

Abstract
Hypertension induces considerable cardiac remodelling, such as hypertrophy, interstitial fibrosis, and abnormal activity of the cardiac sympathetic nervous system, which are established risk factors in several highly dangerous heart diseases, such as ventricular fibrillation and congestive heart failure. All these risk factors and heart diseases are studied extensively in isolation, but to our knowledge, there is no comprehensive review of their interactions. At the same time, there is growing evidence suggesting that such interactions are numerous and that a successful therapy against a particular condition may have unexpectedly weak effects on mortality, as treated patients may die of a different cause exacerbated by the therapy. In this article, we present a multiscale review of the literature focusing on the relationships between the above‐mentioned risk factors and heart diseases, and introduce a framework that gives insight into their possible interactions. We use this framework to demonstrate that conditions such as fibrosis and elevated activity of the sympathetic nervous system may be compensatory, rather than purely pathological, mechanisms in certain contexts. Finally, we show why the described mechanisms are relevant not only in hypertension, but also in the case of healed myocardial infarction.
Keywords: arrhythmia, extracellular matrix, heart failure, hypertension
Abbreviations
- Ang‐II
angiotensin‐II
- CSNS
cardiac sympathetic nervous stimulation
- ECM
extracellular matrix
- MMP
matrix metalloproteinase
- PEA
pulseless electrical activity
- SCD
sudden cardiac death
- TGF‐β
transforming growth factor β
- VF
ventricular fibrillation
Introduction
Hypertension is a major clinical condition that promotes extensive cardiac remodelling, acting as a contributing factor in both systolic and diastolic dysfunction, arrhythmias and symptomatic heart failure, which are among the main sources of mortality worldwide (Drazner, 2011). Through an analysis of the literature, we highlight the connections between hypertension‐induced remodelling and heart diseases, with a focus on the links between arrhythmogenesis, systolic and diastolic heart failure. Systolic heart failure, also known as heart failure with reduced ejection fraction, is characterized by poor contractile function and is usually associated with myocardial dilatation. Diastolic heart failure, also known as heart failure with preserved ejection fraction, is characterized by poor relaxation of ventricles during diastole (e.g. due to high myocardial stiffness) resulting in poor refilling. We discuss how both types of heart failure may be promoted by hypertension, but via different mechanisms.
We first focus on how hypertension induces structural cardiac remodelling and how this remodelling may be cardioprotective in certain contexts. In the second section, we review studies that suggest that hypertrophy is not a sufficient reaction against hypertension and that interstitial fibrosis and collagen crosslinking may play a key role in protecting the heart from systolic heart failure, even though they promote diastolic dysfunction and thus increase the likelihood of diastolic heart failure. In the third section, we review how cardiac structural remodelling and subsequent myocardial stiffness are linked to cardiac sympathetic nervous stimulation (CSNS) and how this is linked to ventricular fibrillation, pulseless electrical activity, and diastolic heart failure. In the fourth section, we show how the observations emerging from this review are relevant not only to hypertension, but also to understanding myocardial infarction. Ultimately, in the fifth section, we summarize the observations made and discuss their implications.
From hypertension to fibrosis
Left ventricular hypertrophy, defined as an abnormal increase in left ventricular mass, is traditionally considered to be an adaptation to increased cardiac workload in hypertension via minimization of wall stress and normalization of ejection performance (Drazner, 2011). However, it is also associated with adverse effects, which is why it is often labelled as maladaptative (Frey & Olson, 2003).
Less attention seems to be paid to diffuse myocardial fibrosis, characterized by an increased deposition of extracellular matrix (ECM), possibly due to the difficulty of isolating fibrosis from hypertrophy, as they tend to occur together in hypertension. While the compensatory role of hypertrophy is acknowledged in the literature (Drazner, 2011), diffuse fibrosis is almost invariably considered to be pathological, mainly due to increased likelihood of fibrillation and increased ventricular stiffness leading to systolic or diastolic dysfunction (Creemers & Pinto, 2011). Below, we summarize studies pointing towards a cardioprotective role of diffuse fibrosis in protecting the heart from dilatation in a pressure overload setting, possibly because the stiff collagen scaffolding associated with fibrosis may mechanically prevent elongation of myocytes during myocardial dilatation. We also discuss how myocardial stiffening is related to systolic and diastolic heart failure.
