

Abstract
The ubiquitin–proteasome system (UPS) plays a critical role in removing unwanted intracellular proteins and is involved in protein quality control, signalling and cell death. Because the heart is subject to continuous metabolic and mechanical stress, the proteasome plays a particularly important role in the heart, and proteasome dysfunction has been suggested as a causative factor in cardiac dysfunction. Proteasome impairment has been detected in cardiomyopathies, heart failure, myocardial ischaemia, and hypertrophy. Proteasome inhibition is also sufficient to cause cardiac dysfunction in healthy pigs, and patients using a proteasome inhibitor for cancer therapy have a higher incidence of heart failure. In this Topical Review we discuss the experimental data which suggest UPS dysfunction is a common feature of cardiomyopathies, with an emphasis on hypertrophic cardiomyopathy caused by sarcomeric mutations. We also propose potential mechanisms by which cardiomyopathy‐causing mutations may lead to proteasome impairment, such as altered calcium handling and increased oxidative stress due to mitochondrial dysfunction.
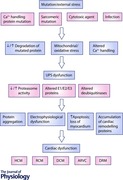
Keywords: cardiomyopathies, dilated cardiomyopathy, hypertrophic cardiomyopathy, proteasome, restrictive cardiomyopathy, sarcomere, ubiquitin
Abbreviations
- ARVC
arrhythmogenic right ventricular cardiomyopathy
- cMyBP‐C
cardiac myosin binding protein C
- DCM
dilated cardiomyopathy
- DRM
desmin‐related myopathy
- FHC
familial hypertrophic cardiomyopathy
- HCM
hypertrophic cardiomyopathy
- NRCM
neonatal rat cardiac myocytes
- PKG
protein kinase G
- RCM
restrictive cardiomyopathy
- SCD
sudden cardiac death
- TnC
troponin C
- TnI
troponin I
- TnT
troponin T
- UPS
ubiquitin–proteasome system
Introduction
Cardiomyopathy refers to a group of diseases that affect the heart muscle. Types of cardiomyopathies include hypertrophic cardiomyopathy (HCM), restrictive cardiomyopathy (RCM), dilated cardiomyopathy (DCM), and arrhythmogenic right ventricular cardiomyopathy (ARVC). Familial HCM is a genetic disease that is inherited in an autosomal dominant manner and is characterized by thickening of the heart muscle, arrhythmias and sudden cardiac death (SCD) (Maron, 2002). Familial HCM is common; it is estimated to affect 1 in 500 people and is the leading cause of sudden death in young athletes (Maron, 2002, 2003). HCM is often caused by mutations in sarcomeric proteins such as myosin heavy chain (MHC), cardiac myosin binding protein C (cMyBP‐C), and cardiac troponin T (TnT) (Gomes et al. 2004; Wu et al. 2012). In DCM the ventricles become dilated and weakened, and cannot pump blood effectively (Araco et al. 2017). Familial DCM is usually inherited in an autosomal dominant manner (Hershberger & Morales, 1993; McNally et al. 2013; Sisakian, 2014). RCM is rare and is characterized by stiffening of the heart muscle that leads to impaired filling (Wu et al. 2015). ARVC is another rare form of cardiomyopathy in which myocardium is replaced by fatty or fibrous tissue, primarily in the right ventricle (Gigli et al. 2016).
The mechanism by which sarcomeric protein mutations cause cardiomyopathies is unclear. Proposed pathological mechanisms of familial cardiomyopathies involve altered cardiac contractility, changes in Ca2+ handling, and altered energy homeostasis (Gomes & Potter, 2004; Frey et al. 2012). Current treatments include β‐adrenergic receptor blockers, Ca2+ channel antagonists, myocardial reduction by surgical or septal ablation, and implanted defibrillators (Maron, 2002). There is growing evidence that ubiquitin–proteasome system (UPS) dysfunction may play a pathogenic role in cardiomyopathies (Predmore et al. 2010; Schlossarek & Carrier, 2011; Day, 2013). Proper UPS function is critical to the function of the cell, as it plays important regulatory roles through degradation of proteins involved in signal transduction as well as quality control through breakdown of misfolded or damaged proteins. UPS dysfunction is believed to play a pathogenic role in many cardiac diseases (Wang & Robbins, 2006; Patterson et al. 2007) and may be a unifying factor of cardiomyopathies.
The sarcomere is the basic contractile unit of striated muscle (Marques & de Oliveira, 2016). It is made up of thick filaments, composed mainly of myosin and cardiac myosin binding protein C (cMyBP‐C), and thin filaments, composed mainly of actin, tropomyosin and troponin. Cardiac muscle contraction is initiated by an action potential that causes an influx of Ca2+ into the cytoplasm (Bers, 2000). At resting Ca2+ levels, the thin filament proteins troponin I (TnI) and tropomyosin block myosin binding sites on actin. Upon an increase in cytosolic Ca2+ levels the sarcomere is activated: TnI and tropomyosin (Tm) are shifted out of myosin binding sites and the head of myosin binds to the thin filaments, forming cross‐bridges. ATP is hydrolysed by myosin to activate the high energy conformation of the myosin head. The myosin head interacts with actin, allowing the head to rotate and resulting in sarcomere shortening. In addition to TnI and tropomyosin, the interaction between myosin and actin is mediated by other proteins such as troponin C (TnC), troponin T (TnT) and cMyBP‐C (de Tombe, 2003).
The UPS is responsible for degrading most intracellular proteins, both native and misfolded, in a process that normally involves polyubiquitination followed by degradation by the proteasome (Li et al. 2011a). Polyubiquitination occurs via the ubiquitination pathway (Fig. 1). The E1 ubiquitin‐activating enzyme utilizes ATP to activate ubiquitin and transfers it to an E2 ubiquitin‐conjugating enzyme. An E3 ubiquitin ligase, which may be a multi‐protein complex, interacts with both the E2 enzyme and the target protein, assisting or directly catalysing the transfer of ubiquitin from the E2 to a lysine residue on the protein substrate. The proteasome is composed of a multi‐subunit 20S core that contains the catalytic activity in the β1, β2, and β5 subunits. Each of these subunits has a different type of proteolytic activity; β1 has caspase‐like activity, β2 has trypsin‐like activity, and β5 has chymotrypsin‐like activity. These subunits work together to cleave proteasome substrates into short peptides, which may be further processed by downstream proteases (Reits et al. 2004). The catalytic sites are located inside the barrel‐like structure of the 20S core, and substrate entry into the proteasome is regulated by caps that bind the core particle. 19S regulatory particles associate with one or both ends of the 20S core to form the 26S proteasome, the most common form of the proteasome (Stadtmueller & Hill, 2011). The 19S cap recognizes ubiquitinated proteins and threads them into the core in an ATP‐dependent manner (Stadtmueller & Hill, 2011). The 20S core can also be capped by alternative regulatory particles such as the 11S regulatory particle, which is ATP‐independent and thought to be involved in the degradation of peptides or partially unfolded proteins. The 11S‐capped proteasome plays an important role in the degradation of oxidized proteins (Jung et al. 2014). Through targeted degradation of proteins that are no longer required by the cell, the proteasome plays a role in virtually all cellular processes (Wang et al. 2011).
Figure 1. Schematic diagram of the UPS.
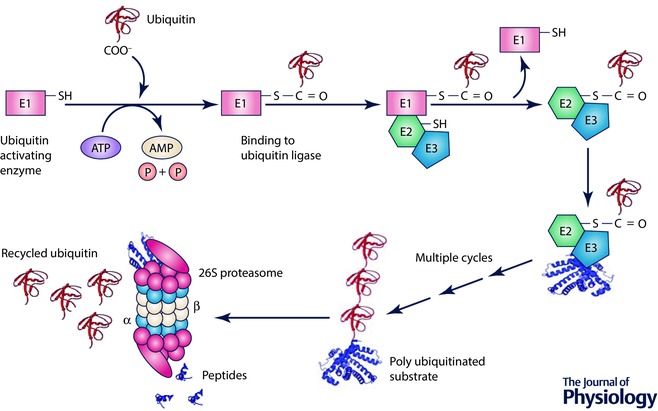
Ubiquitin is conjugated to target proteins by an E1 ubiquitin‐activating enzyme, E2 ubiquitin‐conjugating enzyme and E3 ubiquitin ligase. The ubiquitinated substrate is delivered to the 26S proteasome, which degrades the protein.
Ubiquitin–proteasome system dysfunction
UPS dysfunction in heart disease
The UPS is especially important in the heart, as cardiomyocytes are particularly prone to protein damage due to constant mechanical and metabolic stress (Wang & Robbins, 2006; Patterson et al. 2007). Additionally, the heart has very limited capacity for self‐renewal, meaning that cell death, which may result from impaired proteasome function, can be highly detrimental to the health of the organ (Patterson et al. 2007; Wang, 2013). The importance of proper proteasome function is highlighted by the fact that proteasome inhibition is sufficient to cause cardiac dysfunction in healthy pigs (Herrmann et al. 2013). Accumulation of ubiquitinated proteins, increases and decreases in proteasome activity, and changes in expression of the proteasome and other UPS proteins such as E2 and E3 enzymes have been observed in several experimental and human heart diseases (Weekes et al. 2003; Mearini et al. 2008; Schlossarek & Carrier, 2011; Tian et al. 2012; Day, 2013). Many E3 ligases are known to target proteins associated with HCM and DCM (Table 1). The E3 enzyme, MuRF1 (TRIM63), has also been implicated in the pathogenesis of HCM, as transgenic mice expressing mutations in MuRF1 identified in HCM patients (A48V and I130 M, and a deletion variant Q247*) develop cardiac hypertrophy (Chen et al. 2012). Expression of these MuRF1 mutants in adult cardiomyocytes caused reduced ubiquitination and UPS‐mediated degradation of the thick filament proteins myosin heavy chain 6 and cMyBP‐C. While mutations in MuRF1 and MuRF2 have been identified that contribute to the severity of HCM (Su et al. 2014), a patient with protein accumulation in muscle fibres and HCM had a homozygous TRIM63 (encodes MuRF1) null mutation in combination with the heterozygous TRIM54 (encodes MuRF3) mutation (Olive et al. 2015). The results from Olive et al. 2015 also suggested that MuRF1 and MuRF3 are important for positioning thick filaments in the sarcomeres.
Table 1.
Muscle specific E3 ligases and their putative targets in heart
| Cardiac E3 ubiquitin ligases | Specific targets | Known pathways affected | References |
|---|---|---|---|
| Arkadia | Smad7 | Myocardial fibrosis, TGF‐β signalling | He et al. (2011) |
| Ankyrin repeat‐ and SOCS box‐containing protein 2β (Asb2β) | Desmin | — | Thottakara et al. (2015) |
| Atrogin1/muscle atrophy F‐box (MAFbx) | Akt, calcineurin, truncated M7t‐cMyBP‐C | Calcineurin, FoxO, JNK, p53 signalling, hypertrophy pathway | Li et al. (2004, 2007), Mearini et al. (2010) |
| Carboxy terminus of Hsp70‐interacting protein (CHIP) | p53, oestrogen receptor‐α | Apoptosis | Fan et al. (2005), Naito et al. (2010), Le et al. (2012) |
| Casitas b‐lineage lymphoma (c‐Cbl) | FAK, paxillin, troponin I | — | Rafiq et al. (2012) |
| Cellular inhibitor of apoptosis 1 (cIAP1) | Caspase 3/9 | Apoptosis | Zhao et al. (2015b) |
| Cellular inhibitor of apoptosis 2 (cIAP2) | Caspase 3/7 | Apoptosis | Huang et al. (2000) |
| F‐box and leucine‐rich repeat protein 22 (Fbxl22) | α‐Actinin‐2, filamin C | — | Spaich et al. (2012) |
| E3 ubiquitin‐protein ligase Itchy homolog (ITCH) | Thioredoxin‐interacting protein (TXNIP) | Reactive oxygen species‐induced cardiotoxicity through the thioredoxin system | Otaki et al. (2016) |
| Muscle ring finger protein 1 (MuRF1) | Troponin I, β‐MHC, MLC‐2 | JNK signalling, atrophy and hypertrophy pathways | Kedar et al. (2004), Witt et al. (2005), Pan et al. (2016) |
| Muscle ring finger protein 2 (MuRF2) | ND | — | Witt et al. (2005) |
| Muscle ring finger protein 3 (MuRF3) | β‐MHC, four‐and‐a‐half LIM domain (FHL2) and γ‐filamin | — | Fielitz et al. (2007) |
| Murine double minute 2 (MDM2) | T cap, P53 | Apoptosis, hypertrophy pathway | Tian et al. (2006), Toth et al. (2006) |
| Neural precursor cell expressed developmentally down‐regulated protein 4‐2 (Nedd 4‐2) | Human ether‐à‐go‐go‐related gene (hERG) potassium channels, VEGF, Na+ and K+ channels | — | Abriel et al. (2000), Murdaca et al. (2004), Jespersen et al. (2007), Cui & Zhang (2013), Lamothe & Zhang, (2013) |
| Parkin | VDAC1 | Mitochondrial homeostasis | Watanabe et al. (2014) |
| RING finger protein RNF207 | ND | Action potential duration | Roder et al. (2014) |
| Tripartite motif 21 (TRIM21) | p62 (ubiquitination prevents its dimerization and sequestration) | Hypertrophy pathway | Pan et al. (2016) |
| Tripartite motif 32 (TRIM32) | ND | Hypertrophy pathway | Chen et al. (2016) |
| Seven in absentia homolog 2 (Siah2) | PHD1, PHD3 | Hypoxia pathway | Nakayama et al. (2004) |
| SMAD ubiquitination regulatory factor 1 (Smurf1) | TGFβ receptor | Myocardial fibrosis | Wang et al. (2012) |
| S‐phase kinase‐associated protein 2 (Skp2) | ND | FoxO signalling | Pramod & Shivakumar (2014) |
| SMAD ubiquitination regulatory factor 2 (Smurf2) | TGFβ receptor | Myocardial fibrosis | Cunnington et al. (2009) |
| Mothers against decapentaplegic homolog 7 (Smad7)/Smurf2 | TGFβ receptor | Myocardial fibrosis | Kavsak et al. (2000) |
| TNF receptor‐associated factor 6 (TRAF6) | ND | — | Zhang et al. (2014) |
| E3 component N‐recognin (UBR) 3 | Voltage‐gated Na+ channel Nav 1.5 | — | Zhao et al. (2015a) |
| UBR 6 | Voltage‐gated Na+ channel Nav 1.5 | — | Zhao et al. (2015a) |
| X‐linked inhibitor of proteolysis (XIAP) | Caspase 3 | Apoptosis | Suzuki et al. (2001) |
ND, not determined.
