003). The imbalance between oxygen demand and supply as a potential mechanism in the pathophysiology of heart failure: the role of microvascular growth and abnormalities. Microcirculation 10, 113–126. [DOI] [PubMed] [Google Scholar]
- Callis TE, Pandya K, Hee YS, Tang RH, Tatsuguchi M, Huang ZP, Chen JF, Deng Z, Gunn B, Shumate J, Willis MS, Selzman CH & Wang DZ (2009). MicroRNA‐208a is a regulator of cardiac hypertrophy and conduction in mice. J Clin Invest 119, 2772–2786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carè A, Catalucci D, Felicetti F, Bonci D, Addario A, Gallo P, Bang ML, Segnalini P, Gu Y, Dalton ND, Elia L, Latronico MV, Høydal M, Autore C, Russo MA, Dorn GW 2nd, Ellingsen O, Ruiz‐Lozano P, Peterson KL, Croce CM, Peschle C & Condorelli G (2007). MicroRNA‐133 controls cardiac hypertrophy. Nat Med 13, 613–618. [DOI] [PubMed] [Google Scholar]
- Castaldi A, Zaglia T, Di Mauro V, Carullo P, Viggiani G, Borile G, Di Stefano B, Schiattarella GG, Gualazzi MG, Elia L, Stirparo GG, Colorito ML, Pironti G, Kunderfranco P, Esposito G, Bang ML, Mongillo M, Condorelli G & Catalucci D (2014). MicroRNA‐133 modulates theβ1‐adrenergic receptor transduction cascade. Circ Res 115, 273–283. [DOI] [PubMed] [Google Scholar]
- Cech TR & Steitz JA (2014). The noncoding RNA revolution – trashing old rules to forge new ones. Cell 157, 77–94. [DOI] [PubMed] [Google Scholar]
- Chan YC, Banerjee J, Choi SY & Sen CK (2012). miR‐210: The master hypoxamir. Microcirculation 19, 215–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L (2016). The biogenesis and emerging roles of circular RNAs. Nat Rev Mol Cell Biol 17, 205–211. [DOI] [PubMed] [Google Scholar]
- Cheng Y, Liu X, Zhang S, Lin Y, Yang J & Zhang C (2009). MicroRNA‐21 protects against the H2O2‐induced injury on cardiac myocytes via its target gene PDCD4. J Mol Cell Cardiol 47, 5–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Climent M, Quintavalle M, Miragoli M, Chen J, Condorelli G & Elia L (2015). TGFβ triggers miR‐143/145 transfer from smooth muscle cells to endothelial cells, thereby modulating vessel stabilization. Circ Res 116, 1753–1764. [DOI] [PubMed] [Google Scholar]
- Corsten MF, Dennert R, Jochems S, Kuznetsova T, Devaux Y, Hofstra L, Wagner DR, Staessen JA, Heymans S & Schroen B (2010). Circulating MicroRNA‐208b and MicroRNA‐499 reflect myocardial damage in cardiovascular disease. Circ Cardiovasc Genet 3, 499–506. [DOI] [PubMed] [Google Scholar]
- da Costa Martins PA, Salic K, Gladka MM, Armand A‐S, Leptidis S, el Azzouzi H, Hansen A, Coenen‐de Roo CJ, Bierhuizen MF, van der Nagel R, van Kuik J, de Weger R, de Bruin A, Condorelli G, Arbones ML, Eschenhagen T & De Windt LJ (2010). MicroRNA‐199b targets the nuclear kinase Dyrk1a in an auto‐amplification loop promoting calcineurin/NFAT signaling. Nat Cell Biol 12, 1220–1227. [DOI] [PubMed] [Google Scholar]
- Das S, Bedja D, Campbell N, Dunkerly B, Chenna V, Maitra A & Steenbergen C (2014). miR‐181c regulates the mitochondrial genome, bioenergetics, and propensity for heart failure in vivo . PLoS One 9, e96820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derda AA, Thum S, Lorenzen JM, Bavendiek U, Heineke J, Keyser B, Stuhrmann M, Givens RC, Kennel PJ, Christian Schulze P, Widder JD, Bauersachs J & Thum T (2015). Blood‐based microRNA signatures differentiate various forms of cardiac hypertrophy. Int J Cardiol 196, 115–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devaux Y, Zangrando J, Schroen B, Creemers EE, Pedrazzini T, Chang C‐P, Dorn GW, Thum T & Heymans S (2015). Long noncoding RNAs in cardiac development and ageing. Nat Rev Cardiol 12, 415–425. [DOI] [PubMed] [Google Scholar]
