

Abstract
The heart has the ability to adjust to changing mechanical loads. The Frank–Starling law and the Anrep effect describe exquisite intrinsic mechanisms the heart has for autoregulating the force of contraction to maintain cardiac output under changes of preload and afterload. Although these mechanisms have been known for more than a century, their cellular and molecular underpinnings are still debated. How does the cardiac myocyte sense changes in preload or afterload? How does the myocyte adjust its response to compensate for such changes? In cardiac myocytes Ca2+ is a crucial regulator of contractile force and in this review we compare and contrast recent studies from different labs that address these two important questions. The ‘dimensionality’ of the mechanical milieu under which experiments are carried out provide important clues to the location of the mechanosensors and the kinds of mechanical forces they can sense and respond to. As a first approximation, sensors inside the myocyte appear to modulate reactive oxygen species while sensors on the cell surface appear to also modulate nitric oxide signalling; both signalling pathways affect Ca2+ handling. Undoubtedly, further studies will add layers to this simplified picture. Clarifying the intimate links from cellular mechanics to reactive oxygen species and nitric oxide signalling and to Ca2+ handling will deepen our understanding of the Frank–Starling law and the Anrep effect, and also provide a unified view on how arrhythmias may arise in seemingly disparate diseases that have in common altered myocyte mechanics.
Keywords: calcium signalling, cardiac arrhythmia, cardiac myocytes, heart disease, mechanotransduction, muscle mechanics, nitric oxide synthase, reactive oxygen species
Abbreviations
- CaMKII
Ca2+–calmodulin‐dependent protein kinase II
- NOS
nitric oxide synthase
- eNOS
endothelial NOS
- MCT
mechano‐chemo‐transduction
- nNOS
neuronal NOS
- NO
nitric oxide
- NOS
nitric oxide synthase
- NOX2
nicotinamide adenine dinucleotide phosphate oxidase 2
- ROS
reactive oxygen species
- X‐ROS
NOX2‐derived ROS
Knowledge gap in mechano‐chemo‐transduction
In every heartbeat, cardiac myocytes generate mechanical force to pump blood into circulation to meet the body's demands for oxygen and fuel. The heart must continuously adjust the force of contraction to compensate for changes in mechanical load resulting from changes in posture, physical activity and emotional state. The Frank–Starling law and the Anrep effect describe the intrinsic auto‐regulatory mechanisms that enable the heart to respond and adapt to changes in preload and afterload to maintain adequate cardiac output. Changes in preload or afterload must, by Laplace's law, change the wall stress borne by the cardiac myocytes. The Frank–Starling law and the Anrep effect were discovered more than a century ago yet their cellular and molecular bases remain unclear despite intensive study (de Tombe et al. 2010; Bollensdorff et al. 2011; Cingolani et al. 2013). A critical knowledge gap is how the myocyte senses mechanical load and transduces the load information to biochemical reactions to affect cell function, a process called mechano‐chemo‐transduction (MCT). Deciphering the key molecules and the signalling pathways in MCT is necessary for understanding how the heart auto‐regulates contractility under physiological loading, but also for understanding how arrhythmias and perhaps cardiomyopathies develop under excessive pathological loading (Knoll et al. 2002).
During contraction, myocytes in situ experience longitudinal shortening, transverse thickening, and shearing strains (Waldman et al. 1985). The in situ mechanical environment of cardiac myocytes is complex because regional differences exist in anatomical geometry and cell orientations, and there is considerable mechanical interaction between cells (Waldman et al. 1985; Young et al. 2001). The mechanical environment of isolated single cardiac myocytes is simpler and can be experimentally controlled to allow detailed study of the molecular mechanisms underlying MCT. Here we review the recent work in developing new experimental tools to capture the acute MCT process in action during beat‐to‐beat myocyte contraction. Because of space constraints, it is impossible to reference all relevant original articles in the field. We will focus on addressing how the ‘dimensionality’ of the three‐dimensional mechanical milieu of the adult cardiac myocyte reveals different modes of mechanosensing and mechanotransduction.
Classes of mechanosensors and their cellular locations
Two different classes of mechanosensors for cardiac myocytes are readily conceivable. One class, based on changing the area and thickness of the lipid bilayer that make up the sarcolemma when the myocyte is stretched, involves changes in surface charge density and specific capacitance that would change the electric field (from Gauss's law) and the transmembrane voltage (Shapiro et al. 2012). The other class comprises proteins (ion channels, enzymes, etc.) that change activity (conductance, enzymatic activity, etc.) resulting from conformational changes induced by mechanical forces imposed on the myocyte. In this paper we focus on this latter class of mechanosensors. We find it conceptually useful to think of the mechanosensors as molecular ‘springs’ that are either wholly inside the myocyte (S1 and S2 in Fig. 1) or having a part on the surface (S3 and S4). We refer to S1 and S2 as internal mechanosensors and S3 and S4 as surface mechanosensors. For clarity S1 and S2 are drawn as separate sensors to detect orthogonal stresses, but a single sensor arranged at some intermediate angle can respond to both directions of stress. As the myocyte is stretched or contracts by a fraction, εm, the sensor (we assume) experiences its own strain, εs. Strain is defined by change in length divided by original length, ε = ΔL/L 0 (reference cell and mechanosensors shown are in the left panel of Fig. 1). The negative myocyte strain, εm, during contraction is often called the fractional shortening. Fractional shortening at the whole cell or sarcomere level is routinely measured in the lab but sensor strain, εs, at the molecular level is admittedly a hypothetical – but physically plausible – construct.
Figure 1. Location of mechanosensors and how they change during contraction.