Proteasome inhibition is sufficient to cause cardiac dysfunction
Hearts of 3‐month‐old healthy pigs treated with twice‐weekly injections of the proteasome inhibitor MLN‐273 had 77% lower chymotrypsin‐like proteasome activity and increased levels of ubiquitinated proteins relative to the control group. After 11 weeks of treatment, cardiac output was lower, left ventricular mass was higher, and the hearts showed perivascular and interstitial fibrosis in the inhibitor‐treated group. Overall, the hearts showed alterations consistent with a hypertrophic–restrictive cardiomyopathy phenotype (Herrmann et al. 2013). Proteasome inhibition is also sufficient to promote maladaptive cardiac remodelling and cardiac dysfunction in stressed mouse hearts (Tang et al. 2010). In humans, the use of the proteasome inhibitor bortezomib (Velcade) for cancer treatment has been associated with an increased risk of adverse cardiac events, including congestive heart failure (Voortman & Giaccone, 2006; Enrico et al. 2007; Hacihanefioglu et al. 2008). Even the second generation of proteasome inhibitors (e.g. carfilzomib) has also been reported to show clinical cardiotoxicity (Grandin et al. 2015). Together these results indicate a causative role of UPS impairment in cardiac dysfunction.
UPS dysfunction in cMyBP‐C‐related HCM
Cardiac myosin binding protein C (cMyBP‐C) is a thick filament‐associated protein that interacts with myosin, actin and titin, and plays a role in regulating muscle contraction (Barefield & Sadayappan, 2010). Mutations in cMyBP‐C are the leading cause of familial hypertrophic cardiomyopathy (FHC), accounting for an estimated 42% of FHC cases, and are typically associated with a favourable prognosis (Richard et al. 2003). Several recent studies have shown that proteasome function is impaired in HCM caused by several cMyBP‐C mutations (Table 2). In HCM caused by certain cMyBP‐C mutations, the mutated protein is degraded by the proteasome at a faster rate than the wild‐type protein, which leads to UPS dysfunction (Sarikas et al. 2005; Bahrudin et al. 2008; Vignier et al. 2009; Schlossarek et al. 2012a). This phenomenon was discovered after several studies on cMyBP‐C mutations predicted to produce truncated cMyBP‐C showed none or low levels of the expected protein in tissues or cells (Rottbauer et al. 1997; Flavigny et al. 1999; Yang et al. 1999; Moolman et al. 2000). This finding contradicted the poison polypeptide hypothesis, which suggested that the mutated protein incorporates into the sarcomere and acts as a dominant negative (Rottbauer et al. 1997). It was later demonstrated that the absence of truncated cMyBP‐C is due to its rapid degradation by the proteasome. Sarikas et al. (2005) showed that the low levels of two cMyBP‐C truncation mutations, M6t (3% truncated) and M7t (80% truncated), which were present at 70% and 11%, respectively, of WT levels when expressed in neonatal rat cardiac myocytes (NRCMs), was due to rapid proteasomal degradation. M6t incorporated weakly into the sarcomere, while M7t was misincorporated at the Z disk and was present in ubiquitin aggregates. Treating with a proteasome inhibitor (lactacystin or MG132) raised levels of the two truncated proteins to WT levels, while the lysosomal inhibitor bafilomycin had little effect (Sarikas et al. 2005).
Table 2.
Summary of cMyBP‐C mutants on UPS function
| Mutation | Model/tissue | Effect on cMyBP‐C | Effect on UPS | Reference |
|---|---|---|---|---|
| Splice donor site mutation (G→A at position 1 of 5′ splice donor sequence) resulting in deletion of 160 bp exon and premature termination of translation | Endomyocardial biopsies from left ventricular myocardium of family members with FHC | Western blots showed none of the expected truncation mutant and somewhat reduced levels of WT protein | Not investigated | Rottbauer et al. (1997) |
| Guanine nucleotide insertion in exon 25 (codon 791) resulting in the loss of 40 bases | Myectomy tissue from family members with FHC | Western blots showed none of the expected truncation mutant | Not investigated | Moolman et al. (2000) |
| cMyBP‐C lacking the 240 nucleotides at the 3′ end of the cDNA that constitute the myosin binding domain | Cardiac tissue from transgenic mice | Very low levels of the truncated protein were expressed | Not investigated | Yang et al. (1999) |
| Glu258Lys | Myectomy tissue from FHC patient | The mutant mRNA level was moderately decreased relative to WT. The cMyBP‐C content relative to actin was significantly lower (present at 85% of donor control levels) | Not investigated | Marston et al. (2009) |
| Arg502Trp | Myectomy tissue from FHC patient | The cMyBP‐C content relative to actin was significantly lower (present at 82% of donor control levels). | Not investigated | Marston et al. (2009) |
| Intron17 donor site A>T+4 resulting in truncation in C3 domain | Myectomy tissue from FHC patients | None of expected the 52 kDa protein was detected by Western blot with antibody to N terminus. The cMyBP‐C content relative to actin was significantly lower (present at 68–83% of donor control levels) | Not investigated | Marston et al. (2009) |
| InsG2374 resulting in truncation in C5 domain | Myectomy tissue from FHC patient | The mutant mRNA level was moderately decreased relative to WT. None of the expected 90kDa protein was detected by Western blot with antibody to N terminus. The cMyBP‐C content relative to actin was significantly lower (present at 81% of donor control levels) | Not investigated | Marston et al. (2009) |
| T>A 2604, delC 2605 resulting in truncation in C7 domain | Myectomy tissue from FHC patient | The mutant mRNA level was moderately decreased relative to WT. None of the expected 97 kDa protein was detected by Western blot with antibody to N terminus. The cMyBP‐C content relative to actin was significantly lower (present at 65% of donor control levels) | Not investigated | Marston et al. (2009) |
| delCT 2864/5 resulting in truncation in C7 domain | Myectomy tissue from FHC patient | None of the expected 114 kDa protein was detected by Western blot with antibody to N terminus. The cMyBP‐C content relative to actin was significantly lower (present at 65% of donor control levels) | Not investigated | Marston et al. (2009) |
| Arg1271stop resulting in truncation in C10 domain | Myectomy tissue from FHC patient | The mutant mRNA level was not affected relative to WT. None of the expected 140 kDa protein was detected by Western blot with antibody to N terminus. The cMyBP‐C content relative to actin was significantly lower (present at 77% of donor control levels) | Not investigated | Marston et al. (2009) |
| c.2373dupG resulting in truncation after C5 domain | Cardiac tissue from the left ventricular septum of FHC patients | The mutant mRNA made up 23% of the total cMyBP‐C mRNA. None of the expected 93kDa protein was detected by Western blot with antibody to N terminus | Not investigated | van Dijk et al. (2009) |
| c.2864_2865delCT resulting in truncation at the end of the C8 domain | Cardiac tissue from the left ventricular septum of FHC patients | The mutant mRNA made up 20% of the total cMyBP‐C mRNA. None of the expected 116 kDa protein was detected by Western blot with antibody to N terminus | Not investigated | van Dijk et al. (2009) |
| c.2373dupG, c.2864_2865delCT, splice site mutation c.927 to 2A>G, splice site mutation c.1458 to 1G>C | Cardiac tissue from the left ventricular septum of FHC patients | Samples from patients with FHC due to one of four different cMyBP‐C mutations had 33% lower levels of cMyBP‐C protein relative to non‐failing donor hearts | Not investigated | van Dijk et al. (2012) |
| G→A transition on the last nucleotide of exon 6 | Homozygous knock‐in mice | Homozygous KI mice show 80% lower mRNA than in WT mice | The mRNA levels of five E3 ubiquitin ligases were lower in KI mice relative to WT mice: Nedd4, Ube3c, Mdm2, Trim32, and Asb2β, which showed the greatest reduction (37% lower than WT) | Thottakara et al. (2015) |
| 12bp duplication/4bp deletion in exon 33 which causes frameshift that results in 19 novel amino acids and premature stop codon (3% truncation) | Expressed in neonatal rat cardiac myocytes (NRCMs) | The resulting mutant protein (M6t) was present at 70% of WT levels and incorporated weakly into the sarcomere | Treating with a proteasome inhibitor raised levels of the truncated protein to WT levels | Sarikas et al. (2005) |
| G→A transition at the last nucleotide of exon 6 that leads to skipping of exon 6 (80% truncated) | Expressed in NRCMs | The resulting mutant protein (M7t) was present at 11% of WT levels, misincorporated at the Z disk, and was present in ubiquitin aggregates | The mutant formed aggregates and inhibited the breakdown of other proteasome substrates, leading to proteasome dysfunction. Treating with a proteasome inhibitor raised levels of the truncated protein to WT levels | Sarikas et al. (2005) |
| G→A transition on the last nucleotide of exon 6 | Heterozygous knock‐in mice | 50% and 80% lower mRNA levels than in WT for heterzygotes and homozygotes, respectively. The single G>A change gave rise to three mRNAs: missense, nonsense, and deletion/insertion. Levels of one of these mRNAs were increased by inhibition of nonsense‐mediated mRNA decay | Levels of the proteins encoded by the other mRNAs were increased by proteasomal inhibition | Vignier et al. (2009) |
| G→A transition on the last nucleotide of exon 6 | Ventricular tissue from heterozygous knock‐in mice | Described above | The proteasome was impaired with age as shown by accumulation of the UPS reporter UbG76V‐GFP and lower chymotrypsin‐like activities. Ubiquitinated proteins were higher than in WT at 1 year of age. Adrenergic stress led to proteasome dysfunction | Schlossarek et al. (2012a, b ) |
| Glu334Lys | Mutation identified in Japanese FHC patient. Expressed in COS‐7, NRCMs, and HL‐1 cells | Protein level was approximately half that of WT‐cMyBP‐C, despite similar mRNA levels. Treatment with proteasome inhibitors restored protein levels to WT levels | Expression of E334K‐cMyBP‐C led to decreased chymotrypsin‐like proteasome activity by approximately 50%. Another study showed that the trypsin‐ and caspase‐like activities were decreased by E334K expression as well | Bahrudin et al. (2008), Bahrudin et al. (2011) |
| ∆2864‐2865GC (resulting in 16.2% truncation), ∆Lys814, Gln998Glu, Thr1046Met | Mutation identified in Japanese FHC patient | Protein levels not affected | No effect on proteasome activity. Other UPS functions not investigated | Bahrudin et al. (2008) |
Vignier, et al. developed a cMyBP‐C knock‐in mouse with a point mutation associated with poor prognosis in humans (Vignier et al. 2009). The mutation, a G→A transition in a donor splice site sequence, resulted in 50% and 80% lower total cMyBP‐C mRNA levels than in WT for heterozygotes and homozygotes, respectively. Interestingly, the single G→A change gave rise to three mRNAs: missense, nonsense, and deletion/insertion. Inhibition of nonsense‐mediated mRNA decay increased the levels of nonsense mRNAs only, but not the other mRNAs. Protein levels of cMyBP‐C were lower in heterozygous and homozygous mice, and proteasome inhibition increased the protein levels of cMyBP‐C. These results suggested that nonsense‐mediated mRNA decay as well as the proteasome was responsible for lower levels of mutant cMyBP‐C for this mutation.