- Duisters RF, Tijsen AJ, Schroen B, Leenders JJ, Lentink V, vanderMade I, Herias V, vanLeeuwen RE, Schellings MW, Barenbrug P, Maessen JG, Heymans S, Pinto YM & Creemers EE (2009). miR‐133and miR‐30 regulate connective tissue growth factor: implications for a role of micrornas in myocardial matrix remodelling. Circ Res 104, 170–178. [DOI] [PubMed] [Google Scholar]
- EL Andaloussi SE, Mäger I, Breakefield XO & Wood MJ (2013). Extracellular vesicles: biology and emerging therapeutic opportunities. Nat Rev Drug Discov 12, 347–357. [DOI] [PubMed] [Google Scholar]
- El Azzouzi H, Leptidis S, Dirkx E, Hoeks J, van Bree B, Brand K, McClellan EA, Poels E, Sluimer JC, van den Hoogenhof MM, Armand AS, Yin X, Langley S, Bourajjaj M, Olieslagers S, Krishnan J, Vooijs M, Kurihara H, Stubbs A, Pinto YM, Krek W, Mayr M, da Costa Martins PA, Schrauwen P & De Windt LJ (2013). The hypoxia‐inducible microRNA cluster miR‐199a∼214 targets myocardial PPARδ and impairs mitochondrial fatty acid oxidation. Cell Metab 18, 341–354. [DOI] [PubMed] [Google Scholar]
- Elia L, Contu R, Quintavalle M, Varrone F, Chimenti C, Russo MA, Cimino V, De Marinis L, Frustaci A, Catalucci D & Condorelli G (2009). Reciprocal regulation of microrna‐1 and insulin‐like growth factor‐1 signal transduction cascade in cardiac and skeletal muscle in physiological and pathological conditions. Circulation 120, 2377–2385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epelman S, Lavine KJ, Beaudin AE, Sojka DK, Carrero JA, Calderon VB, Brija T, Gautier EL, Ivanov S, Satpathy AT, Schilling JD, Schwendener R, Sergin I, Razani B, Forsberg EC, Yokoyama WM, Unanue ER, Colonna M, Randolph GJ & Mann DL (2014). Embryonic and adult‐derived resident cardiac macrophages are maintained through distinct mechanisms at steady state and during inflammation. Immunity 40, 91–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friese RS Altshuler AE, Zhang K, Miramontes‐Gonzalez JP, Hightower CM, Jirout ML, Salem RM, Gayen JR, Mahapatra NR, Biswas N, Cale M, Vaingankar SM, Kim HS, Courel M, Taupenot L, Ziegler MG, Schork NJ, Pravenec M, Mahata SK, Schmid‐Schönbein GW & O'Connor DT (2013). MicroRNA‐22 and promoter motif polymorphisms at the Chga locus in genetic hypertension: Functional and therapeutic implications for gene expressionand the pathogenesis of hypertension. Hum Mol Genet 22, 3624–3640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia NA, Ontoria‐Oviedo I, González‐King H, Diez‐Juan A & Sepúlveda P (2015). Glucose starvation in cardiomyocytes enhances exosome secretion and promotes angiogenesis in endothelial cells. PLoS One 10, e0138849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghildiyal M & Zamore PD (2009). Small silencing RNAs: an expanding universe. Nat Rev Genet 10, 94–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grote P, Wittler L, Hendrix D, Koch F, Währisch S, Beisaw A, Macura K, Bläss G, Kellis M, Werber M & Herrmann BG (2013). The tissue‐specific lncRNA Fendrr is an essential regulator of heart and body wall development in the mouse. Dev Cell 24, 206–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurha P, Abreu‐Goodger C, Wang T, Ramirez MO, Drumond AL, Van Dongen S, Chen Y, Bartonicek N, Enright AJ, Lee B, Kelm RJ, Reddy AK, Taffet GE, Bradley A, Wehrens XH, Entman ML & Rodriguez A (2012). Targeted deletion of MicroRNA‐22 promotes stress‐induced cardiac dilation and contractile dysfunction. Circulation 125, 2751–2761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halkein J, Tabruyn SP, Ricke‐Hoch M, Haghikia A, Nguyen NQN, Scherr M, Castermans K, Malvaux L, Lambert V, Thiry M, Sliwa K, Noel A, Martial JA, Hilfiker‐Kleiner D & Struman I (2013). MicroRNA‐146a is a therapeutic target and biomarker for peripartum cardiomyopathy. J Clin Invest 123, 2143–2154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han P, Hang CT, Yang J & Chang CP (2011). Chromatin remodelling in cardiovascular development and physiology. Circ Res 108, 378–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han P, Li W, Lin C‐H, Yang J, Shang C, Nurnberg ST, Jin KK, Xu W, Lin C‐Y, Lin C‐J, Xiong Y, Chien H‐C, Zhou B, Ashley E, Bernstein D, Chen P‐S, Chen H‐SV, Quertermous T & Chang C‐P (2014). A long noncoding RNA protects the heart from pathological hypertrophy. Nature 514, 102–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen TB, Jensen TI, Clausen BH, Bramsen JB, Finsen B, Damgaard CK & Kjems J (2013). Natural RNA circles function as efficient microRNA sponges. Nature 495, 384–388. [DOI] [PubMed] [Google Scholar]