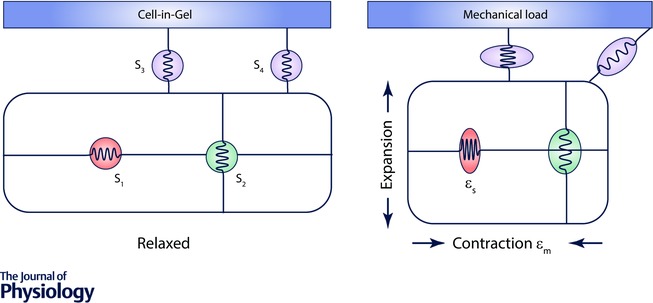
Internal mechanosensors S1 and S2 respond to orthogonal strains. Surface sensor S3 lies on the neutral plane. εm is the myocyte strain (fractional shortening) and εs is the sensor strain. Left panel depicts a myocyte embedded in‐gel at resting state. Right panel depicts a myocyte in‐gel at contracted state when the internal and surface mechanosensors are under mechanical loading.
Cell strain and sensor strain
To approximate the three‐dimensional (3‐D) mechanical environment, we developed the cell‐in‐gel system that embeds single myocytes in a 3‐D viscoelastic hydrogel (Jian et al. 2014). The contracting myocyte does work on the gel because the boronate‐crosslinked polyvinyl alcohol gel matrix binds to cell surface glycans (Luo et al. 2009). Mechanical analysis shows that the cell experiences longitudinal tension (along the cell's long axis), transverse compression (along the short axis) and surface traction forces (vector sum of the shear and normal forces) in this system (Shaw et al. 2013). A contracting myocyte must pull against the gel in the longitudinal direction and, because of the isovolumic constraint, must push against the gel in the transverse direction. In this gel environment, contraction or stretching of the myocyte (by εm) is expected to cause strains in both internal and surface mechanosensors (S1–S4 in Fig. 1). S3 and S4 bind to the gel and so experience strain when the cell contracts or stretches. However, S3 and S4 can respond differently because of their different longitudinal positions on the cell. During contraction both S3 and S4 are compressed but only S4 experiences shearing (the end attached to the myocyte moves to the left). S3 on the cell's neutral plane (located at a point along the long axis, near the centre, where no displacement occurs during contraction or stretch) experiences no shear. We do not think that surface mechanosensors primarily sense shear for two reasons. First, a straightforward geometrical argument shows that the sensor strain due to shear only (no transverse compression or elongation) is proportional to the square of the myocyte strain εs ∝ εm 2. Thus for typical fractional shortening of ∼10%, the sensor strain would be only ∼1%. This low sensitivity would make sensors depending on shear unlikely to be effective load sensors. Second, because surface displacement (not strain) varies linearly from zero at the neutral plane to maximal at the ends of the cell, there would be a gradient of sensor strain. Assuming that sensor strain translates to some effect on Ca2+ (see below) one would then expect a Ca2+ gradient along the longitudinal axis. We observed no such gradient (Jian et al. 2014), so we think that – at least in the cell‐in‐gel system – either the surface mechanosensors are not responding primarily to shear or the stress from sensors is somehow integrated and evenly distributed throughout the cell perhaps because of the tensegrity structure of myocyte (White, 2011) .
On the other hand, when the myocyte contracts or is stretched the cell surface moves transversely uniformly so the surface mechanosensors are compressed or stretched to the same extent εs everywhere along the longitudinal axis. Furthermore, in this case, sensor strain is proportional to myocyte strain, εs ∝ εm. The high sensitivity and longitudinal uniformity of response make us favour the interpretation that surface mechanosensors are compressed or stretched in the gel or in situ.
Our working hypothesis is that the magnitude of the sensor strain is positively related to the magnitude of the intracellular signal: bigger sensor strain, bigger signal. This hypothesis is saved from being a mere platitude because it dissociates sensor strain from cell strain. Because of this dissociation, equal cell strains need not produce the same sensor strains in different experimental systems. This difference can be exploited to distinguish surface and internal mechanosensors.
Interrogating different mechano‐chemo‐transduction pathways
The relaxed myocyte is depicted in the top left panel (Load‐free) of Fig. 2. In this reference state neither the cell nor the mechanosensors are strained. The perfect circles represent this strain‐free state of the mechanosensors. Different experimental systems will cause different strains on the mechanosensors. Physiological saline solution offers little mechanical resistance (load‐free) to the contracting myocyte (Fig. 2, Load‐free, right panel) so the surface mechanosensors experience little or no strain while the internal mechanosensors are strained (note the change from circles to ellipses).
Figure 2. How different experimental systems distinguish between surface and internal mechanosensors.
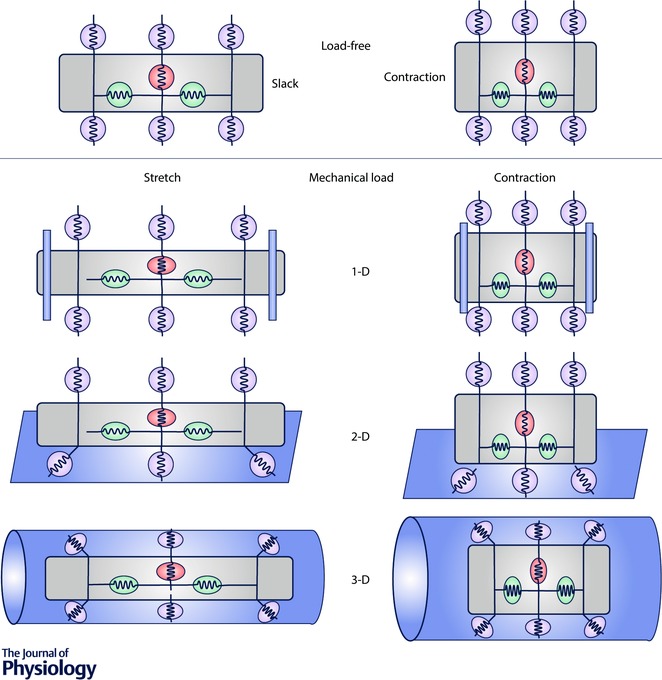
Surface (yellow) and internal (red and green) mechanosensors are depicted as springs. As the myocyte is stretched (left column) or contracts (right column) surface mechanosensors experience different strains (change in length divided by original length) depending on experimental system. The ellipticity of the mechanosensor represents the degree of strain. A perfect circle indicates zero strain. In the 1‐D system, the blue cylinders at the cell edges are the glass rods or carbon fibres. In the 2‐D system the blue sheet represents the stretchable membrane. In the 3‐D system, the blue cylinder represents the hydrogel.