An HCM‐causing missense mutation in cMyBP‐C (E334K, identified in a Japanese HCM patient) has also been identified that is degraded at an accelerated rate (Bahrudin et al. 2008). When E334K‐cMyBP‐C was expressed in COS‐7 and NRCMs, the protein level was approximately half that of WT‐cMyBP‐C, despite similar mRNA levels (Bahrudin et al. 2008). Treatment with proteasome inhibitors (lactacystin or MG132) restored protein levels of E334K‐cMyBP‐C to WT levels, while treatment with the lysosomal inhibitor chloroquine had no effect. In NRCMs, expression of E334K‐cMyBP‐C led to decreased chymotrypsin‐like proteasome activity by approximately 50%. Another study showed that the trypsin‐ and caspase‐like activities of the proteasome were decreased by E334K expression as well (Bahrudin et al. 2008). E334K expression also led to increased levels of the pro‐apoptotic proteins p53, Bax and cytochrome c and lower levels of anti‐apoptotic proteins. Staining with annexin V suggested more E334K‐transfected cells were apoptotic than WT‐transfected cells. It may be noted that not all cMyBP‐C mutants are degraded at an accelerated rate by the proteasome; different mechanisms are in play depending on the specific mutation (Table 2).
Schlossarek et al. investigated effects on the UPS using the heterozygous knock‐in (KI) for the mutated cMyBP‐C developed by Vignier et al., which reflects what occurs genetically in HCM patients, and a heterozygous MyBP‐C knock‐out (KO) (Vignier et al. 2009; Schlossarek et al. 2012a). The KO mice have left ventricular hypertrophy and reduced fractional shortening and serve as a model of cMyBP‐C insufficiency (Carrier et al. 2004). Both models were compared with WT mice and exhibited cardiac hypertrophy; the KO mice at 2 weeks and later, and KI at birth and later. The proteasome was shown to be impaired with age in the KI mice only, as shown by the accumulation of the UPS reporter UbG76V‐GFP and lower chymotrypsin‐like activities. Ubiquitinated proteins were higher in both KO and KI than in WT at 1 year of age. Accumulation of autophagy‐related proteins such as beclin‐1 suggests defects in the autophagy–lysosome pathway in both KO and KI mice (Schlossarek et al. 2012a; Lin et al. 2015). Adrenergic stress due to treatment with a combination of isoprenaline and phenylephrine led to septal hypertrophy in both models, but proteasome dysfunction only in the KI mice (Schlossarek et al. 2012b).
UPS dysfunction in troponin T‐related HCM
Mutations in TnT account for approximately 7% of HCM cases (Richard et al. 2003). We recently showed that UPS function is impaired in hearts of 3‐month‐old mice with severe TnT‐related HCM (Gilda et al. 2016). Transgenic mice expressing human cardiac TnT with the I79N or R278C mutation were compared with mice expressing WT human TnT. The I79N mutation is myofilament Ca2+ sensitizing and associated with severe, early onset HCM and sudden cardiac death. The R278C mutation does not affect myofilament Ca2+ sensitivity and is associated with mild, late onset heart disease. Proteomics carried out on these transgenic mouse models showed that expression of several proteasome subunits was affected, with decreases in subunit levels in R278C hearts relative to WT and higher levels in I79N than R278C hearts, suggesting differential regulation of the proteasome in I79N and R278C hearts. Western blotting was used to verify these changes, and showed a similar pattern of expression. When proteasome activity was measured, 20S and 26S chymotrypsin‐like proteasome activity and immunoproteasome activity (β1i and β5i) were lower in hearts of I79N mice relative to WT mice. The immunoproteasome is an alternative form of the proteasome in which the catalytic β1, β2 and β5 subunits are swapped out for the β1i, β2i and β5i inducible subunits in response to pro‐inflammatory cytokines or oxidative stress. The inducible subunits have altered cleavage specificity that favours the generation of antigenic peptides and an increased ability to degrade oxidized proteins. Decreased proteasome activity in I79N hearts was accompanied by higher levels of ubiquitinated and oxidized proteins. The only change in proteasome activity in R278C hearts was decreased β1i immunoproteasome activity.
In I79N mice, proteomics and metabolomics also suggested accelerated energy production, as shown by increased levels of enzymes and metabolites in the glycolytic pathway, tricarboxylic acid cycle and the electron transport chain, and upregulated expression of antioxidant enzymes. Previous studies have shown that energy depletion occurs in HCM, and impaired energy metabolism has been suggested as an important factor in HCM. These findings suggest that I79N heart cells are subject to higher stress and may upregulate antioxidants to cope with these changes. It is possible that accelerated energy production may lead to mitochondrial dysfunction and increased ROS production. This could lead to higher levels of oxidized proteins for the protein to degrade, as well as direct damage to proteasome subunits if they are oxidatively modified.
The proteasome is also impaired in mice with the HCM‐causing F110I TnT mutation. The F110I mutation increases myofilament Ca2+ sensitivity, though to a lesser extent than the I79N mutation (Hernandez et al. 2005). In hearts of 3‐month‐old F110I mice, 20S proteasome activity was decreased and the levels of ubiquitinated proteins were increased (Gomes, 2009). Although no other research related to proteasome dysfunction in TnT‐related cardiomyopathies has been published, one study in Drosophila showed that TnT with a mutation that causes muscle defects was rapidly degraded (Fyrberg et al. 1990). In flies homozygous for three different mutations in TnT, a splice donor mutation, an intronic deletion, and S311F, levels of TnT protein were reduced. Reduced TnT protein levels of the splice donor and intronic deletion mutants may be due to lower mRNA levels as a result of splicing defects. Lower levels of the S311 F mutant may be due to accelerated proteasomal degradation, but this study did not investigate the role of the proteasome.
UPS perturbations in dilated cardiomyopathy
In DCM the chambers of the heart become enlarged, and systolic function is typically reduced (McNally et al. 2013). In the past DCM has been associated with a poor prognosis, though advances in diagnosis treatment have improved outlook for DCM patients (Keeling et al. 1995; Franklin & Burch, 2000; Kubo et al. 2008; Hazebroek et al. 2012). Mutations in titin, a large sarcomeric protein that acts as a stretch sensor, are a major cause of DCM (Herman et al. 2012; McNally et al. 2013; Begay et al. 2015). While few studies have been conducted on the UPS in DCM, certain investigations indicate proteasome impairment as well as alterations in the ubiquitination pathway in DCM hearts. Interestingly, loss or mutation of different proteins in the ubiquitination pathway has been implicated in the pathogenesis of DCM (Xiong et al. 2007; Al‐Yacoub et al. 2016). A homozygous mutation (Gly243Arg) in the cardiac E3 ligase, FBXO32 (Atrogin 1/MAFbx), was recently found to be associated with DCM (Watanabe et al. 2014; Al‐Yacoub et al. 2016). There are also reports of protein aggregation in DCM, which may be linked to UPS function (Hamada et al. 2004; Gianni et al. 2010).
A study on bovine DCM revealed that levels of ubiquitin carboxyl‐terminal hydrolase (UCH), a deubiquitinating protein, were greatly elevated in DCM hearts, and levels of ubiquitinated proteins were generally higher (Weekes et al. 1999). Hearts of human DCM patients had higher levels of E1 and E2 enzymes, and levels of ubiquitinated proteins were twofold higher than ischaemic hearts and fivefold higher than control donor hearts (Weekes et al. 2003). Levels of UCH were also higher in human DCM hearts, in agreement with the study on bovine DCM. Proteasome activity was not measured in this study, but these findings showed UPS alterations at the level of the ubiquitination pathway. In other patients with DCM, proteasome levels were increased, and oxidative stress appeared to be increased (Otsuka et al. 2010). Immunohistochemistry showed that expression of proteasome and ubiquitin was enhanced, and they were present in large granular structures relative to control hearts. Ubiquitin‐positive granular structures were likely to be due to the accumulation and aggregation of ubiquitinated proteins. Products of lipid and carbohydrate oxidation were also higher in DCM hearts. Levels of superoxide dismutase‐1 (SOD1) were higher as well, consistent with increased oxidative stress in DCM hearts. A separate study comparing cardiac tissue from DCM patients and donors showed that levels of polyubiquitinated proteins were increased in DCM hearts, as well as the chymotrypsin‐like activity of the proteasome (Birks et al. 2008).
Inflammation, autoimmunity, and compromised protein quality control have been linked to DCM. As the UPS is involved in the immune response and inflammation, Voigt et al. (2010) investigated anti‐proteasomal immunity in hearts of DCM patients. They found that autoimmune responses to proteasome in the heart were increased in DCM patients, predominantly towards 20S α subunits, and the levels of antibodies to proteasome were particularly enhanced in advanced heart failure (Voigt et al. 2010). This suggests that in DCM patients altered proteasome structures are resulting in the generation of antibodies to these ‘foreign’ proteins.
Desmin‐related myopathy
Desmin‐related myopathy (DRM) is a disease associated with mutations in desmin or associated proteins (Paulin & Li, 2004; Goldfarb & Dalakas, 2009). Desmin is an intermediate filament expressed in cardiac, skeletal and smooth muscle that plays an important role in maintaining the contractile apparatus (Paulin & Li, 2004; Goldfarb & Dalakas, 2009). DRM patients typically have skeletal muscle weakness and cardiomyopathy, and respiratory weakness is also commonly observed (van Spaendonck‐Zwarts et al. 2011). Causes of death in DRM patients include sudden cardiac death and heart failure (van Spaendonck‐Zwarts et al. 2011). DRM‐causing mutations in desmin cause improper folding, which contributes to the accumulation of unfolded proteins and pre‐amyloid oligomer formation (McLendon & Robbins, 2011). In line with this, DRM can also be caused by mutations in αB‐crystallin (CryAB), a chaperone that folds desmin (McLendon & Robbins, 2011). The proteasome plays an important role in removing misfolded proteins, and can itself be inhibited by protein aggregates. In mice expressing a mutated form of desmin that is linked to DRM (deletion of 7 amino acids, R173–E179), cardiac UPS function is impaired, as shown by increased levels of the UPS reporter GFPdgn (Liu et al. 2006a). UPS impairment was likely to be due to impaired delivery of substrates into the proteasome, as the proteolytic activities were found to be increased rather than decreased, and subunits of the 19S cap were depleted (Liu et al. 2006a). In cultured neonatal rat ventricular myocytes, overexpressing mutated desmin caused desmin‐positive aggregates to form and disrupted proteasome function, as shown by the accumulation of the proteasome reporter GFPu (Liu et al. 2006b). Co‐expressing the chaperones CryAB or Hsp70 significantly reduced desmin aggregation and reduced or completely abolished GFPu accumulation, demonstrating that desmin aggregation is necessary to cause proteasome dysfunction (Liu et al. 2006b). The proteasome is also impaired in DRM mice with the R120G mutation in CryAB (Chen et al. 2005). There was an accumulation of ubiquitinated proteins and GFPdgn reporter in the heart, while proteolytic activities of the proteasome were increased and subunit expression was decreased (Chen et al. 2005). This is similar to findings in DRM mice with desmin mutations and suggests the defect occurs in substrate delivery. These studies demonstrate that desmin mutants impair the proteasome, which may contribute to further protein aggregation and cardiac dysfunction in DRM.
UPS perturbations in restrictive cardiomyopathy
The R145W mutation in TnI causes restrictive cardiomyopathy and has been associated with sudden cardiac death (van den Wijngaard et al. 2011). This mutation increases the Ca2+ sensitivity of force and ATPase activity (Gomes et al. 2005; Wen et al. 2009). Proteomic analysis of hearts from 3‐month‐old mice expressing R145W‐TnI found that several pathways were affected, including increases in enzymes involved in ATP production and stress‐related proteins. No change was observed in the levels of two proteasome subunits: PSMA6 and Rpt1. Importantly, proteolytic assays showed that proteasome activity was lower in R145W mice than WT mice, indicating proteasome impairment (Cui et al. 2013b).
UPS dysfunction in right ventricular dysfunction
Effects on the UPS were investigated in mice with right ventricular hypertrophy/failure due to pulmonary artery constriction, and the chymotrypsin‐like activity of the proteasome was found to be decreased, while the proteasome subunit Rpt5 as well as UCHL1 deubiquitinase and Smurf1 E3 ligase were increased (Rajagopalan et al. 2013). Levels of polyubiquitinated proteins were also increased in conjunction with impaired proteasome function (Rajagopalan et al. 2013). Levels of the pro‐apoptotic protein Bax were higher, while the anti‐apoptotic protein Bcl‐2 was lower (Rajagopalan et al. 2013). An increase in ubiquitinated proteins as well as changes in several E3 ligases, including Mdm2 and E6AP, was observed in a feline right ventricular pressure overload model (Balasubramanian et al. 2006).
Potential mechanisms for UPS dysfunction
There are multiple mechanisms by which mutations in sarcomeric proteins could lead to UPS dysfunction, such as altered degradation of the mutated protein, altered calcium handling, and mitochondrial dysfunction. Possible mechanisms to explain how HCM mutations in sarcomeric proteins cause proteasome dysfunction are shown in Fig. 2 and discussed below.
Figure 2. Schematic diagram showing the potential mechanisms by which familial cardiomyopathy mutations in sarcomeric proteins may cause proteasome dysfunction.