- Heymans S, Corsten MF, Verhesen W, Carai P, van Leeuwen RE, Custers K, Peters T, Hazebroek M, Stöger L, Wijnands E, Janssen BJ, Creemers EE, Pinto YM, Grimm D, Schürmann N, Vigorito E, Thum T, Stassen F, Yin X, Mayr M, de Windt LJ, Lutgens E, Wouters K, de Winther MP, Zacchigna S, Giacca M, van Bilsen M, Papageorgiou AP & Schroen B (2013). Macrophage MicroRNA‐155 promotes cardiac hypertrophy and failure. Circulation 128, 1420–1432. [DOI] [PubMed] [Google Scholar]
- Hu S, Huang M, Li Z, Jia F, Ghosh Z, Lijkwan MA, Fasanaro P, Sun N, Wang X, Martelli F, Robbins RC & Wu JC (2010). MicroRNA‐210 as a novel therapy for treatment of ischemic heart disease. Circulation 122 (Suppl.), S124–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang ZP, Chen J, Seok HY, Zhang Z, Kataoka M, Hu X & Wang DZ (2013). MicroRNA‐22 regulates cardiac hypertrophy and remodelling in response to stress. Circ Res 112, 1234–1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jentzsch C, Leierseder S, Loyer X, Flohrschütz I, Sassi Y, Hartmann D, Thum T, Laggerbauer B & Engelhardt S (2012). A phenotypic screen to identify hypertrophy‐modulating microRNAs in primary cardiomyocytes. J Mol Cell Cardiol 52, 13–20. [DOI] [PubMed] [Google Scholar]
- Karakikes I, Chaanine AH, Kang S, Mukete BN, Jeong D, Zhang S, Hajjar RJ & Lebeche D (2013). Therapeutic cardiac‐targeted delivery of miR‐1 reverses pressure overload‐induced cardiac hypertrophy and attenuates pathological remodelling. J Am Heart Assoc 2, e000078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kehat I & Molkentin JD (2010). Molecular pathways underlying cardiac remodelling during pathophysiological stimulation. Circulation 122, 2727–2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan MAF, Reckman YJ, Aufiero S, van den Hoogenhof MMGG, van der Made I, Beqqali A, Koolbergen DR, Rasmussen TB, van der Velden J, Creemers EE & Pinto YM (2016). RBM20 regulates circular RNA production from the Titin gene. Circ Res 119, 996–1003. [DOI] [PubMed] [Google Scholar]
- Klattenhoff CA, Scheuermann JC, Surface LE, Bradley RK, Fields PA, Steinhauser ML, Ding H, Butty VL, Torrey L, Haas S, Abo R, Tabebordbar M, Lee RT, Burge CB & Boyer LA (2013). Braveheart, a long noncoding RNA required for cardiovascular lineage commitment. Cell 152, 570–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinjan DA & Van Heyningen V (2005). Long‐range control of gene expression: emerging mechanisms and disruption in disease. Am J Hum Genet 76, 8–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knezevic I, Patel A, Sundaresan NR, Gupta MPM, Solaro RJ, Nagalingam RS & Gupta MPM (2012). A novel cardiomyocyte‐enriched MicroRNA, miR‐378, targets insulin‐like growth factor 1 receptor: Implications in postnatal cardiac remodelling and cell survival. J Biol Chem 287, 12913–12926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konstam MA, Kramer DG, Patel AR, Maron MS & Udelson JE (2011). Left ventricular remodelling in heart failure: current concepts in clinical significance and assessment. JACC Cardiovasc Imaging 4, 98–108. [DOI] [PubMed] [Google Scholar]