3‐D cell‐in‐gel system
The polyvinyl alcohol matrix in the 3‐D cell‐in‐gel system can be tuned to have Young's modulus between 1 and 20 kPa and viscosity of about 1 Pa s (at a strain rate of 1 s–1). These values are comparable to myocardial stiffness and viscosities measured in the human heart (Pislaru et al. 2014). Figure 2 (3‐D) depicts the myocyte in the 3‐D gel (blue cylinder). Because of the gel's viscoelasticity the cell does work against the gel during contraction (Shaw et al. 2013) and presumably part of the work goes into compressing and shearing the surface mechanosensors (right panel). Note the variation in shearing along the long axis of the cell. The mechanosensor at the centre of the cell (the neutral plane) undergoes no shear while those at the edge undergo the most shear. The internal mechanosensors experience the same kinds of strain (but perhaps of different magnitude) as in load‐free conditions. Experimental evidence from our lab and others suggests that compression of the surface mechanosensors activates nitric oxide synthase (NOS).
Myocytes contracting in‐gel showed an increased magnitude of Ca2+ transient in systole, compared with load‐free cell contraction in bathing solution (Jian et al. 2014). This increase of the Ca2+ transient enhances contractility to compensate for the greater mechanical load, possibly contributing to the Anrep effect. However, the MCT also caused spontaneous Ca2+ sparks during diastole that sometimes arise synchronously to form Ca2+ waves (Awasthi et al. 2015), which could trigger afterdepolarizations and premature action potentials that provide substrate for cardiac arrhythmias (Kass & Tsien, 1982; Berlin et al. 1989; Katra & Laurita, 2005). MCT in the cell‐in‐gel system involves several important signalling pathways. Both of the constitutive NOS isoforms – neuronal NOS (nNOS or NOS1) and endothelial NOS (eNOS or NOS3) – participate in elevating the systolic Ca2+ transient; however, only nNOS plays a critical role in the induction of diastolic Ca2+ sparks and waves. We also found that nicotinamide adenine dinucleotide phosphate oxidase 2 (NOX2), whose importance in mechanosensing was identified by Prosser et al. (2011), is also involved in generating Ca2+ sparks and waves. We cannot rule out the possibility that surface mechanosensors may also activate reactive oxygen species (ROS) production, since the surface sensors have not been studied in isolation. Furthermore, downstream from NOX2 and NOS signalling, Ca2+–calmodulin‐dependent protein kinase II (CaMKII) was found to mediate the MCT effect on Ca2+ handling (Jian et al. 2014).
Comparison with 1‐D systems
An important experimental method for studying MCT uses a pair of carbon fibres or glass rods attached to the myocyte to stretch the cell along the long axis (Le Guennec et al. 1990; Alvarez et al. 1999; Calaghan & White, 2004; Iribe et al. 2009; Prosser et al. 2011). We refer to this system, shown in Fig. 2 as a 1‐D stretching system because mechanical loading on the cell is directed primarily along one dimension. A critical difference between the 3‐D cell‐in‐gel system and the 1‐D system is the mechanical environment of the myocyte. In the cell‐in‐gel system the myocyte pulls or pushes against the gel during stretch or contraction so the surface mechanosensors may be stretched or compressed. In contrast, in a 1‐D system the myocyte is bathed in physiological saline solution, which is not elastic and has very low viscosity compared to the gel (1 Pa s vs. ∼10−3 Pa s, the viscosity of water). Therefore, the surface mechanosensors are unlikely to experience significant strain (small εs) even though myocyte strain (εm, fractional shortening) might be large. This dissociation between myocyte strain and surface mechanosensor strain is illustrated in Fig. 2 by the surface mechanosensors remaining circular despite stretching or contraction in the 1‐D system.
Using the 1‐D system, Iribe et al. (2009) found that stretching the myocyte at resting state induced spontaneous Ca2+ sparks. Prosser et al. further discovered that the MCT is mediated by NOX2‐derived ROS (X‐ROS) signalling (Prosser et al. 2011), and cyclic stretching of the resting myocyte elevates the ROS level (Prosser et al. 2013). Importantly, the non‐specific NOS inhibitor N ω‐nitro‐l‐arginine methyl ester (l‐NAME) had no effect on the stretch‐induced Ca2+ sparks (Iribe et al. 2009). By contrast, l‐NAME abolished the Ca2+ sparks in the cell‐in‐gel system (Jian et al. 2014). On the other hand, we also found that X‐ROS signalling is present in myocytes contracting in gel. The differences and similarities between the 1‐D stretching system versus the 3‐D cell‐in‐gel system point to distinct signalling pathways activated by internal and surface mechanosensors. Internal mechanosensors are presumably activated during stretching or contraction in both 1‐D and 3‐D systems but the surface mechanosensors are likely to be activated only in the 3‐D system. Therefore, these data suggest that X‐ROS signalling is activated by internal mechanosensors while NO signalling is activated by surface mechanosensors.
The idea that surface mechanosensors activate NOS is further supported by the work of Petroff et al. (2001), who embedded myocytes in agarose and then encapsulated the cell and agarose in a deformable tube. In this experimental setting, because agarose is inelastically deformable and does not bind to biomolecules to any significant extent (Freifelder, 1982), stretching the tube squeezes the encapsulated cells to exert compressive stress on the cell and presumably the surface mechanosensors as illustrated in Fig. 2 (3‐D system, left). Petroff et al. (2001) found that stretching the agarose tube caused spontaneous Ca2+ sparks in resting myocytes, and also increased Ca2+ transient in electrically stimulated myocytes, and this mechanotransduction is mediated by eNOS‐dependent NO signalling.