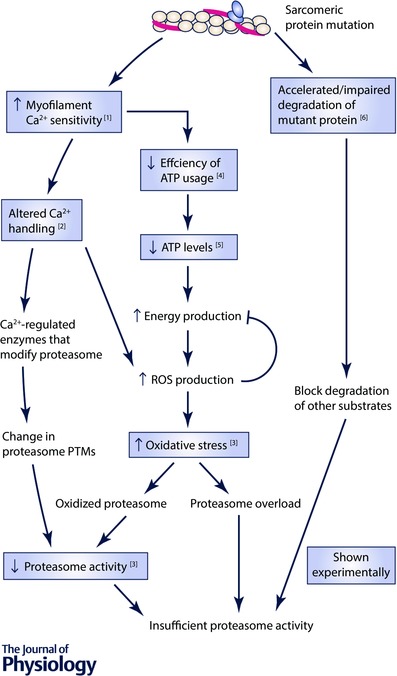
Boxes show changes that have been observed experimentally and the numbers in these boxes correspond to the following references: 1, Baudenbacher et al. (2008); 2, Schober et al. (2012); 3, Gilda et al. (2016); 4, Miller et al. (2001); 5, JE Gilda & AV Gomes, unpublished data; 6, Bahrudin et al. (2008). PTMs, post‐translational modifications.
Altered proteasomal degradation of mutant sarcomeric protein
Abnormal sarcomeric proteins may be degraded by the proteasome at an increased rate, thereby overloading the proteasome and leading to insufficient activity to break down other UPS substrates (Sarikas et al. 2005; Bahrudin et al. 2008). Mutants may also be resistant to degradation, and clog the proteasome, preventing entry of other proteasome substrates. Sarikas et al.’s investigation showed that the cMyBP‐C M7t mutant formed aggregates and inhibited the breakdown of other proteasome substrates, leading to proteasome dysfunction (Sarikas et al. 2005). These aggregates were ubiquitin‐positive suggesting that they contained the mutant cMyBP‐C that may be ubiquitin bound, but not degraded. The presence of ubiquitin‐positive aggregates is a prominent feature of most neurodegenerative disorders, where misfolded proteins and aggregates are proposed to overload the proteasome and/or block the pore (Ciechanover & Brundin, 2003). Misfolded proteins may accumulate due to increased abundance as the result of genetic mutations, aging and/or cellular stress and are associated with proteotoxicity (McLendon & Robbins, 2015). Both cytosolic and nuclear proteasomes participate in the degradation of misfolded proteins (von Mikecz et al. 2008; McLendon & Robbins, 2015). Since excellent recent reviews concerning the presence of aggresomes and proteotoxicity in cardiomyopathies exist this aspect will not be covered in significant detail in this review (Willis & Patterson, 2013; McLendon & Robbins, 2015). In addition to the UPS, several mechanisms have evolved to remove or repair misfolded proteins, such as autophagy, which clears misfolded proteins and aggregates, and chaperones, which repair or protect misfolded proteins from degradation. It is not known if mutations in sarcomeric proteins other than cMyBP‐C, such as actin and troponin I, affect their rate of degradation by the proteasome.
Altered calcium handling
Another potential mechanism is that alterations in intracellular Ca2+ concentrations may affect proteasome activity. Most cardiomyopathy‐causing sarcomeric protein mutations increase Ca2+ sensitivity of muscle contraction, and mutations that sensitize the myofilament to the effects of Ca2+ are associated with a high risk of SCD (Huke & Knollmann, 2010). TnC, the Ca2+‐binding subunits of the troponin complex, is a major cytosolic Ca2+ buffer, and increases in myofilament Ca2+ sensitivity have been shown to alter Ca2+ handling in the cell. Increased myofilament Ca2+ sensitivity leads to slow Ca2+ transient decays and increased diastolic Ca2+ concentrations (Knollmann et al. 2003; Sirenko et al. 2006; Schober et al. 2012; Wu et al. 2012). One potential mechanism by which altered Ca2+ concentrations could affect proteasome activity is by affecting the activity of Ca2+‐regulated proteins that associate with and modify the proteasome. Calmodulin, a Ca2+‐binding protein, has been shown to interact with 26S proteasome subunits non‐ATPase regulatory subunit 2 and non‐ATPase regulatory subunit 12 (Shen et al. 2005). Interaction of calmodulin with either or both proteasome subunits could potentially directly affect proteasome activity. Calmodulin also interacts with the proteasome associating protein RAD23 homolog B (Shen et al. 2005).
Post‐translational modifications of the proteasome, and phosphorylation in particular, are very important in regulating the activity and stability of the proteasome. Many subunits of the proteasome are endogenously phosphorylated, and over 400 proteasome phosphorylation sites have been identified in total (Gomes et al. 2006, 2009; Drews et al. 2007; Cui et al. 2013a). Proteasome activity is regulated by enzymes such as protein phosphatase 2A (PP2A), which decreases proteasome phosphorylation and activity, and protein kinase A (PKA), which enhances proteasome phosphorylation and activity (Marambaud et al. 1996; Zong et al. 2006; Zhang et al. 2007). In addition to these enzymes, several other kinases have been identified that phosphorylate the proteasome and modulate its activity and/or stability, such as casein kinase 2, p38 MAPK, polo‐like kinase, c‐Abl tyrosine kinase, and Arg tyrosine kinase (Castano et al. 1996; Feng et al. 2001; Bose et al. 2004; Liu et al. 2006 c; Lee et al. 2010). Certain Ca2+‐dependent enzymes have been shown to associate with and modify the proteasome, such as calcium/calmodulin‐dependent protein kinase II (CaMKII) and calcineurin (Li et al. 2011b; Djakovic et al. 2012). In neurons, CaMKIIα phosphorylates Rpt6, a subunit of the 19S proteasome regulatory particle, leading to increased proteasome activity (Djakovic et al. 2012). The regulatory subunit of calcineurin, a phosphatase regulated by Ca2+/calmodulin, has been shown to interact with the 20S proteasome subunit PSMA7 (α4) and stimulate proteasome activity (Li et al. 2011b). In cardiomyocytes, protein kinase G (PKG) has been shown to positively regulate proteasomal function (Ranek et al. 2013). The same group found that parasympathetic muscarinic 2 receptor activation stimulates cardiac proteasome activity in a PKG dependent manner (Ranek et al. 2014). Reduced parasympathetic tone as well as reduced PKG activity are linked to cardiac malfunction, and may serve as a possible cause of impaired proteasomal degradation in cardiomyopathies. It is possible that the altered Ca2+ levels in the cell, which occur as a result of myofilament Ca2+‐sensitizing mutations such as the I79N mutation in TnT, may affect the activity of these and other Ca2+‐regulated enzymes that modify the proteasome.
Mitochondrial dysfunction and oxidative stress
Another potential mechanism by which increased Ca2+ concentrations could affect proteasome activity is by inducing mitochondrial dysfunction, which could lead to increased reactive oxygen species (ROS) production (Celsi et al. 2009). Calcium plays an important role in regulating mitochondrial function. Studies have shown that when mitochondria are exposed to high concentrations of Ca2+ in the cell, Ca2+ is pumped into the mitochondria through the Ca2+ uniporter (Bianchi et al. 2004). Increases in cytosolic Ca2+ levels can lead to mitochondrial matrix Ca2+ overload which results in increased production of ROS (Peng & Jou, 2010). Mitochondria produce ROS such as superoxide, hydrogen peroxide, and hydroxyl radicals, as well as reactive nitrogen species. Mitochondrial ROS is mainly produced from complexes I and III of the electron transport chain during oxidative phosphorylation. High levels of ROS are detrimental to the cell, as they can react with proteins and other molecules in the cell and damage them. Mitochondrial dysfunction has been reported in mice and patients with HCM (Vakrou & Abraham, 2014). In cardiac cells with altered Ca2+ handling, it is possible that mitochondrial Ca2+ levels and ROS production are affected. We found that in 3‐month I79N mouse hearts, levels of oxidized proteins were increased (Gilda et al. 2016). Increased levels of ROS could lead to oxidative protein damage and an overload of proteasome substrates, and consequent proteasome functional insufficiency. ROS may also directly react with proteasome subunits and damage the proteasome, leading to decreased activity.
Subunits of the proteasome can be oxidatively damaged, leading to lower proteasome activity (Aiken et al. 2011). Oxidation of the Rpt3 subunit of the 19S regulatory particle leads to decreased proteasome activity (Ishii et al. 2005). Oxidation of the 20S proteasome has also been shown to impair the proteolytic activity of the proteasome (Bulteau et al. 2002). Breusing et al. (2009) showed that with age, protein oxidation increases and proteasome activity decreases. Impaired docking of the 19S to the 20S, which would reduce polyubiquitin‐dependent degradation, was observed in human end‐stage heart failure (Day et al. 2013). Our lab recently showed that treating H9c2 cardiac cells with the NSAID meclofenamate sodium resulted in elevated levels of mitochondrial ROS, increased oxidation of 19S proteasome subunits, altered 19S and 20S association, and decreased proteasome activity (Ghosh et al. 2016).
Impaired energy metabolism has also been suggested as a unifying factor of HCM, and myofilament Ca2+‐sensitizing mutations lead to decreased efficiency of ATP usage. Recent proteomics and metabolomics data suggest that energy production pathways are accelerated in hearts of mice with the I79N mutation in TnT (Gilda et al. 2016), which may lead to enhanced energy production via the electron transport chain and subsequently increased mitochondrial ROS production. It may also be noted that the UPS utilizes ATP; hence ATP depletion could potentially impair the UPS by a more direct route, though further investigation is needed. Unpublished results from our laboratory suggest that ATP levels in the hearts are significantly lower in I79N transgenic mice compared with WT mice. Hearts from mice with the R92W and R92L mutations in TnT utilize more ATP during muscle contraction and have lower levels of ATP and higher levels of ADP and Pi (He et al. 2007). H9c2 cardiac cells subjected to severe ATP depletion showed stress‐associated proteotoxicity and cell death (Kabakov et al. 2002). As such, lower ATP levels as observed in the I79N hearts are also likely to contribute to UPS dysfunction.
Can increasing proteasome activity improve cardiac function?
Enhancing proteasome function by overexpression of the proteasome subunit PA28α decreased cardiac hypertrophy and increased the lifespan of R120G‐CryAB mice (Li et al. 2011a). Enhancement of proteasome function by PA28α overexpression also reduces the size of infarct and protects against cardiac dysfunction in mice with ischaemia‐reperfusion injury (Li et al. 2011a). Enhancing proteasome function in mice by overexpression of the proteasome regulatory subunit PA28α partially attenuated right ventricular failure and improved survival (Rajagopalan et al. 2013). Activation of the proteasome by pharmacological means (e.g. via PDE5 inhibition/PKG activation) was found to protect against DRM disease progression in mice (Ranek et al. 2013). These results all suggest that enhancing proteasome function can have a cardioprotective effect.
Mechanistic links between proteasome dysfunction and cardiomyopathy
The UPS plays an important role in most cellular processes, and is particularly important in the heart; therefore UPS impairment may lead to cardiac dysfunction through multiple mechanisms.
Electrophysiological dysfunction
Bahrudin et al. hypothesized that since UPS impairment led to an accumulation of pro‐apoptotic proteins, it may also lead to accumulation of other proteins that it normally degrades, such as ion channels and Ca2+ handling proteins, and this may contribute to the electrophysiological dysfunction and the arrhythmias observed in patients with the E334K mutation in cMyBP‐C. In HL‐1 cells and NRCMs, expression of E334K‐cMyBP‐C led to lower proteasome activity and significantly higher levels of Kv1.5, Nav1.5, Cav3.2, Hcn4, Cav1.2, Serca, RyR2 and Ncx1 compared with their levels in WT‐cMyBP‐C‐expressing cells, despite similar mRNA levels (Bahrudin et al. 2011). Expression of E334K‐cMyBP‐C led to higher Ca2+ transients and longer action potential durations (Bahrudin et al. 2011). Thus, proteasome impairment could in part explain some of the electrophysiological dysfunction in cardiomyopathy patients.
Proteasome inhibition and apoptosis
Proteasome inhibition has been shown to induce apoptosis in several different cell types (Ding et al. 2007; Pandit & Gartel, 2011; Tsuchiya et al. 2011). Proteasome inhibition is often associated with an increase in the levels of pro‐apoptotic proteins. Levels of pro‐apoptotic proteins have been shown to be increased in cardiomyopathies. The apoptosis regulator p53 was shown to be upregulated in DCM (Birks et al. 2008). The E334K mutation in cMyBP‐C, which leads to reduced proteasome activity, increased the levels of pro‐apoptotic proteins (p53, Bax and cytochrome c) and decreased levels of anti‐apoptotic proteins (Bcl‐2 and Bcl‐XL) (Bahrudin et al. 2008). Sarikas et al. (2005) observed an increased rate of apoptosis in M7t‐cMyBP‐C‐expressing NRCMs.