- Lee JH, Gao C, Peng G, Greer C, Ren S, Wang Y & Xiao X (2011). Analysis of transcriptome complexity through RNA sequencing in normal and failing murine hearts. Circ Res 109, 1332–1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leptidis S, el Azzouzi H, Lok SI, de Weger R, Olieslagers S, Kisters N, Silva GJ, Heymans S, Cuppen E, Berezikov E, de Windt LJ & da Costa Martins P (2013). A Deep sequencing approach to uncover the miRNOME in the human heart. PLoS One 8, e57800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lips DJ, DeWindt LJ, Van Kraaij DJW & Doevendans PA (2003). Molecular determinants of myocardial hypertrophy and failure: Alternative pathways for beneficial and maladaptive hypertrophy. Eur Heart J 24, 883–896. [DOI] [PubMed] [Google Scholar]
- Matkovich SJ, Van Booven DJ, Youker KA, Torre‐Amione G, Diwan A, Eschenbacher WH, Dorn LE, Watson MA, Margulies KB & Dorn GW (2009). Reciprocal regulation of myocardial microRNAs and messenger RNA in human cardiomyopathy and reversal of the microRNA signature by biomechanical support. Circulation 119, 1263–1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matkovich SJ, Edwards JR, Grossenheider TC, de Guzman Strong C & Dorn GW (2014). Epigenetic coordination of embryonic heart transcription by dynamically regulated long noncoding RNAs. Proc Natl Acad Sci USA 111, 12264–12269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matkovich SJ, Hu Y, Eschenbacher WH, Dorn LE & Dorn GW (2012). Direct and indirect involvement of MicroRNA‐499 in clinical and experimental cardiomyopathy. Circ Res 111, 521–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matkovich SJ, Wang W, Tu Y, Eschenbacher WH, Dorn LE, Condorelli G, Diwan A, Nerbonne JM & Dorn GW (2010). MicroRNA‐133a protects against myocardial fibrosis and modulates electrical repolarization without affecting hypertrophy in pressure‐overloaded adult hearts. Circ Res 106, 166–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Memczak S, Jens M, Elefsinioti A, Torti F, Krueger J, Rybak A, Maier L, Mackowiak SD, Gregersen LH, Munschauer M, Loewer A, Ziebold U, Landthaler M, Kocks C, le Noble F & Rajewsky N (2013). Circular RNAs are a large class of animal RNAs with regulatory potency. Nature 495, 333–338. [DOI] [PubMed] [Google Scholar]
- Nakagawa S (2016). Lessons from reverse‐genetic studies of lncRNAs. Biochim Biophys Acta 1859, 177–183. [DOI] [PubMed] [Google Scholar]
- Papageorgiou N, Tsalamandris S, Giolis A & Tousoulis D (2015). MicroRNAs in cardiovascular disease: perspectives and reality. Cardiol Rev 24, 110–118. [DOI] [PubMed] [Google Scholar]
- Patrick DM, Montgomery RL, Qi X, Obad S, Kauppinen S, Hill JA, Van Rooij E & Olson EN (2010). Stress‐dependent cardiac remodelling occurs in the absence of microRNA‐21 in mice. J Clin Invest 120, 3912–3916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennacchio LA, Ahituv N, Moses AM, Prabhakar S, Nobrega MA, Shoukry M, Minovitsky S, Dubchak I, Holt A, Lewis KD, Plajzer‐Frick I, Akiyama J, De Val S, Afzal V, Black BL, Couronne O, Eisen MB, Visel A & Rubin EM (2006). In vivo enhancer analysis of human conserved non‐coding sequences. Nature 444, 499–502. [DOI] [PubMed] [Google Scholar]
- Qu S, Yang X, Li X, Wang J, Gao Y, Shang R, Sun W, Dou K & Li H (2015). Circular RNA: A new star of noncoding RNAs. Cancer Lett 365, 141–148. [DOI] [PubMed] [Google Scholar]
- Roncarati R, Viviani Anselmi C, Losi MA, Papa L, Cavarretta E, Da Costa Martins P, Contaldi C, Saccani Jotti G, Franzone A, Galastri L, Latronico MVG, Imbriaco M, Esposito G, De Windt L, Betocchi S & Condorelli G (2014). Circulating miR‐29a, among other up‐regulated microRNAs, is the only biomarker for both hypertrophy and fibrosis in patients with hypertrophic cardiomyopathy. J Am Coll Cardiol 63, 920–927. [DOI] [PubMed] [Google Scholar]