Comparison with 2‐D systems
Investigation using a 2D‐stretching system provides further evidence to support the idea that surface mechanosensors work via NOS. Garbincius & Michele (2015) cultured adult mouse ventricular myocytes (treated with blebbistatin to impair contraction) on laminin‐coated membrane, and then stretched the membrane to apply shear stress on cells (Fig. 2, 2‐D). After cyclically stretching the membrane at 1 Hz for 1 h, they measured the intracellular NO level, and found that shear stress increased nNOS‐derived NO. An important difference between the 2‐D and 3‐D systems is that in the former, the surface mechanosensors not facing the membrane are exposed to bath solution and experience very little strain. Thus in the 2‐D system, mechanotransduction signals come from internal mechanosensors and only a fraction of the surface mechanosensors, while in the 3‐D system all surface mechanosensors contribute to signalling.
As described above, different experimental systems can be used to interrogate different mechanosensors and MCT pathways. The 1‐D systems can be used to interrogate the internal mechanosensors separate from the surface mechanosensors; it is designed for studying adult ventricular myocytes (rod‐shaped cells). The 2‐D systems can be used to interrogate the surface mechanosensors, but it is difficult to attach adult cardiac myocytes on a 2‐D substrate. The 2‐D systems are often used for cultured cells such as neonatal ventricular myocytes that can attach to the deformable membrane. The advantages of the 3‐D cell‐in‐gel system are that the gel is easy to use, can embed freshly isolated adult cardiac myocytes and any cell type, and resembles more closely the 3‐D in situ mechanical environment. Because many heart diseases occur in adult hearts and especially in ageing hearts, it is important to control mechanical load on adult cardiac myocytes in studying mechanotransduction and heart disease mechanisms.
Mapping MCT pathways: internal mechanosensors and surface mechanosensors
What are the mechanosensors that sense different types of mechanical stresses? The mechanical structure of striated myocytes is composed of a sarcomeric cytoskeleton network with a remarkable axial and transverse order of filaments that form crystalline‐like lattices (Gautel & Djinovic‐Carugo, 2016). This intracellular mechanical network is also linked through the transmembrane costamere structure to the extracellular matrix (Ervasti, 2003). During the cardiac cycle, contraction and stretching of myocytes exert mechanical stresses on all these mechanical scaffolds. Based on the location of molecules in the myocyte's architecture, it is plausible that surface traction including normal and shear stresses can be sensed by surface mechanosensors, and tension and compression can act on internal mechanosensors and load‐bearing molecules in the sarcomeric cytoskeleton network.
For example, the giant protein titin spans half of the sarcomere to connect z‐disk, I‐band, A‐band and M‐line structures (Labeit et al. 1990; Bang et al. 2001). When stretched during diastolic filling, the extensible I‐band segment of titin lengthens and develops passive tension (Granzier & Irving, 1995). When relaxed after contraction, the Ig domains of titin recoil and provide restoring forces (Helmes et al. 1996; Preetha et al. 2005). Thus titin can sense both tension and compression during myocyte lengthening and contraction in the cardiac cycle. The passive force of titin is dependent on its compliance, which is regulated by phosphorylation of the N2B spring element by PKA (Yamasaki et al. 2002) and CaMKIIδ (Hidalgo et al. 2013; Perkin et al. 2015). Because the CaMKIIδ activity is upregulated by MCT in the cell‐in‐gel system (Jian et al. 2014), mechanical loading is expected to reduce myocyte stiffening due to passive force generation by titin. Furthermore, titin mutations are the most common cause of dilated cardiomyopathy (Gigli et al. 2016), highlighting the essential role of titin in tension‐bearing and mechanotransduction.
Another important load‐bearing molecule in the cytoskeleton network is the microtubule, which provides a compression‐resisting structure in the myocyte. It was shown that microtubules affect the longitudinal shear stiffness, and this is consistent with microtubules serving as compressive resistance struts within a tensegrity model (White, 2011). Khairallah et al. (2012) showed that X‐ROS amplified Ca2+ influx through stretch‐activated channels in skeletal muscle of mdx mice (a model Duchenne muscular dystrophy). They also showed that MCT by X‐ROS signalling depends on microtubules. Disrupting the dense microtubular network in adult mdx mice resulted in little ROS production and Ca2+ influx. A recent seminal study by Robison et al. (2016) showed that detyrosinated microtubules interact with sarcomere to form a load‐bearing structure parallel to the myofilament. The microtubules buckle during myocyte contraction to resist compression; increased detyrosination of microtubules enhances their binding to the sarcomere and stiffen the myocyte in some forms of heart disease. Disrupting microtubules was found to impair the MCT through the NOX2–ROS pathway in the 1‐D system (Prosser et al. 2011), again demonstrating a significant role of microtubules in MCT.
While stretch and compression can be sensed by internal mechanosensors, surface traction and compression can be sensed by cell‐surface mechanosensors. As an illustrative example, dystroglycans outwardly link to the extracellular matrix and inwardly link to dystrophin to form the dystrophin glycoprotein complex (DGC). The DGC, together with the integrin–talin–vinculin complex, form the costamere structure that links myofibrils and Z‐disks to the sarcolemma and forms transmembrane linkages to the extracellular matrix (Anastasi et al. 2009). Costameres serve as radiating beams in the mechanical scaffold to provide structural support during beat‐to‐beat contraction (Danowski et al. 1992). In muscular dystrophy, mutations in the DGC lead to skeletal and cardiac muscle dysfunction, arrhythmias and sudden death (Judge et al. 2011; Groh, 2012; Fayssoil et al. 2013).
The link DGC to NOS has been well established in skeletal and cardiac muscle. In cardiomyocytes, nNOS is found to bind to α1‐syntrophin in the DGC (Williams et al. 2006) and to dystrophin (Lai et al. 2013). Studies using a dystrophin‐null mouse model (mdx) found that the mdx hearts have decreased nNOS, unchanged eNOS, and increased iNOS expression (Bia et al. 1999), and are extremely vulnerable to pressure overload (Kamogawa et al. 2001). Expression of the nNOS transgene could prevent the progressive ventricular fibrosis and greatly reduced myocarditis of mdx mice (Wehling‐Henricks et al. 2005). Mechanistic study in the 2‐D stretching system (using a membrane coated with laminin that binds to dystroglycans and exerts shear stress during stretching) shows that cyclic stretch increased nNOS‐derived NO in ventricular myocytes; in contrast, the stretch‐activated NO synthesis was impaired in the mdx model (Garbincius & Michele, 2015). Most recently, Casadei and colleagues found the dystrophin–nNOS link in atrial myocytes. In their study, microRNA‐31 repression of dystrophin translation caused nNOS translocation and depletion in human and goat atrial fibrillation (Reilly et al. 2016).