Besides other models described earlier, a cMyBP‐C KI mouse model of HCM showed reduced proteasome activity and decreased mRNA levels of the muscle‐specific E3 ligases, Asb2β (Thottakara et al. 2015). In neonatal mouse cardiomyocytes Asb2β was found to target desmin for degradation (Thottakara et al. 2015). Reduced proteasome activity has also been observed in explanted failing hearts and myectomy samples from HCM patients when compared with samples from non‐failing hearts (Predmore et al. 2010). Increased protein expression of p53 in patient HCM samples suggest that apoptosis may play a key role in HCM pathogenesis (Predmore et al. 2010). In these human heart samples from HCM patients, reduced proteasome activity was not due to changes in proteasome protein content suggesting that post‐translational modifications of proteasomes may be responsible for the impaired protein degradation in HCM (Predmore et al. 2010). Mutations in cMyBP‐C accounted for most of the HCM patients used in the Predmore et al. studies, but one TnT mutant (D86A) patient and one Tm (I284V) mutant patient were also part of the HCM group investigated (Predmore et al. 2010). These studies provided the first suggestion that patients with HCM associated with TnT and Tm mutations may also affect proteasome function. As even small increases in apoptosis can cause cardiac dysfunction, it is possible that one mechanism by which proteasome impairment leads to cardiac dysfunction is through increased apoptosis. Altered Ca2+ handling may also be linked to the increased apoptosis; increased Ca2+ influx may lead to sarcoplasmic reticulum Ca2+ overload and cell death by the mitochondrial death pathway.
Proteasome inhibition and calcineurin
Another mechanism could be the activation of the calcineurin–nuclear factor of activated T‐cells (NFAT) pathway by proteasome inhibition (Tang et al. 2010). Both calcineurin and NFAT are degraded by the UPS, and in hearts with impaired proteasome activity, such as in the D7‐desmin mouse hearts, the levels of calcineurin and NFAT may be increased (Tang et al. 2010). Increased calcineurin activity is physiologically important since calcineurin has been shown to be a key regulator of cardiomyocyte hypertrophy (Chen et al. 2016).
Conclusions
The current experimental data suggest that UPS dysfunction may be a hallmark of cardiomyopathies. It is likely that in some cases UPS dysfunction in cardiomyopathies may be caused directly by changes in the ability of the UPS system to degrade the mutant protein (as observed with some cMyBP‐C mutants). It is possible that direct effects of changes in Ca2+ signalling resulting in changes in proteasome activity through interacting partners or post‐translational modifications may also be important in cardiomyopathies. Limited experimental data also suggest that indirect effects of changes in Ca2+ signalling could lead to mitochondrial dysfunction that increases ROS and affects proteasome function via post‐translational modifications. Increased levels of ROS and/or decreased ATP levels may also affect UPS components other than the proteasome. UPS dysfunction could lead to myriad cellular problems including proteotoxicity, mitochondrial dysfunction and apoptosis. Proteotoxicity, mitochondrial dysfunction and apoptosis are each individually associated with cell death and cardiovascular disease, but cardiomyopathies are likely to involve more than one and possibly all three of these processes.
Future studies
Studies have shown that familial cardiomyopathy‐causing mutations in cMyBP‐C, TnT and TnI impair proteasome activity (Bahrudin et al. 2008; Cui et al. 2013b; Gilda et al. 2016). Further studies are needed to determine whether the UPS is affected in cardiomyopathies caused by other mutations in these sarcomeric proteins as well as other sarcomeric proteins, such as α‐tropomyosin and myosin heavy chain. As experimental evidence for the cause of UPS dysfunction as well as the mechanism(s) by which UPS dysfunction may cause cardiomyopathies is still in its infancy, significant studies to further investigate the cause and effect of UPS dysfunction in cardiomyopathies are needed. In particular, the molecular effects of altered Ca2+ levels in cardiac cells as well as the role of mitochondrial dysfunction need to be investigated in cardiomyopathies. Studies are needed to determine if the interaction between calmodulin and proteasome affects proteasome function, and if this plays a role in cardiomyopathies. It is unknown if cardiomyopathy‐causing mutations in other sarcomeric proteins besides cMyBP‐C affect their rate of degradation by the proteasome. A major area of future study should be devoted to investigating the effects of enhancing proteasome function on cardiovascular function in cardiomyopathies, especially under conditions related to increased stress, such as under elevated adrenergic stress.
Additional information
Competing interests
The authors have no competing interests.
Author contributions
Both authors have approved the final version of the manuscript and agree to be accountable for all aspects of the work. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
This work was partly funded by NIH HL096819 and AHA 16GRNT31350040 grants (A.V.G.).
Biographies
Jennifer Gilda is a doctoral candidate at the University of California, Davis. One of her major research interests is the involvement of the proteasome in familial cardiomyopathies. Using proteomics, metabolomics, mouse models, and biochemical techniques, she has contributed to our understanding of how the ubiquitin‐proteasome system and other pathways are affected in troponin‐related cardiomyopathies.

Aldrin Gomes is an Associate Professor at the University of California, Davis. His major interest is determining the molecular mechanisms of signal transduction, particularly in the role of proteostasis (protein homeostasis) in cardiovascular diseases. His background is in biochemistry, proteomics, and striated muscle physiology.
References
- Abriel H, Kamynina E, Horisberger JD & Staub O (2000). Regulation of the cardiac voltage‐gated Na+ channel (H1) by the ubiquitin‐protein ligase Nedd4. FEBS Lett 466, 377–380. [DOI] [PubMed] [Google Scholar]
- Aiken CT, Kaake RM, Wang X & Huang L (2011). Oxidative stress‐mediated regulation of proteasome complexes. Mol Cell Proteomics 10, R110.006924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al‐Yacoub N, Shaheen R, Awad SM, Kunhi M, Dzimiri N, Nguyen HC, Xiong Y, Al‐Buraiki J, Al‐Habeeb W, Alkuraya FS & Poizat C (2016). FBXO32, encoding a member of the SCF complex, is mutated in dilated cardiomyopathy. Genome Biol 17, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araco M, Merlo M, Carr‐White G & Sinagra G (2017). Genetic bases of dilated cardiomyopathy. J Cardiovasc Med 18, 123–130. [DOI] [PubMed] [Google Scholar]
- Bahrudin U, Morikawa K, Takeuchi A, Kurata Y, Miake J, Mizuta E, Adachi K, Higaki K, Yamamoto Y, Shirayoshi Y, Yoshida A, Kato M, Yamamoto K, Nanba E, Morisaki H, Morisaki T, Matsuoka S, Ninomiya H & Hisatome I (2011). Impairment of ubiquitin‐proteasome system by E334K cMyBPC modifies channel proteins, leading to electrophysiological dysfunction. J Mol Biol 413, 857–878. [DOI] [PubMed] [Google Scholar]
- Bahrudin U, Morisaki H, Morisaki T, Ninomiya H, Higaki K, Nanba E, Igawa O, Takashima S, Mizuta E, Miake J, Yamamoto Y, Shirayoshi Y, Kitakaze M, Carrier L & Hisatome I (2008). Ubiquitin‐proteasome system impairment caused by a missense cardiac myosin‐binding protein C mutation and associated with cardiac dysfunction in hypertrophic cardiomyopathy. J Mol Biol 384, 896–907. [DOI] [PubMed] [Google Scholar]
- Balasubramanian S, Mani S, Shiraishi H, Johnston RK, Yamane K, Willey CD, Cooper GT, Tuxworth WJ & Kuppuswamy D (2006). Enhanced ubiquitination of cytoskeletal proteins in pressure overloaded myocardium is accompanied by changes in specific E3 ligases. J Mol Cell Cardiol 41, 669–679. [DOI] [PubMed] [Google Scholar]
- Barefield D & Sadayappan S (2010). Phosphorylation and function of cardiac myosin binding protein‐C in health and disease. J Mol Cell Cardiol 48, 866–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baudenbacher F, Schober T, Pinto JR, Sidorov VY, Hilliard F, Solaro RJ, Potter JD & Knollmann BC (2008). Myofilament Ca2+ sensitization causes susceptibility to cardiac arrhythmia in mice. J Clin Invest 118, 3893–3903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begay RL, Graw S, Sinagra G, Merlo M, Slavov D, Gowan K, Jones KL, Barbati G, Spezzacatene A, Brun F, Di Lenarda A, Smith JE, Granzier HL, Mestroni L & Taylor M (2015). Role of titin missense variants in dilated cardiomyopathy. J Am Heart Assoc 4, e002645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bers DM (2000). Calcium fluxes involved in control of cardiac myocyte contraction. Circ Res 87, 275–281. [DOI] [PubMed] [Google Scholar]
- Bianchi K, Rimessi A, Prandini A, Szabadkai G & Rizzuto R (2004). Calcium and mitochondria: mechanisms and functions of a troubled relationship. Biochim Biophys Acta 1742, 119–131. [DOI] [PubMed] [Google Scholar]
- Birks EJ, Latif N, Enesa K, Folkvang T, Luong LA, Sarathchandra P, Khan M, Ovaa H, Terracciano CM, Barton PJ, Yacoub MH & Evans PC (2008). Elevated p53 expression is associated with dysregulation of the ubiquitin‐proteasome system in dilated cardiomyopathy. Cardiovasc Res 79, 472–480. [DOI] [PubMed] [Google Scholar]
- Bose S, Stratford FL, Broadfoot KI, Mason GG & Rivett AJ (2004). Phosphorylation of 20S proteasome alpha subunit C8 (alpha7) stabilizes the 26S proteasome and plays a role in the regulation of proteasome complexes by gamma‐interferon. Biochem J 378, 177–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breusing N, Arndt J, Voss P, Bresgen N, Wiswedel I, Gardemann A, Siems W & Grune T (2009). Inverse correlation of protein oxidation and proteasome activity in liver and lung. Mech Ageing Dev 130, 748–753. [DOI] [PubMed] [Google Scholar]
- Bulteau AL, Szweda LI & Friguet B (2002). Age‐dependent declines in proteasome activity in the heart. Arch Biochem Biophys 397, 298–304. [DOI] [PubMed] [Google Scholar]
- Carrier L, Knoll R, Vignier N, Keller DI, Bausero P, Prudhon B, Isnard R, Ambroisine ML, Fiszman M, Ross J Jr, Schwartz K & Chien KR (2004). Asymmetric septal hypertrophy in heterozygous cMyBP‐C null mice. Cardiovasc Res 63, 293–304. [DOI] [PubMed] [Google Scholar]
- Castano JG, Mahillo E, Arizti P & Arribas J (1996). Phosphorylation of C8 and C9 subunits of the multicatalytic proteinase by casein kinase II and identification of the C8 phosphorylation sites by direct mutagenesis. Biochem 35, 3782–3789. [DOI] [PubMed] [Google Scholar]
- Celsi F, Pizzo P, Brini M, Leo S, Fotino C, Pinton P & Rizzuto R (2009). Mitochondria, calcium and cell death: a deadly triad in neurodegeneration. Biochim Biophys Acta 1787, 335–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Huang J, Ji Y, Zhang X, Wang P, Deng K, Jiang X, Ma G & Li H (2016). Tripartite motif 32 prevents pathological cardiac hypertrophy. Clin Sci (Lond) 130, 813–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q, Liu JB, Horak KM, Zheng H, Kumarapeli AR, Li J, Li F, Gerdes AM, Wawrousek EF & Wang X (2005). Intrasarcoplasmic amyloidosis impairs proteolytic function of proteasomes in cardiomyocytes by compromising substrate uptake. Circ Res 97, 1018–1026. [DOI] [PubMed] [Google Scholar]
- Chen SN, Czernuszewicz G, Tan Y, Lombardi R, Jin J, Willerson JT & Marian AJ (2012). Human molecular genetic and functional studies identify TRIM63, encoding Muscle RING Finger Protein 1, as a novel gene for human hypertrophic cardiomyopathy. Circ Res 111, 907–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciechanover A & Brundin P (2003). The ubiquitin proteasome system in neurodegenerative diseases: sometimes the chicken, sometimes the egg. Neuron 40, 427–446. [DOI] [PubMed] [Google Scholar]