Systems‐level evidence
In an experimental study that used aortic banding to induce pressure overload in rats, Woodiwiss et al. (2001) divided treated animals into two groups, those with non‐failing hearts and those with hearts failing due to dilatation and systolic dysfunction. The authors showed a marked decrease in collagen crosslinking in the group with failing hearts, while total collagen content and the degree of hypertrophy were similar between the two groups. The first important observation in this study is that hypertrophy itself does not seem to be sufficient protection against persistent hypertension. A second observation is that the component of fibrotic remodelling that might offer protection against dilatation is collagen crosslinking, which has been shown to be the main factor in determining myocardial stiffness (Badenhorst et al. 2003), rather than total collagen content. Therefore, if there is to be a protective effect of fibrosis, the collagen produced needs to be appropriately crosslinked to prevent dilatation.
The work by Janicki and Spinale is another demonstration of the importance of fibrosis, showing that in the Syrian hamster, matrix metalloproteinase (MMP) activity and subsequent decrease of collagen volume fraction paralleled significant ventricular dilatation and heart failure, presumably due to systolic dysfunction (Janicki et al. 2006). MMP activity and its effect (breaking collagen and collagen crosslinks) has also been demonstrated in human studies of heart failure (Gunja‐Smith et al. 1996) and these studies therefore provide support for the view that stiff collagen scaffolding is a barrier to dilatation.
Kim et al. (2000) produced a genetically modified mouse model that manifests a chronic overexpression of MMP‐1. The modified animals, without an artificial increase in blood pressure, demonstrated hypertrophy and increased collagen content with subsequent reduction of collagen content, dilatation, and loss of systolic and diastolic function.
Using a different genetic modification, Takeda et al. (2010) investigated the effect of fibroblast‐specific deletion of the Klf5 gene in murine hearts subject to previous transverse aortic constriction, demonstrating that fibroblasts (and Klf5) play a key role in response to pressure overload. While fibroblast‐specific deletion of Klf5 ameliorated fibrosis and hypertrophy following pressure overload, it also led to left ventricular dilatation and early death.
An alternative style of inducing ventricular dilatation and subsequent heart failure is rapid pacing, as shown by McElmurray et al. (1999) in pigs. In their study, the authors show that inhibition of MMP increases myocardial stiffness, ameliorates the degree of ventricular dilatation and leads to a decrease in fractional shortening. This study is important as it shows that a possible benefit of increased cardiac stiffness may not be bound to dilatation necessarily due to hypertension, but also due to other mechanisms.
Another study of MMP inhibitors is the one by Peterson et al. (2001), in a rat model of heart failure, where the inhibition of MMP prevented development of heart failure due to dilatation. This study suggests the importance of the time scale of collagen remodelling during heart failure, since the collagen content increases in failing hearts. This observation raises the question: if fibrosis offers protection against dilatation, why is collagen abundant in dilated cardiomyopathy? The answer might be, as mentioned above, that the collagen crosslinking is the key factor, provided that there is enough collagen to be crosslinked. Indeed Gunja‐Smith et al. (1996) showed that crosslinking is decreased in dilated cardiomyopathy, most likely due to the activity of MMPs. Therefore, we might hypothesize that the overproduction of collagen in failing hearts is a dysfunctional remnant of an originally cardioprotective mechanism (increasing collagen content and collagen crosslinking to increase ventricular stiffness), where, for a reason not yet understood, the formation of crosslinks is now diminished, even while collagen continues to be produced.
Interpreting the above‐summarized studies together, we can see that the two variants of heart failure (with reduced or preserved systolic function) may be viewed as two sides of the same coin: the ventricular stiffness. That is, if the heart does not stiffen in the presence of pressure overload, it is likely to dilate due to increased stretch; however, if it does stiffen it will nevertheless diminish the capability of the ventricle to relax, which is why the heart may develop diastolic dysfunction and eventually diastolic heart failure.