In the cell‐in‐gel system, because the cell‐surface glycans are tethered to the gel by boronate–polyethylene glycol crosslinking (Luo et al. 2009), surface traction imposes mechanical stress on glycosylated molecules including dystroglycans. Indeed, downstream in the dystrophin glycoprotein complex, dystrophin‐associated nNOS was found critical for mediating MCT induction of Ca2+ sparks in ventricular myocytes (Jian et al. 2014).
So far our focus has been on the MCT pathways involving NOS and NOX2 signalling to the Ca2+ handling system, but any conformational change in a protein (channel, enzyme, transporter, etc.) could potentially alter its activity. Thus, in principle, many proteins could sense mechanical force. For example, some ion channels such as stretch‐activated channels (Guharay & Sachs, 1984; Calaghan & White, 2004), the sodium channel Nav1.5 (Morris & Juranka, 2007), the Na+/H+ exchanger (Calaghan & White, 2004; Alvarez et al. 1999), the canonical transient receptor potential (TRPC6) channel (Seo et al. 2014) and the TRP vanilloid type 2 (TRPV2) channel (Lorin et al. 2015) are expressed in heart and are found to be mechanosensitive. Their localizations in different subcellular domains may position them to sense different mechanical stresses. Nav1.5 is on the intercalated disk and surface membrane but absent from transverse tubules (t‐tubules) (Westenbroek et al. 2013); TRPC6 (Dyachenko et al. 2009), TRPV2 (Lorin et al. 2015) and the Na+/H+ exchanger (Yang et al. 2002) are in the t‐tubules but not on the surface sarcolemma. However, it is still far from clear how the menagerie of mechanosensitive molecules is integrated so that the heart can respond to changes in mechanical loads. Further studies can be facilitated by development of innovative techniques to control mechanical stresses on the myocytes.
A mechano‐centric view of arrhythmias
Mechanical stresses are associated with various heart diseases. For example, hypertension, muscular dystrophy and dilated cardiomyopathy are seemingly disparate diseases but they share two common features. First, the risk of arrhythmias is high in these diseases (hypertension: McLenachan et al. 1987; Hennersdorf & Strauer, 2001; Saadeh & Jones, 2001; dilated cardiomyopathy: Meinertz et al. 1984; Neri et al. 1986; muscular dystrophies: McNally & MacLeod, 2005; Groh, 2012). Second, all these diseases involve altered myocyte mechanics. Hypertension exerts chronic pressure overload on ventricular wall. Muscular dystrophies involve mutations in the MCT machinery. Dilated cardiomyopathy involves high ventricular wall tension because of thinning of the wall and increased chamber radius. The high wall tension demands that myocytes contract more strongly to maintain a given stroke volume.
We described MCT mechanisms by which myocytes could sense mechanical load and alter Ca2+ dynamics to compensate for loading. The MCT pathways provide autoregulation of Ca2+ signalling and contractility in normal hearts, but may cause Ca2+ dysregulation in diseased hearts. As discussed, excessive mechanical loading can cause a high rate of diastolic Ca2+ sparks, which could trigger spontaneous Ca2+ waves (Cheng et al. 1993, 1996; Izu et al. 2001, 2006; Thul et al. 2012) leading to arrhythmogenic activities (Kass & Tsien, 1982; Berlin et al. 1989; Katra & Laurita, 2005). Interestingly, hypertensive patients also experience greater variability in mean arterial pressure than normotensives (Mancia et al. 2003). The many incidences of elevated blood pressure fluctuations on an already increased tension provide many chances for high frequency spontaneous Ca2+ sparks and waves to occur that could lead to arrhythmias. Like the lottery, the more times you play the more chances to lose. In the same vein, muscular dystrophy mutations could disrupt MCT pathways to cause Ca2+ dysregulation and arrhythmogenic activities. Various mutations also present opportunities to understand what happens when parts of the MCT pathways become compromised. Dilated cardiomyopathy inevitably involves high wall tension caused by dilation of the ventricular chamber and thinning of the wall, which is also expected to cause high frequency of Ca2+ sparks and waves that provide substrate to cardiac arrhythmias.
A mechano‐centric view of arrhythmias, although perhaps parochial, highlights the significant impact of mechanical stresses on setting up conditions for arrhythmias by MCT effects on Ca2+ handling. This viewpoint suggests that novel antiarrhythmic drugs could target molecules in the MCT pathways such as nNOS or X‐ROS to correct the Ca2+ dysregulation engendered by excessive mechanical loading. Understanding the MCT pathways and the key molecular determinants may hold the key to developing drug therapies to reduce arrhythmias and cardiomyopathy in patients with hypertension, muscular dystrophy and dilated cardiomyopathy.
Additional information
Author contribution
Y.C.‐I.: Conception and design; financial support; administrative support; provision of study materials or patients; collection and assembly of data; data analysis and interpretation; manuscript writing. L.I.: conception and design; financial support; administrative support; provision of study materials or patients; collection and assembly of data; data analysis and interpretation; manuscript writing. Both authors have approved the final version of the manuscript and agree to be accountable for all aspects of the work. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
This work was supported by grants from the National Institutes of Health (NIH) R01HL90880 to L.T.I. and Y.C.‐I., American Heart Association 14GRNT20510041 to Y.C.‐I., and NIH R01HL123526 to Y.C.‐I. and L.T.I.
Biography
Ye Chen‐Izu and Leighton T. Izu collaborate in an interdisciplinary team to combine experiments with mathematical modeling for in‐depth analyses of the interaction of three dynamics systems – electrical, Ca2+ signaling, and mechanical systems – that govern cardiac function and heart diseases.

Contributor Information
Ye Chen‐Izu, Email: ychenizu@ucdavis.edu.