- van Rooij E, Sutherland LB, Liu N, Williams AH, McAnally J, Gerard RD, Richardson JA & Olson EN (2006). A signature pattern of stress‐responsive microRNAs that can evoke cardiac hypertrophy and heart failure. Proc Natl Acad Sci USA 103, 18255–18260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Rooij E, Sutherland LB, Qi X, Richardson JA, Hill J & Olson EN (2007). Control of stress‐dependent cardiac growth and gene expression by a microRNA. Science (80‐ ) 316, 575–579. [DOI] [PubMed] [Google Scholar]
- van Rooij E, Sutherland LB, Thatcher JE, DiMaio JM, Naseem RH, Marshall WS, Hill JA & Olson EN (2008). Dysregulation of microRNAs after myocardial infarction reveals a role of miR‐29 in cardiac fibrosis. Proc Natl Acad Sci USA 105, 13027–13032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salomon C, Ryan J, Sobrevia L, Kobayashi M, Ashman K, Mitchell M & Rice GE (2013). Exosomal signaling during hypoxia mediates microvascular endothelial cell migration and vasculogenesis. PLoS One 8, e68451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salzman J (2016). Circular RNA expression: Its Potential regulation and function. Trends Genet 32, 309–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salzman J, Chen RE, Olsen MN, Wang PL & Brown PO (2013). Cell‐type specific features of circular RNA expression. PLoS Genet 9, e1003777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu I & Minamino T (2016). Physiological and pathological cardiac hypertrophy. J Mol Cell Cardiol 97, 245–262. [DOI] [PubMed] [Google Scholar]
- Shiojima I, Sato K, Izumiya Y, Schiekofer S, Ito M, Liao R, Colucci WS & Walsh K (2005). Disruption of coordinated cardiac hypertrophy and angiogenesis contributes to the transition to heart failure. J Clin Invest 115, 2108–2118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simone NL, Soule BP, Ly D, Saleh AD, Savage JE, Degraff W, Cook J, Harris CC, Gius D & Mitchell JB (2009). Ionizing radiation‐induced oxidative stress alters miRNA expression. PLoS One 4, e6377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatsuguchi M, Seok HY, Callis TE, Thomson JM, Chen JF, Newman M, Rojas M, Hammond SM & Wang DZ (2007). Expression of microRNAs is dynamically regulated during cardiomyocyte hypertrophy. J Mol Cell Cardiol 42, 1137–1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thum T, Gross C, Fiedler J, Fischer T, Kissler S, Bussen M, Galuppo P, Just S, Rottbauer W, Frantz S, Castoldi M, Soutschek J, Koteliansky V, Rosenwald A, Basson MA, Licht JD, Pena JT, Rouhanifard SH, Muckenthaler MU, Tuschl T, Martin GR, Bauersachs J & Engelhardt S (2008). MicroRNA‐21 contributes to myocardial disease by stimulating MAP kinase signaling in fibroblasts. Nature 456, 980–984. [DOI] [PubMed] [Google Scholar]
- Thum T, Galuppo P, Wolf C, Fiedler J, Kneitz S, Van Laake LW, Doevendans PA, Mummery CL, Borlak J, Haverich A, Gross C, Engelhardt S, Ertl G & Bauersachs J (2007). MicroRNAs in the human heart: A clue to fetal gene reprogramming in heart failure. Circulation 116, 258–267. [DOI] [PubMed] [Google Scholar]
- Topkara VK, Evans S, Zhang W, Epelman S, Staloch L, Barger PM & Mann DL (2011). Therapeutic targeting of innate immunity in the failing heart. J Mol Cell Cardiol 51, 594–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu Y, Wan L, Bu L, Zhao D, Dong D, Huang T, Cheng Z & Shen B (2013). MicroRNA‐22 downregulation by atorvastatin in a mouse model of cardiac hypertrophy: A new mechanism for antihypertrophic intervention. Cell Physiol Biochem 31, 997–1008. [DOI] [PubMed] [Google Scholar]
- Ucar A Gupta SK, Fiedler J, Erikci E, Kardasinski M, Batkai S, Dangwal S, Kumarswamy R, Bang C, Holzmann A, Remke J, Caprio M, Jentzsch C, Engelhardt S, Geisendorf S, Glas C, Hofmann TG, Nessling M, Richter K, Schiffer M, Carrier L, Napp LC, Bauersachs J, Chowdhury K & Thum T (2012). The miRNA‐212/132 family regulates both cardiac hypertrophy and cardiomyocyte autophagy. Nat Commun 3, 1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valadi H, Ekström K, Bossios A, Sjöstrand M, Lee JJ & Lötvall JO (2007). Exosome‐mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat Cell Biol 9, 654–659. [DOI] [PubMed] [Google Scholar]