- Cui Z, Scruggs SB, Gilda JE, Ping P & Gomes AV (2013a). Regulation of cardiac proteasomes by ubiquitination, SUMOylation, and beyond. J Mol Cell Cardiol 71, 32–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui Z, Venkatraman G, Hwang SM & Gomes AV (2013b). Effect of the Troponin I Restrictive Cardiomyopathy Mutation R145W on Protein Expression in Murine Murine Hearts. Biophys J 104, p312a. [Google Scholar]
- Cui Z & Zhang S (2013). Regulation of the human ether‐a‐go‐go‐related gene (hERG) channel by Rab4 protein through neural precursor cell‐expressed developmentally down‐regulated protein 4‐2 (Nedd4‐2). J Biol Chem 288, 21876–21886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunnington RH, Nazari M & Dixon IM (2009). c‐Ski, Smurf2, and Arkadia as regulators of TGF‐beta signalling: new targets for managing myofibroblast function and cardiac fibrosis. Can J Physiol Pharmacol 87, 764–772. [DOI] [PubMed] [Google Scholar]
- Day SM (2013). The ubiquitin proteasome system in human cardiomyopathies and heart failure. Am J Physiol Heart Circ Physiol 304, H1283–H1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day SM, Divald A, Wang P, Davis F, Bartolone S, Jones R & Powell SR (2013). Impaired assembly and post‐translational regulation of 26S proteasome in human end‐stage heart failure. Circ Heart Fail 6, 544–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Tombe PP (2003). Cardiac myofilaments: mechanics and regulation. J Biomech 36, 721–730. [DOI] [PubMed] [Google Scholar]
- Ding WX, Ni HM, Chen X, Yu J, Zhang L & Yin XM (2007). A coordinated action of Bax, PUMA, and p53 promotes MG132‐induced mitochondria activation and apoptosis in colon cancer cells. Mol Cancer Ther 6, 1062–1069. [DOI] [PubMed] [Google Scholar]
- Djakovic SN, Marquez‐Lona EM, Jakawich SK, Wright R, Chu C, Sutton MA & Patrick GN (2012). Phosphorylation of Rpt6 regulates synaptic strength in hippocampal neurons. J Neurosci 32, 5126–5131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drews O, Wildgruber R, Zong C, Sukop U, Nissum M, Weber G, Gomes AV & Ping P (2007). Mammalian proteasome subpopulations with distinct molecular compositions and proteolytic activities. Mol Cell Proteomics 6, 2021–2031. [DOI] [PubMed] [Google Scholar]
- Enrico O, Gabriele B, Nadia C, Sara G, Daniele V, Giulia C, Antonio S & Mario P (2007). Unexpected cardiotoxicity in haematological bortezomib treated patients. Br J Haematol 138, 396–397. [DOI] [PubMed] [Google Scholar]
- Fan M, Park A & Nephew KP (2005). CHIP (carboxyl terminus of Hsc70‐interacting protein) promotes basal and geldanamycin‐induced degradation of estrogen receptor‐α. Mol Endocrinol 19, 2901–2914. [DOI] [PubMed] [Google Scholar]
- Feng Y, Longo DL & Ferris DK (2001). Polo‐like kinase interacts with proteasomes and regulates their activity. Cell Growth Differ 12, 29–37. [PubMed] [Google Scholar]
- Fielitz J, van Rooij E, Spencer JA, Shelton JM, Latif S, van der Nagel R, Bezprozvannaya S, de Windt L, Richardson JA, Bassel‐Duby R & Olson EN (2007). Loss of muscle‐specific RING‐finger 3 predisposes the heart to cardiac rupture after myocardial infarction. Proc Natl Acad Sci USA 104, 4377–4382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flavigny J, Souchet M, Sebillon P, Berrebi‐Bertrand I, Hainque B, Mallet A, Bril A, Schwartz K & Carrier L (1999). COOH‐terminal truncated cardiac myosin‐binding protein C mutants resulting from familial hypertrophic cardiomyopathy mutations exhibit altered expression and/or incorporation in fetal rat cardiomyocytes. J Mol Biol 294, 443–456. [DOI] [PubMed] [Google Scholar]
- Franklin O & Burch M (2000). Dilated cardiomyopathy in childhood. Images Paediatr Cardiol 2, 3–10. [PMC free article] [PubMed] [Google Scholar]
- Frey N, Luedde M & Katus HA (2012). Mechanisms of disease: hypertrophic cardiomyopathy. Nat Rev Cardiol 9, 91–100. [DOI] [PubMed] [Google Scholar]
- Fyrberg E, Fyrberg CC, Beall C & Saville DL (1990). Drosophila melanogaster troponin‐T mutations engender three distinct syndromes of myofibrillar abnormalities. J Mol Biol 216, 657–675. [DOI] [PubMed] [Google Scholar]
- Ghosh R, Hwang SM, Cui Z, Gilda JE & Gomes AV (2016). Different effects of the nonsteroidal anti‐inflammatory drugs meclofenamate sodium and naproxen sodium on proteasome activity in cardiac cells. J Mol Cell Cardiol 94, 131–144. [DOI] [PubMed] [Google Scholar]
- Gianni D, Li A, Tesco G, McKay KM, Moore J, Raygor K, Rota M, Gwathmey JK, Dec GW, Aretz T, Leri A, Semigran MJ, Anversa P, Macgillivray TE, Tanzi RE & del Monte F (2010). Protein aggregates and novel presenilin gene variants in idiopathic dilated cardiomyopathy. Circulation 121, 1216–1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gigli M, Begay RL, Morea G, Graw SL, Sinagra G, Taylor MR, Granzier H & Mestroni L (2016). A review of the giant protein titin in clinical molecular diagnostics of cardiomyopathies. Front Cardiovasc Med 3, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilda JE, Lai X, Witzmann FA & Gomes AV (2016). Delineation of molecular pathways involved in cardiomyopathies caused by troponin T mutations. Mol Cell Proteomics 15, 1962–1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldfarb LG & Dalakas MC (2009). Tragedy in a heartbeat: malfunctioning desmin causes skeletal and cardiac muscle disease. J Clin Invest 119, 1806–1813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes A (2009). Proteasome dysfunction in troponin related cardiomyopathies. Biophys J 96, p372a. [Google Scholar]
- Gomes AV, Barnes JA, Harada K & Potter JD (2004). Role of troponin T in disease. Mol Cell Biochem 263, 115–129. [DOI] [PubMed] [Google Scholar]
- Gomes AV, Liang J & Potter JD (2005). Mutations in human cardiac troponin I that are associated with restrictive cardiomyopathy affect basal ATPase activity and the calcium sensitivity of force development. J Biol Chem 280, 30909–30915. [DOI] [PubMed] [Google Scholar]
- Gomes AV & Potter JD (2004). Molecular and cellular aspects of troponin cardiomyopathies. Ann NY Acad Sci 1015, 214–224. [DOI] [PubMed] [Google Scholar]
- Gomes AV, Young GW, Wang Y, Zong C, Eghbali M, Drews O, Lu H, Stefani E & Ping P (2009). Contrasting proteome biology and functional heterogeneity of the 20 S proteasome complexes in mammalian tissues. Mol Cell Proteomics 8, 302–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes AV, Zong C, Edmondson RD, Li X, Stefani E, Zhang J, Jones RC, Thyparambil S, Wang GW, Qiao X, Bardag‐Gorce F & Ping P (2006). Mapping the murine cardiac 26S proteasome complexes. Circ Res 99, 362–371. [DOI] [PubMed] [Google Scholar]
- Grandin EW, Ky B, Cornell RF, Carver J & Lenihan DJ (2015). Patterns of cardiac toxicity associated with irreversible proteasome inhibition in the treatment of multiple myeloma. J Card Fail 21, 138–144. [DOI] [PubMed] [Google Scholar]
- Hacihanefioglu A, Tarkun P & Gonullu E (2008). Acute severe cardiac failure in a myeloma patient due to proteasome inhibitor bortezomib. Int J Hematol 88, 219–222. [DOI] [PubMed] [Google Scholar]
- Hamada H, Suzuki M, Yuasa S, Mimura N, Shinozuka N, Takada Y, Nishino T, Nakaya H, Koseki H & Aoe T (2004). Dilated cardiomyopathy caused by aberrant endoplasmic reticulum quality control in mutant KDEL receptor transgenic mice. Mol Cell Biol 24, 8007–8017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hazebroek M, Dennert R & Heymans S (2012). Idiopathic dilated cardiomyopathy: possible triggers and treatment strategies. Neth Heart J 20, 332–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He H, Javadpour MM, Latif F, Tardiff JC & Ingwall JS (2007). R‐92L and R‐92W mutations in cardiac troponin T lead to distinct energetic phenotypes in intact mouse hearts. Biophys J 93, 1834–1844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He X, Gao X, Peng L, Wang S, Zhu Y, Ma H, Lin J & Duan DD (2011). Atrial fibrillation induces myocardial fibrosis through angiotensin II type 1 receptor‐specific Arkadia‐mediated downregulation of Smad7. Circ Res 108, 164–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herman DS, Lam L, Taylor MR, Wang L, Teekakirikul P, Christodoulou D, Conner L, DePalma SR, McDonough B, Sparks E, Teodorescu DL, Cirino AL, Banner NR, Pennell DJ, Graw S, Merlo M, Di Lenarda A, Sinagra G, Bos JM, Ackerman MJ, Mitchell RN, Murry CE, Lakdawala NK, Ho CY, Barton PJ, Cook SA, Mestroni L, Seidman JG & Seidman CE (2012). Truncations of titin causing dilated cardiomyopathy. N Engl J Med 366, 619–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez OM, Szczesna‐Cordary D, Knollmann BC, Miller T, Bell M, Zhao J, Sirenko SG, Diaz Z, Guzman G, Xu Y, Wang Y, Kerrick WG & Potter JD (2005). F110I and R278C troponin T mutations that cause familial hypertrophic cardiomyopathy affect muscle contraction in transgenic mice and reconstituted human cardiac fibres. J Biol Chem 280, 37183–37194. [DOI] [PubMed] [Google Scholar]
- Herrmann J, Wohlert C, Saguner AM, Flores A, Nesbitt LL, Chade A, Lerman LO & Lerman A (2013). Primary proteasome inhibition results in cardiac dysfunction. Eur J Heart Fail 15, 614–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hershberger RE & Morales A (1993). Dilated Cardiomyopathy Overview. In GeneReviews, ed. Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH, Bird TD, Fong CT, Mefford HC, Smith RJH. & Stephens K. University of Washington, Seattle, Seattle. [Google Scholar]
- Huang H, Joazeiro CA, Bonfoco E, Kamada S, Leverson JD & Hunter T (2000). The inhibitor of apoptosis, cIAP2, functions as a ubiquitin‐protein ligase and promotes in vitro monoubiquitination of caspases 3 and 7. J Biol Chem 275, 26661–26664. [DOI] [PubMed] [Google Scholar]
- Huke S & Knollmann BC (2010). Increased myofilament Ca2+‐sensitivity and arrhythmia susceptibility. J Mol Cell Cardiol 48, 824–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishii T, Sakurai T, Usami H & Uchida K (2005). Oxidative modification of proteasome: identification of an oxidation‐sensitive subunit in 26 S proteasome. Biochemistry 44, 13893–13901. [DOI] [PubMed] [Google Scholar]
- Jespersen T, Membrez M, Nicolas CS, Pitard B, Staub O, Olesen SP, Baro I & Abriel H (2007). The KCNQ1 potassium channel is down‐regulated by ubiquitylating enzymes of the Nedd4/Nedd4‐like family. Cardiovasc Res 74, 64–74. [DOI] [PubMed] [Google Scholar]
- Jung T, Hohn A & Grune T (2014). The proteasome and the degradation of oxidized proteins: Part II – protein oxidation and proteasomal degradation. Redox Biol 2, 99–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabakov AE, Budagova KR, Latchman DS & Kampinga HH (2002). Stressful preconditioning and HSP70 overexpression attenuate proteotoxicity of cellular ATP depletion. Am J Physiol Cell Physiol 283, C521–C534. [DOI] [PubMed] [Google Scholar]
- Kavsak P, Rasmussen RK, Causing CG, Bonni S, Zhu H, Thomsen GH & Wrana JL (2000). Smad7 binds to Smurf2 to form an E3 ubiquitin ligase that targets the TGF beta receptor for degradation. Mol Cell 6, 1365–1375. [DOI] [PubMed] [Google Scholar]
- Kedar V, McDonough H, Arya R, Li HH, Rockman HA & Patterson C (2004). Muscle‐specific RING finger 1 is a bona fide ubiquitin ligase that degrades cardiac troponin I. Proc Natl Acad Sci USA 101, 18135–18140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keeling PJ, Goldman JH, Slade AK, Elliott PM, Caforio AL, Poloniecki J & McKenna WJ (1995). Prognosis of idiopathic dilated cardiomyopathy. J Card Fail 1, 337–345. [DOI] [PubMed] [Google Scholar]
- Knollmann BC, Kirchhof P, Sirenko SG, Degen H, Greene AE, Schober T, Mackow JC, Fabritz L, Potter JD & Morad M (2003). Familial hypertrophic cardiomyopathy‐linked mutant troponin T causes stress‐induced ventricular tachycardia and Ca2+‐dependent action potential remodelling. Circ Res 92, 428–436. [DOI] [PubMed] [Google Scholar]