Leighton T. Izu, Email: ltizu@ucdavis.edu
References
- Alvarez BV, Perez NG, Ennis IL, Camilion de Hurtado MC & Cingolani HE (1999). Mechanisms underlying the increase in force and Ca2+ transient that follow stretch of cardiac muscle: a possible explanation of the Anrep effect. Circ Res 85, 716–722. [DOI] [PubMed] [Google Scholar]
- Anastasi G, Cutroneo G, Gaeta R, Di Mauro D, Arco A, Consolo A, Santoro G, Trimarchi F & Favaloro A (2009). Dystrophin‐glycoprotein complex and vinculin‐talin‐integrin system in human adult cardiac muscle. Int J Mol Med 23, 149–159. [PubMed] [Google Scholar]
- Awasthi S, Izu L, Mao Z, Jian Z, Landas T, Lerner A, Shimkunas R, Woldeyesus RA, Bossuyt J, Wood BM, Chen Y‐J, Matthews DL, Lieu DK, Chiamvimonvat N, Lam KS, Chen‐Izu Y & Chan JW (2015). Multimodal SHG‐2PF imaging of microdomain Ca2+‐contraction coupling in live cardiac myocytes. Circ Res 118, e19–e28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bang ML, Centner T, Fornoff F, Geach AJ, Gotthardt M, McNabb M, Witt CC, Labeit D, Gregorio CC, Granzier H & Labeit S (2001). The complete gene sequence of titin, expression of an unusual approximately 700‐kDa titin isoform, and its interaction with obscurin identify a novel Z‐line to I‐band linking system. Circ Res 89, 1065–1072. [DOI] [PubMed] [Google Scholar]
- Berlin JR, Cannell MB & Lederer WJ (1989). Cellular origins of the transient inward current in cardiac myocytes. Role of fluctuations and waves of elevated intracellular calcium. Circ Res 65 115–126. [DOI] [PubMed] [Google Scholar]
- Bia BL, Cassidy PJ, Young ME, Rafael JA, Leighton B, Davies KE, Radda GK & Clarke K (1999). Decreased myocardial nNOS, increased iNOS and abnormal ECGs in mouse models of Duchenne muscular dystrophy. J Mol Cell Cardiol 31, 1857–1862. [DOI] [PubMed] [Google Scholar]
- Bollensdorff C, Lookin O & Kohl P (2011). Assessment of contractility in intact ventricular cardiomyocytes using the dimensionless ‘Frank‐Starling Gain’ index. Pflugers Arch 462, 39–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calaghan S & White E (2004). Activation of Na+‐H+ exchange and stretch‐activated channels underlies the slow inotropic response to stretch in myocytes and muscle from the rat heart. J Physiol 559, 205–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng H, Lederer MR, Lederer WJ & Cannell MB (1996). Calcium sparks and [Ca2+]i waves in cardiac myocytes. Am J Physiol Cell Physiol 270, C148–C159. [DOI] [PubMed] [Google Scholar]
- Cheng H, Lederer WJ & Cannell MB (1993). Calcium sparks: elementary events underlying excitation‐contraction coupling in heart muscle. Science 262, 740–744. [DOI] [PubMed] [Google Scholar]
- Cingolani HE, Pérez NG, Cingolani OH & Ennis IL (2013). The Anrep effect: 100 years later. Am J Physiol Heart Circ Physiol 304, H175–H182. [DOI] [PubMed] [Google Scholar]
- Danowski BA, Imanaka‐Yoshida K, Sanger JM & Sanger JW (1992). Costameres are sites of force transmission to the substratum in adult rat cardiomyocytes. J Cell Biol 118, 1411–1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Tombe PP, Mateja RD, Tachampa K, Ait Mou Y, Farman GP & Irving TC (2010). Myofilament length dependent activation. J Mol Cell Cardiol 48, 851–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyachenko V, Husse B, Rueckschloss U & Isenberg G (2009). Mechanical deformation of ventricular myocytes modulates both TRPC6 and Kir2.3 channels. Cell Calcium 45, 38–54. [DOI] [PubMed] [Google Scholar]
- Ervasti JM ( 2003). Costameres: the Achilles' heel of Herculean muscle. J Biol Chem 278, 13591–13594. [DOI] [PubMed] [Google Scholar]
- Fayssoil A, Renault G, Guerchet N, Marchiol‐Fournigault C, Fougerousse F & Richard I (2013). Cardiac characterization of mdx mice using high‐resolution Doppler echocardiography. J Ultrasound Med 32, 757–761. [DOI] [PubMed] [Google Scholar]
- Freifelder D (1982). Physical Biochemistry: Applications to Biochemistry and Molecular Biology, 2nd edn WH Freeman, San Francisco. [Google Scholar]
- Garbincius JF & Michele DE (2015). Dystrophin‐glycoprotein complex regulates muscle nitric oxide production through mechanoregulation of AMPK signaling. Proc Natl Acad Sci USA 112, 13663–13668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautel M & Djinovic‐Carugo K (2016). The sarcomeric cytoskeleton: from molecules to motion. J Exp Biol 219, 135–145. [DOI] [PubMed] [Google Scholar]
- Gigli M, Begay RL, Morea G, Graw SL, Sinagra G, Taylor MR, Granzier H & Mestroni L (2016). A Review of the Giant Protein Titin in Clinical Molecular Diagnostics of Cardiomyopathies. Front Cardiovasc Med 3, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granzier HL & Irving TC (1995). Passive tension in cardiac muscle: contribution of collagen, titin, microtubules, and intermediate filaments. Biophys J 68, 1027–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groh WJ ( 2012). Arrhythmias in the muscular dystrophies. Heart Rhythm 9, 1890–1895. [DOI] [PubMed] [Google Scholar]
- Guharay F & Sachs F (1984). Stretch‐activated single ion channel currents in tissue‐cultured embryonic chick skeletal muscle. J Physiol 352, 685–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helmes M, Trombitas K & Granzier H (1996). Titin develops restoring force in rat cardiac myocytes. Circ Res 79, 619–626. [DOI] [PubMed] [Google Scholar]