- Valiente‐Alandi I, Schafer AE & Blaxall BC (2016). Extracellular matrix‐mediated cellular communication in the heart. J Mol Cell Cardiol 91, 228–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varrone F, Gargano B, Carullo P, Di Silvestre D, De Palma A, Grasso L, Di Somma C, Mauri P, Benazzi L, Franzone A, Jotti GS, Bang ML, Esposito G, Colao A, Condorelli G & Catalucci D (2013). The circulating level of FABP3 is an indirect biomarker of microRNA‐1. J Am Coll Cardiol 61, 88–95. [DOI] [PubMed] [Google Scholar]
- Viereck J Kumarswamy R, Foinquinos A, Xiao K, Avramopoulos P, Kunz M, Dittrich M, Maetzig T, Zimmer K, Remke J, Just A, Fendrich J, Scherf K, Bolesani E, Schambach A, Weidemann F, Zweigerdt R, de Windt LJ, Engelhardt S, Dandekar T, Batkai S & Thum T (2016). Long noncoding RNA Chast promotes cardiac remodelling. Sci Transl Med 8, 326ra22. [DOI] [PubMed] [Google Scholar]
- Waldenström A, Gennebäck N, Hellman U & Ronquist G (2012). Cardiomyocyte microvesicles contain DNA/RNA and convey biological messages to target cells. PLoS One 7, e34653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G‐K, Zhu J‐Q, Zhang J‐T, Li Q, Li Y, He J, Qin Y‐W & Jing Q (2010). Circulating microRNA: a novel potential biomarker for early diagnosis of acute myocardial infarction in humans. Eur Heart J 31, 659–666. [DOI] [PubMed] [Google Scholar]
- Wang J, Song Y, Zhang Y, Xiao H, Sun Q, Hou N, Guo S, Wang Y, Fan K, Zhan D, Zha L, Cao Y, Li Z, Cheng X, Zhang Y & Yang X (2012). Cardiomyocyte overexpression of miR‐27b induces cardiac hypertrophy and dysfunction in mice. Cell Res 22, 516–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K, Liu F, Zhou LY, Long B, Yuan SM, Wang Y, Liu CY, Sun T, Zhang XJ & Li PF (2014a). The long noncoding RNA CHRF regulates cardiac hypertrophy by targeting miR‐489. Circ Res 114, 1377–1388. [DOI] [PubMed] [Google Scholar]
- Wang K, Long B, Zhou L‐Y, Liu F, Zhou Q‐Y, Liu C‐Y, Fan Y‐Y & Li P‐F (2014b). CARL lncRNA inhibits anoxia‐induced mitochondrial fission and apoptosis in cardiomyocytes by impairing miR‐539‐dependent PHB2 downregulation. Nat Commun 5, 3596. [DOI] [PubMed] [Google Scholar]
- Wilusz JE & Sharp PA (2013). A circuitous route to noncoding RNA. Science 340, 440–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu XD, Song XW, Li Q, Wang GK, Jing Q & Qin YW (2012). Attenuation of microRNA‐22 derepressed PTEN to effectively protect rat cardiomyocytes from hypertrophy. J Cell Physiol 227, 1391–1398. [DOI] [PubMed] [Google Scholar]
- Yang K‐C, Yamada KA, Patel AY, Topkara VK, George I, Cheema FH, Ewald GA, Mann DL & Nerbonne JM (2014). Deep RNA sequencing reveals dynamic regulation of myocardial noncoding RNAs in failing human heart and remodelling with mechanical circulatory support. Circulation 129, 1009–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Ago T, Zhai P, Abdellatif M & Sadoshima J (2011). Thioredoxin 1 negatively regulates angiotensin II‐induced cardiac hypertrophy through upregulation of miR‐98/let‐7. Circ Res 108, 305–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Cappello T & Wang L (2015). Emerging role of microRNAs in lipid metabolism. Acta Pharm Sin B 5, 145–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Huang X‐R, Wei L‐H, Chung AC, Yu C‐M & Lan H‐Y (2014). miR‐29b as a therapeutic agent for angiotensin II‐induced cardiac fibrosis by targeting TGF‐β/Smad3 signaling. Mol Ther 22, 974–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziaeian B & Fonarow GC (2016). Epidemiology and aetiology of heart failure. Nat Rev Cardiol 13, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]