- Kubo T, Matsumura Y, Kitaoka H, Okawa M, Hirota T, Hamada T, Hitomi N, Hoshikawa E, Hayato K, Shimizu Y, Yamasaki N, Yabe T, Nishinaga M, Takata J & Doi Y (2008). Improvement in prognosis of dilated cardiomyopathy in the elderly over the past 20 years. J Cardiol 52, 111–117. [DOI] [PubMed] [Google Scholar]
- Lamothe SM & Zhang S (2013). The serum‐ and glucocorticoid‐inducible kinases SGK1 and SGK3 regulate hERG channel expression via ubiquitin ligase Nedd4‐2 and GTPase Rab11. J Biol Chem 288, 15075–15084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le NT, Takei Y, Shishido T, Woo CH, Chang E, Heo KS, Lee H, Lu Y, Morrell C, Oikawa M, McClain C, Wang X, Tournier C, Molina CA, Taunton J, Yan C, Fujiwara K, Patterson C, Yang J & Abe J (2012). p90RSK targets the ERK5‐CHIP ubiquitin E3 ligase activity in diabetic hearts and promotes cardiac apoptosis and dysfunction. Circ Res 110, 536–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SH, Park Y, Yoon SK & Yoon JB (2010). Osmotic stress inhibits proteasome by p38 MAPK‐dependent phosphorylation. J Biol Chem 285, 41280–41289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li HH, Kedar V, Zhang C, McDonough H, Arya R, Wang DZ & Patterson C (2004). Atrogin‐1/muscle atrophy F‐box inhibits calcineurin‐dependent cardiac hypertrophy by participating in an SCF ubiquitin ligase complex. J Clin Invest 114, 1058–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li HH, Willis MS, Lockyer P, Miller N, McDonough H, Glass DJ & Patterson C (2007). Atrogin‐1 inhibits Akt‐dependent cardiac hypertrophy in mice via ubiquitin‐dependent coactivation of Forkhead proteins. J Clin Invest 117, 3211–3223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Horak KM, Su H, Sanbe A, Robbins J & Wang X (2011a). Enhancement of proteasomal function protects against cardiac proteinopathy and ischemia/reperfusion injury in mice. J Clin Invest 121, 3689–3700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li N, Zhang Z, Zhang W & Wei Q (2011b). Calcineurin B subunit interacts with proteasome subunit alpha type 7 and represses hypoxia‐inducible factor‐1α activity via the proteasome pathway. Biochem Biophys Res Commun 405, 468–472. [DOI] [PubMed] [Google Scholar]
- Lin F, Ghislat G, Luo S, Renna M, Siddiqi F & Rubinsztein DC (2015). XIAP and cIAP1 amplifications induce Beclin 1‐dependent autophagy through NFκB activation. Hum Mol Genet 24, 2899–2913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Chen Q, Huang W, Horak KM, Zheng H, Mestril R & Wang X (2006a). Impairment of the ubiquitin‐proteasome system in desminopathy mouse hearts. FASEB J 20, 362–364. [DOI] [PubMed] [Google Scholar]
- Liu J, Tang M, Mestril R & Wang X (2006b). Aberrant protein aggregation is essential for a mutant desmin to impair the proteolytic function of the ubiquitin‐proteasome system in cardiomyocytes. J Mol Cell Cardiol 40, 451–454. [DOI] [PubMed] [Google Scholar]
- Liu X, Huang W, Li C, Li P, Yuan J, Li X, Qiu XB, Ma Q & Cao C (2006. c). Interaction between c‐Abl and Arg tyrosine kinases and proteasome subunit PSMA7 regulates proteasome degradation. Molecular Cell 22, 317–327. [DOI] [PubMed] [Google Scholar]
- Marambaud P, Wilk S & Checler F (1996). Protein kinase A phosphorylation of the proteasome: a contribution to the alpha‐secretase pathway in human cells. J Neurochem 67, 2616–2619. [DOI] [PubMed] [Google Scholar]
- Maron BJ (2002). Hypertrophic cardiomyopathy: a systematic review. JAMA 287, 1308–1320. [DOI] [PubMed] [Google Scholar]
- Maron BJ (2003). Sudden death in young athletes. N Engl J Med 349, 1064–1075. [DOI] [PubMed] [Google Scholar]
- Marques MA & de Oliveira GA (2016). Cardiac troponin and tropomyosin: structural and cellular perspectives to unveil the hypertrophic cardiomyopathy phenotype. Front Physiol 7, 429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marston S, Copeland O, Jacques A, Livesey K, Tsang V, McKenna WJ, Jalilzadeh S, Carballo S, Redwood C & Watkins H (2009). Evidence from human myectomy samples that MYBPC3 mutations cause hypertrophic cardiomyopathy through haploinsufficiency. Circ Res 105, 219–222. [DOI] [PubMed] [Google Scholar]
- McLendon PM & Robbins J (2011). Desmin‐related cardiomyopathy: an unfolding story. Am J Physiol Heart Circ Physiol 301, H1220–H1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLendon PM & Robbins J (2015). Proteotoxicity and cardiac dysfunction. Circ Res 116, 1863–1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNally EM, Golbus JR & Puckelwartz MJ (2013). Genetic mutations and mechanisms in dilated cardiomyopathy. J Clin Invest 123, 19–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mearini G, Gedicke C, Schlossarek S, Witt CC, Kramer E, Cao P, Gomes MD, Lecker SH, Labeit S, Willis MS, Eschenhagen T & Carrier L (2010). Atrogin‐1 and MuRF1 regulate cardiac MyBP‐C levels via different mechanisms. Cardiovasc Res 85, 357–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mearini G, Schlossarek S, Willis MS & Carrier L (2008). The ubiquitin‐proteasome system in cardiac dysfunction. Biochim Biophys Acta 1782, 749–763. [DOI] [PubMed] [Google Scholar]
- Miller T, Szczesna D, Housmans PR, Zhao J, de Freitas F, Gomes AV, Culbreath L, McCue J, Wang Y, Xu Y, Kerrick WG & Potter JD (2001). Abnormal contractile function in transgenic mice expressing a familial hypertrophic cardiomyopathy‐linked troponin T (I79N) mutation. J Biol Chem 276, 3743–3755. [DOI] [PubMed] [Google Scholar]
- Moolman JA, Reith S, Uhl K, Bailey S, Gautel M, Jeschke B, Fischer C, Ochs J, McKenna WJ, Klues H & Vosberg HP (2000). A newly created splice donor site in exon 25 of the MyBP‐C gene is responsible for inherited hypertrophic cardiomyopathy with incomplete disease penetrance. Circulation 101, 1396–1402. [DOI] [PubMed] [Google Scholar]
- Murdaca J, Treins C, Monthouel‐Kartmann MN, Pontier‐Bres R, Kumar S, Van Obberghen E & Giorgetti‐Peraldi S (2004). Grb10 prevents Nedd4‐mediated vascular endothelial growth factor receptor‐2 degradation. J Biol Chem 279, 26754–26761. [DOI] [PubMed] [Google Scholar]
- Naito AT, Okada S, Minamino T, Iwanaga K, Liu ML, Sumida T, Nomura S, Sahara N, Mizoroki T, Takashima A, Akazawa H, Nagai T, Shiojima I & Komuro I (2010). Promotion of CHIP‐mediated p53 degradation protects the heart from ischemic injury. Circ Res 106, 1692–1702. [DOI] [PubMed] [Google Scholar]
- Nakayama K, Frew IJ, Hagensen M, Skals M, Habelhah H, Bhoumik A, Kadoya T, Erdjument‐Bromage H, Tempst P, Frappell PB, Bowtell DD & Ronai Z (2004). Siah2 regulates stability of prolyl‐hydroxylases, controls HIF1α abundance, and modulates physiological responses to hypoxia. Cell 117, 941–952. [DOI] [PubMed] [Google Scholar]
- Olive M, Abdul‐Hussein S, Oldfors A, Gonzalez‐Costello J, van der Ven PF, Furst DO, Gonzalez L, Moreno D, Torrejon‐Escribano B, Alio J, Pou A, Ferrer I & Tajsharghi H (2015). New cardiac and skeletal protein aggregate myopathy associated with combined MuRF1 and MuRF3 mutations. Hum Mol Genet 24, 3638–3650. [DOI] [PubMed] [Google Scholar]
- Otaki Y, Takahashi H, Watanabe T, Funayama A, Netsu S, Honda Y, Narumi T, Kadowaki S, Hasegawa H, Honda S, Arimoto T, Shishido T, Miyamoto T, Kamata H, Nakajima O & Kubota I (2016). HECT‐type ubiquitin E3 ligase ITCH interacts with thioredoxin‐interacting protein and ameliorates reactive oxygen species‐induced cardiotoxicity. J Am Heart Assoc 5, e002485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otsuka K, Terasaki F, Shimomura H, Tsukada B, Horii T, Isomura T, Suma H, Shibayama Y & Kitaura Y (2010). Enhanced expression of the ubiquitin‐proteasome system in the myocardium from patients with dilated cardiomyopathy referred for left ventriculoplasty: an immunohistochemical study with special reference to oxidative stress. Heart Vessels 25, 474–484. [DOI] [PubMed] [Google Scholar]
- Pan JA, Sun Y, Jiang YP, Bott AJ, Jaber N, Dou Z, Yang B, Chen JS, Catanzaro JM, Du C, Ding WX, Diaz‐Meco MT, Moscat J, Ozato K, Lin RZ & Zong WX (2016). TRIM21 ubiquitylates SQSTM1/p62 and suppresses protein sequestration to regulate redox homeostasis. Mol Cell 62, 149–151. [DOI] [PubMed] [Google Scholar]
- Pandit B & Gartel AL (2011). Proteasome inhibitors induce p53‐independent apoptosis in human cancer cells. Am J Pathol 178, 355–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson C, Ike C, Willis PW 4th, Stouffer GA & Willis MS (2007). The bitter end: the ubiquitin‐proteasome system and cardiac dysfunction. Circulation 115, 1456–1463. [DOI] [PubMed] [Google Scholar]
- Paulin D & Li Z (2004). Desmin: a major intermediate filament protein essential for the structural integrity and function of muscle. Exp Cell Res 301, 1–7. [DOI] [PubMed] [Google Scholar]
- Peng TI & Jou MJ (2010). Oxidative stress caused by mitochondrial calcium overload. Ann NY Acad Sci 1201, 183–188. [DOI] [PubMed] [Google Scholar]
- Pramod S & Shivakumar K (2014). Mechanisms in cardiac fibroblast growth: an obligate role for Skp2 and FOXO3a in ERK1/2 MAPK‐dependent regulation of p27kip1. Am J Physiol Heart Circ Physiol 306, H844–H855. [DOI] [PubMed] [Google Scholar]
- Predmore JM, Wang P, Davis F, Bartolone S, Westfall MV, Dyke DB, Pagani F, Powell SR & Day SM (2010). Ubiquitin proteasome dysfunction in human hypertrophic and dilated cardiomyopathies. Circulation 121, 997–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rafiq K, Guo J, Vlasenko L, Guo X, Kolpakov MA, Sanjay A, Houser SR & Sabri A (2012). c‐Cbl ubiquitin ligase regulates focal adhesion protein turnover and myofibril degeneration induced by neutrophil protease cathepsin G. J Biol Chem 287, 5327–5339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajagopalan V, Zhao M, Reddy S, Fajardo G, Wang X, Dewey S, Gomes AV & Bernstein D (2013). Altered ubiquitin‐proteasome signalling in right ventricular hypertrophy and failure. Am J Physiol Heart Circ Physiol 305, H551–H562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranek MJ, Kost CK Jr, Hu C, Martin DS & Wang X (2014). Muscarinic 2 receptors modulate cardiac proteasome function in a protein kinase G‐dependent manner. J Mol Cell Cardiol 69, 43–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranek MJ, Terpstra EJ, Li J, Kass DA & Wang X (2013). Protein kinase G positively regulates proteasome‐mediated degradation of misfolded proteins. Circulation 128, 365–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reits E, Neijssen J, Herberts C, Benckhuijsen W, Janssen L, Drijfhout JW & Neefjes J (2004). A major role for TPPII in trimming proteasomal degradation products for MHC class I antigen presentation. Immunity 20, 495–506. [DOI] [PubMed] [Google Scholar]
- Richard P, Charron P, Carrier L, Ledeuil C, Cheav T, Pichereau C, Benaiche A, Isnard R, Dubourg O, Burban M, Gueffet JP, Millaire A, Desnos M, Schwartz K, Hainque B & Komajda M (2003). Hypertrophic cardiomyopathy: distribution of disease genes, spectrum of mutations, and implications for a molecular diagnosis strategy. Circulation 107, 2227–2232. [DOI] [PubMed] [Google Scholar]
- Roder K, Werdich AA, Li W, Liu M, Kim TY, Organ‐Darling LE, Moshal KS, Hwang JM, Lu Y, Choi BR, MacRae CA & Koren G (2014). RING finger protein RNF207, a novel regulator of cardiac excitation. J Biol Chem 289, 33730–33740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rottbauer W, Gautel M, Zehelein J, Labeit S, Franz WM, Fischer C, Vollrath B, Mall G, Dietz R, Kubler W & Katus HA (1997). Novel splice donor site mutation in the cardiac myosin‐binding protein‐C gene in familial hypertrophic cardiomyopathy. Characterization of cardiac transcript and protein. J Clin Invest 100, 475–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarikas A, Carrier L, Schenke C, Doll D, Flavigny J, Lindenberg KS, Eschenhagen T & Zolk O (2005). Impairment of the ubiquitin‐proteasome system by truncated cardiac myosin binding protein C mutants. Cardiovasc Res 66, 33–44. [DOI] [PubMed] [Google Scholar]