- Hennersdorf MG & Strauer BE (2001). Arterial hypertension and cardiac arrhythmias. J Hypertens 19, 167–177. [DOI] [PubMed] [Google Scholar]
- Hidalgo CG, Chung CS, Saripalli C, Methawasin M, Hutchinson KR, Tsaprailis G, Labeit S, Mattiazzi A & Granzier HL (2013). The multifunctional Ca2+/calmodulin‐dependent protein kinase II delta (CaMKIIδ) phosphorylates cardiac titin's spring elements. J Mol Cell Cardiol 54, 90–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iribe G, Ward CW, Camelliti P, Bollensdorff C, Mason F, Burton RAB, Garny A, Morphew MK, Hoenger A, Lederer WJ & Kohl P (2009). Axial stretch of rat single ventricular cardiomyocytes causes an acute and transient increase in Ca2+ spark rate. Circ Res 104, 787–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izu LT, Means SA, Shadid JN, Chen‐Izu Y & Balke CW (2006). Interplay of ryanodine receptor distribution and calcium dynamics. Biophys J 91, 95–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izu LT, Wier WG & Balke CW (2001). Evolution of cardiac calcium waves from stochastic calcium sparks. Biophys J 80, 103–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jian Z, Han H, Zhang T, Puglisi J, Izu LT, Shaw JA, Onofiok E, Erickson JR, Chen Y‐J, Horvath B, Shimkunas R, Xiao W, Li Y, Pan T, Chan J, Banyasz T, Tardiff JC, Chiamvimonvat N, Bers DM, Lam KS & Chen‐Izu Y (2014). Mechanochemotransduction during cardiomyocyte contraction is mediated by localized nitric oxide signaling. Sci Signal 7, ra27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Judge D, Kass D, Thompson WR & Wagner K (2011). Pathophysiology and therapy of cardiac dysfunction in Duchenne muscular dystrophy. Am J Cardiovasc Drugs 11, 287–294. [DOI] [PubMed] [Google Scholar]
- Kamogawa Y, Biro S, Maeda M, Setoguchi M, Hirakawa T, Yoshida H & Tei C (2001). Dystrophin‐deficient myocardium is vulnerable to pressure overload in vivo. Cardiovasc Res 50, 509–515. [DOI] [PubMed] [Google Scholar]
- Kass RS & Tsien RW (1982). Fluctuations in membrane current driven by intracellular calcium in cardiac Purkinje fibers. Biophys J 38, 259–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katra RP & Laurita KR (2005). Cellular mechanism of calcium‐mediated triggered activity in the heart. Circ Res 96, 535–542. [DOI] [PubMed] [Google Scholar]
- Khairallah RJ, Shi G, Sbrana F, Prosser BL, Borroto C, Mazaitis MJ, Hoffman EP, Mahurkar A, Sachs F, Sun Y, Chen Y‐W, Raiteri R, Lederer WJ, Dorsey SG & Ward CW (2012). Microtubules underlie dysfunction in Duchenne muscular dystrophy. Sci Signal 5, ra56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knoll R, Hoshijima M, Hoffman HM, Person V, Lorenzen‐Schmidt I, Bang ML, Hayashi T, Shiga N, Yasukawa H, Schaper W, McKenna W, Yokoyama M, Schork NJ, Omens JH, McCulloch AD, Kimura A, Gregorio CC, Poller W, Schaper J, Schultheiss HP & Chien KR (2002). The cardiac mechanical stretch sensor machinery involves a Z disc complex that is defective in a subset of human dilated cardiomyopathy. Cell 111, 943–955. [DOI] [PubMed] [Google Scholar]
- Labeit S, Barlow DP, Gautel M, Gibson T, Holt J, Hsieh CL, Francke U, Leonard K, Wardale J, Whiting A & Trinick J (1990). A regular pattern of two types of 100‐residue motif in the sequence of titin. Nature 345, 273–276. [DOI] [PubMed] [Google Scholar]
- Lai Y, Zhao J, Yue Y & Duan D (2013). α2 and α3 helices of dystrophin R16 and R17 frame a microdomain in the α1 helix of dystrophin R17 for neuronal NOS binding. Proc Natl Acad Sci USA 110, 525–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Guennec JY, Peineau N, Argibay JA, Mongo KG & Garnier D (1990). A new method of attachment of isolated mammalian ventricular myocytes for tension recording: length dependence of passive and active tension. J Mol Cell Cardiol 22, 1083–1093. [DOI] [PubMed] [Google Scholar]
- Lorin C, Vogeli I & Niggli E (2015). Dystrophic cardiomyopathy: role of TRPV2 channels in stretch‐induced cell damage. Cardiovasc Res 106, 153–162. [DOI] [PubMed] [Google Scholar]
- Luo J, Onofiok E, Shi C, Liu R & Lam KS (2009). A novel hydrogel functionalized with specific peptidomimetic ligands for 2‐D and 3‐D cell culture. Adv Exp Med Biol 611, 19–20. [DOI] [PubMed] [Google Scholar]
- Mancia G, Parati G, Castiglioni P, Tordi R, Tortorici E, Glavina F & Di Rienzo M (2003). Daily life blood pressure changes are steeper in hypertensive than in normotensive subjects. Hypertension 42, 277–282. [DOI] [PubMed] [Google Scholar]
- McLenachan JM, Henderson E, Morris KI & Dargie HJ (1987). Ventricular arrhythmias in patients with hypertensive left ventricular hypertrophy. N Engl J Med 317, 787–792. [DOI] [PubMed] [Google Scholar]
- McNally EM & MacLeod H (2005). Therapy insight: cardiovascular complications associated with muscular dystrophies. Nat Clin Pract Cardiovasc Med 2, 301–308. [DOI] [PubMed] [Google Scholar]
- Meinertz T, Hofmann T, Kasper W, Treese N, Bechtold H, Stienen U, Pop T, Leitner ER, Andresen D & Meyer J (1984). Significance of ventricular arrhythmias in idiopathic dilated cardiomyopathy. Am J Cardiol 53, 902–907. [DOI] [PubMed] [Google Scholar]
- Morris CE & Juranka PF (2007). Nav channel mechanosensitivity: activation and inactivation accelerate reversibly with stretch. Biophys J 93, 822–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neri R, Mestroni L, Salvi A & Camerini F (1986). Arrhythmias in dilated cardiomyopathy. Postgrad Med J 62, 593–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perkin J, Slater R, Del Favero G, Lanzicher T, Hidalgo C, Anderson B, Smith JE 3rd, Sbaizero O, Labeit S & Granzier H (2015). Phosphorylating titin's cardiac N2B element by ERK2 or CaMKIIδ lowers the single molecule and cardiac muscle force. Biophys J 109, 2592–2601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petroff MG, Kim SH, Pepe S, Dessy C, Marban E, Balligand JL & Sollott SJ (2001). Endogenous nitric oxide mechanisms mediate the stretch dependence of Ca2+ release in cardiomyocytes. Nat Cell Biol 3, 867–873. [DOI] [PubMed] [Google Scholar]
- Pislaru C, Urban MW, Pislaru SV, Kinnick RR & Greenleaf JF (2014). Viscoelastic properties of normal and infarcted myocardium measured by a multifrequency shear wave method: comparison with pressure‐segment length method. Ultrasound Med Biol 40, 1785–1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preetha N, Yiming W, Helmes M, Norio F, Siegfried L & Granzier H (2005). Restoring force development by titin/connectin and assessment of Ig domain unfolding. J Muscle Res Cell Motil 26, 307–317. [DOI] [PubMed] [Google Scholar]
- Prosser BL, Ward CW & Lederer WJ (2011). X‐ROS signaling: rapid mechano‐chemo transduction in heart. Science 333, 1440–1445. [DOI] [PubMed] [Google Scholar]
- Prosser BL, Ward CW & Lederer WJ (2013). X‐ROS signalling is enhanced and graded by cyclic cardiomyocyte stretch. Cardiovasc Res 98, 307–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reilly SN, Liu X, Carnicer R, Recalde A, Muszkiewicz A, Jayaram R, Carena MC, Wijesurendra R, Stefanini M, Surdo NC, Lomas O, Ratnatunga C, Sayeed R, Krasopoulos G, Rajakumar T, Bueno‐Orovio A, Verheule S, Fulga TA, Rodriguez B, Schotten U & Casadei B (2016). Up‐regulation of miR‐31 in human atrial fibrillation begets the arrhythmia by depleting dystrophin and neuronal nitric oxide synthase. Sci Transl Med 8, 340ra74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robison P, Caporizzo MA, Ahmadzadeh H, Bogush AI, Chen CY, Margulies KB, Shenoy VB & Prosser BL (2016). Detyrosinated microtubules buckle and bear load in contracting cardiomyocytes. Science 352, aaf0659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saadeh AM & Jones JV (2001). Predictors of sudden cardiac death in never previously treated patients with essential hypertension: long‐term follow‐up. J Hum Hypertens 15, 677–680. [DOI] [PubMed] [Google Scholar]
- Seo K, Rainer PP, Lee DI, Hao S, Bedja D, Birnbaumer L, Cingolani OH & Kass DA (2014). Hyperactive adverse mechanical stress responses in dystrophic heart are coupled to transient receptor potential canonical 6 and blocked by cGMP‐protein kinase G modulation. Circ Res 114, 823–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro MG, Homma K, Villarreal S, Richter CP & Bezanilla F (2012). Infrared light excites cells by changing their electrical capacitance. Nat Commun 3, 736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw J, Izu L & Chen‐Izu Y (2013). Mechanical analysis of single myocyte contraction in a 3‐D elastic matrix. PLoS One 8, e75492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thul R, Coombes S, Roderick HL & Bootman MD (2012). Subcellular calcium dynamics in a whole‐cell model of an atrial myocyte. Proc Natl Acad Sci USA 109, 2150–2155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldman LK, Fung YC & Covell JW (1985). Transmural myocardial deformation in the canine left ventricle. Normal in vivo three‐dimensional finite strains. Circ Res 57, 152–163. [DOI] [PubMed] [Google Scholar]
- Wehling‐Henricks M, Jordan MC, Roos KP, Deng B & Tidball JG (2005). Cardiomyopathy in dystrophin‐deficient hearts is prevented by expression of a neuronal nitric oxide synthase transgene in the myocardium. Hum Mol Genet 14, 1921–1933. [DOI] [PubMed] [Google Scholar]
- Westenbroek RE, Bischoff S, Fu Y, Maier SK, Catterall WA & Scheuer T (2013). Localization of sodium channel subtypes in mouse ventricular myocytes using quantitative immunocytochemistry. J Mol Cell Cardiol 64, 69–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White E ( 2011). Mechanical modulation of cardiac microtubules. Pflugers Arch 462, 177–184. [DOI] [PubMed] [Google Scholar]
- Williams JC, Armesilla AL, Mohamed TMA, Hagarty CL, McIntyre FH, Schomburg S, Zaki AO, Oceandy D, Cartwright EJ, Buch MH, Emerson M & Neyses L (2006). The sarcolemmal calcium pump, α‐1 syntrophin, and neuronal nitric‐oxide synthase are parts of a macromolecular protein complex. J Biol Chem 281, 23341–23348. [DOI] [PubMed] [Google Scholar]
- Yamasaki R, Wu Y, McNabb M, Greaser M, Labeit S & Granzier H (2002). Protein kinase A phosphorylates titin's cardiac‐specific N2B domain and reduces passive tension in rat cardiac myocytes. Circ Res 90, 1181–1188. [DOI] [PubMed] [Google Scholar]
- Yang Z, Pascarel C, Steele DS, Komukai K, Brette F & Orchard CH (2002). Na+‐Ca2+ exchange activity is localized in the T‐tubules of rat ventricular myocytes. Circ Res 91, 315–322. [DOI] [PubMed] [Google Scholar]
- Young AA, Dokos S, Powell KA, Sturm B, McCulloch AD, Starling RC, McCarthy PM & White RD (2001). Regional heterogeneity of function in nonischemic dilated cardiomyopathy. Cardiovasc Res 49, 308–318. [DOI] [PubMed] [Google Scholar]