- Schlossarek S & Carrier L (2011). The ubiquitin‐proteasome system in cardiomyopathies. Curr Opin Cardiol 26, 190–195. [DOI] [PubMed] [Google Scholar]
- Schlossarek S, Englmann DR, Sultan KR, Sauer M, Eschenhagen T & Carrier L (2012a). Defective proteolytic systems in Mybpc3‐targeted mice with cardiac hypertrophy. Basic Res Cardiol 107, 235. [DOI] [PubMed] [Google Scholar]
- Schlossarek S, Schuermann F, Geertz B, Mearini G, Eschenhagen T & Carrier L (2012b). Adrenergic stress reveals septal hypertrophy and proteasome impairment in heterozygous Mybpc3‐targeted knock‐in mice. J Muscle Res Cell Motil 33, 5–15. [DOI] [PubMed] [Google Scholar]
- Schober T, Huke S, Venkataraman R, Gryshchenko O, Kryshtal D, Hwang HS, Baudenbacher FJ & Knollmann BC (2012). Myofilament Ca sensitization increases cytosolic Ca binding affinity, alters intracellular Ca homeostasis, and causes pause‐dependent Ca‐triggered arrhythmia. Circ Res 111, 170–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen X, Valencia CA, Szostak JW, Dong B & Liu R (2005). Scanning the human proteome for calmodulin‐binding proteins. Proc Natl Acad Sci USA 102, 5969–5974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirenko SG, Potter JD & Knollmann BC (2006). Differential effect of troponin T mutations on the inotropic responsiveness of mouse hearts – role of myofilament Ca2+ sensitivity increase. J Physiol 575, 201–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sisakian H (2014). Cardiomyopathies: Evolution of pathogenesis concepts and potential for new therapies. World J Cardiol 6, 478–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spaich S, Will RD, Just S, Kuhn C, Frank D, Berger IM, Wiemann S, Korn B, Koegl M, Backs J, Katus HA, Rottbauer W & Frey N (2012). F‐box and leucine‐rich repeat protein 22 is a cardiac‐enriched F‐box protein that regulates sarcomeric protein turnover and is essential for maintenance of contractile function in vivo. Circ Res 111, 1504–1516. [DOI] [PubMed] [Google Scholar]
- Stadtmueller BM & Hill CP (2011). Proteasome activators. Molecular Cell 41, 8–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su M, Wang J, Kang L, Wang Y, Zou Y, Feng X, Wang D, Ahmad F, Zhou X, Hui R & Song L (2014). Rare variants in genes encoding MuRF1 and MuRF2 are modifiers of hypertrophic cardiomyopathy. Int J Mol Sci 15, 9302–9313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki Y, Nakabayashi Y & Takahashi R (2001). Ubiquitin‐protein ligase activity of X‐linked inhibitor of apoptosis protein promotes proteasomal degradation of caspase‐3 and enhances its anti‐apoptotic effect in Fas‐induced cell death. Proc Natl Acad Sci USA 98, 8662–8667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang M, Li J, Huang W, Su H, Liang Q, Tian Z, Horak KM, Molkentin JD & Wang X (2010). Proteasome functional insufficiency activates the calcineurin‐NFAT pathway in cardiomyocytes and promotes maladaptive remodelling of stressed mouse hearts. Cardiovasc Res 88, 424–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thottakara T, Friedrich FW, Reischmann S, Braumann S, Schlossarek S, Kramer E, Juhr D, Schluter H, van der Velden J, Munch J, Patten M, Eschenhagen T, Moog‐Lutz C & Carrier L (2015). The E3 ubiquitin ligase Asb2β is downregulated in a mouse model of hypertrophic cardiomyopathy and targets desmin for proteasomal degradation. J Mol Cell Cardiol 87, 214–224. [DOI] [PubMed] [Google Scholar]
- Tian LF, Li HY, Jin BF, Pan X, Man JH, Zhang PJ, Li WH, Liang B, Liu H, Zhao J, Gong WL, Zhou T & Zhang XM (2006). MDM2 interacts with and downregulates a sarcomeric protein, TCAP. Biochem Biophys Res Commun 345, 355–361. [DOI] [PubMed] [Google Scholar]
- Tian Z, Zheng H, Li J, Li Y, Su H & Wang X (2012). Genetically induced moderate inhibition of the proteasome in cardiomyocytes exacerbates myocardial ischemia‐reperfusion injury in mice. Circ Res 111, 532–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toth A, Nickson P, Qin LL & Erhardt P (2006). Differential regulation of cardiomyocyte survival and hypertrophy by MDM2, an E3 ubiquitin ligase. J Biol Chem 281, 3679–3689. [DOI] [PubMed] [Google Scholar]
- Tsuchiya T, Bonner HP, Engel T, Woods I, Matsushima S, Ward MW, Taki W, Henshall DC, Concannon CG & Prehn JH (2011). Bcl‐2 homology domain 3‐only proteins Puma and Bim mediate the vulnerability of CA1 hippocampal neurons to proteasome inhibition in vivo. Eur J Neurosci 33, 401–408. [DOI] [PubMed] [Google Scholar]
- Vakrou S & Abraham MR (2014). Hypertrophic cardiomyopathy: a heart in need of an energy bar? Front Physiol 5, 309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Wijngaard A, Volders P, Van Tintelen JP, Jongbloed JD, van den Berg MP, Lekanne Deprez RH, Mannens MM, Hofmann N, Slegtenhorst M, Dooijes D, Michels M, Arens Y, Jongbloed R & Smeets BJ (2011). Recurrent and founder mutations in the Netherlands: cardiac Troponin I (TNNI3) gene mutations as a cause of severe forms of hypertrophic and restrictive cardiomyopathy. Neth Heart J 19, 344–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Dijk SJ, Dooijes D, dos Remedios C, Michels M, Lamers JM, Winegrad S, Schlossarek S, Carrier L, ten Cate FJ, Stienen GJ & van der Velden J (2009). Cardiac myosin‐binding protein C mutations and hypertrophic cardiomyopathy: haploinsufficiency, deranged phosphorylation, and cardiomyocyte dysfunction. Circulation 119, 1473–1483. [DOI] [PubMed] [Google Scholar]
- van Dijk SJ, Paalberends ER, Najafi A, Michels M, Sadayappan S, Carrier L, Boontje NM, Kuster DW, van Slegtenhorst M, Dooijes D, dos Remedios C, ten Cate FJ, Stienen GJ & van der Velden J (2012). Contractile dysfunction irrespective of the mutant protein in human hypertrophic cardiomyopathy with normal systolic function. Circ Heart Fail 5, 36–46. [DOI] [PubMed] [Google Scholar]
- van Spaendonck‐Zwarts KY, van Hessem L, Jongbloed JD, de Walle HE, Capetanaki Y, van der Kooi AJ, van Langen IM, van den Berg MP & van Tintelen JP (2011). Desmin‐related myopathy. Clin Genet 80, 354–366. [DOI] [PubMed] [Google Scholar]
- Vignier N, Schlossarek S, Fraysse B, Mearini G, Kramer E, Pointu H, Mougenot N, Guiard J, Reimer R, Hohenberg H, Schwartz K, Vernet M, Eschenhagen T & Carrier L (2009). Nonsense‐mediated mRNA decay and ubiquitin‐proteasome system regulate cardiac myosin‐binding protein C mutant levels in cardiomyopathic mice. Circ Res 105, 239–248. [DOI] [PubMed] [Google Scholar]
- Voigt A, Bartel K, Egerer K, Trimpert C, Feist E, Gericke C, Kandolf R, Klingel K, Kuckelkorn U, Stangl K, Felix SB, Baumann G, Kloetzel PM & Staudt A (2010). Humoral anti‐proteasomal autoimmunity in dilated cardiomyopathy. Basic Res Cardiol 105, 9–18. [DOI] [PubMed] [Google Scholar]
- von Mikecz A, Chen M, Rockel T & Scharf A (2008). The nuclear ubiquitin‐proteasome system: visualization of proteasomes, protein aggregates, and proteolysis in the cell nucleus. Methods Mol Biol 463, 191–202. [DOI] [PubMed] [Google Scholar]
- Voortman J & Giaccone G (2006). Severe reversible cardiac failure after bortezomib treatment combined with chemotherapy in a non‐small cell lung cancer patient: a case report. BMC Cancer 6, 129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Sun A, Li L, Zhao G, Jia J, Wang K, Ge J & Zou Y (2012). Up‐regulation of BMP‐2 antagonizes TGF‐β1/ROCK‐enhanced cardiac fibrotic signalling through activation of Smurf1/Smad6 complex. J Cell Mol Med 16, 2301–2310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X (2013). Repeated intermittent administration of a ubiquitous proteasome inhibitor leads to restrictive cardiomyopathy. Eur J Heart Fail 15, 597–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Li J, Zheng H, Su H & Powell SR (2011). Proteasome functional insufficiency in cardiac pathogenesis. Am J Physiol Heart Circ Physiol 301, H2207–H2219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X & Robbins J (2006). Heart failure and protein quality control. Circ Res 99, 1315–1328. [DOI] [PubMed] [Google Scholar]
- Watanabe M, Funakoshi T, Unuma K, Aki T & Uemura K (2014). Activation of the ubiquitin‐proteasome system against arsenic trioxide cardiotoxicity involves ubiquitin ligase Parkin for mitochondrial homeostasis. Toxicology 322, 43–50. [DOI] [PubMed] [Google Scholar]
- Weekes J, Morrison K, Mullen A, Wait R, Barton P & Dunn MJ (2003). Hyperubiquitination of proteins in dilated cardiomyopathy. Proteomics 3, 208–216. [DOI] [PubMed] [Google Scholar]
- Weekes J, Wheeler CH, Yan JX, Weil J, Eschenhagen T, Scholtysik G & Dunn MJ (1999). Bovine dilated cardiomyopathy: proteomic analysis of an animal model of human dilated cardiomyopathy. Electrophor 20, 898–906. [DOI] [PubMed] [Google Scholar]
- Wen Y, Xu Y, Wang Y, Pinto JR, Potter JD & Kerrick WG (2009). Functional effects of a restrictive‐cardiomyopathy‐linked cardiac troponin I mutation (R145W) in transgenic mice. J Mol Biol 392, 1158–1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willis MS & Patterson C (2013). Proteotoxicity and cardiac dysfunction—Alzheimer's disease of the heart? N Engl J Med 368, 455–464. [DOI] [PubMed] [Google Scholar]
- Witt SH, Granzier H, Witt CC & Labeit S (2005). MURF‐1 and MURF‐2 target a specific subset of myofibrillar proteins redundantly: towards understanding MURF‐dependent muscle ubiquitination. J Mol Biol 350, 713–722. [DOI] [PubMed] [Google Scholar]
- Wu B, Wang L, Liu Q & Luo Q (2012). Myocardial contractile and metabolic properties of familial hypertrophic cardiomyopathy caused by cardiac troponin I gene mutations: a simulation study. Exp Physiol 97, 155–169. [DOI] [PubMed] [Google Scholar]
- Wu W, Lu CX, Wang YN, Liu F, Chen W, Liu YT, Han YC, Cao J, Zhang SY & Zhang X (2015). Novel phenotype‐genotype correlations of restrictive cardiomyopathy with myosin‐binding protein C (MYBPC3) gene mutations tested by next‐generation sequencing. J Am Heart Assoc 4, e001879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong S, Van Pelt CS, Elizondo‐Fraire AC, Fernandez‐Garcia B & Lozano G (2007). Loss of Mdm4 results in p53‐dependent dilated cardiomyopathy. Circulation 115, 2925–2930. [DOI] [PubMed] [Google Scholar]
- Yang Q, Sanbe A, Osinska H, Hewett TE, Klevitsky R & Robbins J (1999). In vivo modelling of myosin binding protein C familial hypertrophic cardiomyopathy. Circ Res 85, 841–847. [DOI] [PubMed] [Google Scholar]
- Zhang F, Hu Y, Huang P, Toleman CA, Paterson AJ & Kudlow JE (2007). Proteasome function is regulated by cyclic AMP‐dependent protein kinase through phosphorylation of Rpt6. J Biol Chem 282, 22460–22471. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Xu X, Ceylan‐Isik AF, Dong M, Pei Z, Li Y & Ren J (2014). Ablation of Akt2 protects against lipopolysaccharide‐induced cardiac dysfunction: role of Akt ubiquitination E3 ligase TRAF6. J Mol Cell Cardiol 74, 76–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao C, Wang L, Ma X, Zhu W, Yao L, Cui Y, Liu Y, Li J, Liang X, Sun Y, Li L & Chen YH (2015a). Cardiac Nav1.5 is modulated by ubiquitin protein ligase E3 component n‐recognin UBR3 and 6. J Cell Mol Med 19, 2143–2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao D, Sun Y, Wei X, Liang H, Zhao L, Dong X, Chen H, Chen W, Yang J, Wang X, Gao F & Yi W (2015b). cIAP1 attenuates shear stress‐induced hBMSC apoptosis for tissue‐engineered blood vessels through the inhibition of the mitochondrial apoptosis pathway. Life Sci 137, 81–88. [DOI] [PubMed] [Google Scholar]
- Zong C, Gomes AV, Drews O, Li X, Young GW, Berhane B, Qiao X, French SW, Bardag‐Gorce F & Ping P (2006). Regulation of murine cardiac 20S proteasomes: role of associating partners. Circ Res 99, 372–380. [DOI] [PubMed] [Google Scholar]