Cellular‐level evidence
It is well known that the collagen network in heart is not static, but undergoes continuous synthesis and degradation (Bishop & Laurent, 1995). If we focus on studies suggesting a protective effect of fibrosis in hypertension, we might ask if there are any mechanisms on a cellular and subcellular level that would promote such remodelling. Indeed, such mechanisms exist and are extensively researched; advances in this field are summarized in McCain & Parker (2011), which focuses on stretch sensing in myocytes, and Fan et al. (2012), which focuses on stretch sensing in fibroblasts and pro‐fibrotic signalling. In the context of this review, the key points are the following: (1) stretch promotes the release of growth factors such as TGF‐β and angiotensin II (Ruwhof & Laarse, 2000), both of which are pro‐fibrotic (González et al. 2002; Leask & Abraham, 2004); and (2) at the same time, TGF‐β1 upregulates lysyl oxidase, which promotes formation of collagen crosslinks (McCormick & Thomas, 1998). Therefore, there are known mechanisms both for stretch‐upregulated production of collagen and its crosslinking.
The signalling pathways involved in fibrotic remodelling are far from simple and may be cell‐type dependent. In the work of Engebretsen & Tønnessen (2014) in mice, broad β‐inhibition ameliorated fibrosis in pressure overload, but resulted in increased cardiac dilatation and ultimately mortality. Similar results were obtained using a different method of broad β‐signalling inhibition (Koitabashi et al. 2011).
On the other hand, in the study by Koitabashi et al., an inhibition of TGF‐β2 specifically in myocytes did not introduce dilatation, while improving cardiac function. Rainer et al. (2014) have similarly shown that after myocardial infarction, broad TGF‐β‐blockade has increased mortality due to wall rupture after dilatation, while myocyte‐specific TGF‐β‐blockade was beneficial for survival in the short term, decreasing the likelihood of wall rupture. Intriguingly, the existing literature suggests that myocyte‐specific TGF‐β2‐blockade is not only anti‐fibrotic, but also promotes ventricular stiffening: one of the effects of myocyte‐specific TGF‐β‐blockade was observed to be the attenuation of MMP‐9, activation of which is considered to be an important factor in transition from compensated hypertrophy to heart failure due to dilatation (Yabluchanskiy et al. 2013). At the same time, such a transition is mainly characterized by loss of collagen crosslinking, rather than of total collagen content (Woodiwiss et al. 2001). It is also known that MMP‐9 activation plays a role in formation of abdominal aortic aneurysms, a condition caused by insufficient strength of aortic wall (Wilson et al. 2008). We could therefore speculate that MMP‐9 activation decreases tissue stiffness and that its inhibition by myocyte‐specific beta‐blockade allows formation of stronger (presumably more crosslinked) collagen that prevents aortic or myocardial rupture, which provides a mechanism of beneficial effects seen in the studies by Rainer et al. and Koitabashi et al. In addition, a recent study in post‐infarction MMP‐9‐null mice demonstrated attenuated left ventricular dilatation, increased tissue stiffening, and a considerable increase in lysyl oxidase (and thus collagen crosslinking), even though collagen deposition was decreased (Voorhees et al. 2015). A visual summary of the observations above is given in Fig. 1.
Figure 1. An overview of key pro‐fibrotic signals and the crosslink‐breakup signalling of TGF‐β in myocytes.
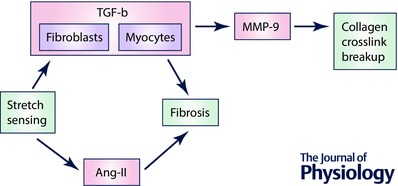
Ang‐II, angiotensin‐II; MMP‐9, matrix metalloproteinase 9; TGF‐β, transforming growth factor β.
The apparently opposite reaction to broad versus myocyte‐specific beta‐blockade suggests that there might be a competition between stretch‐sensing mechanisms in myocytes (promoting crosslink breakup via MMP‐9 activation) and fibroblasts (promoting cardiac stiffening and fibrosis). We might speculate that this competition could underlie how the myocytes sensing functional overload may decrease the myocardial integrity in order to elongate in an originally compensatory role according to the Frank–Starling mechanism; it is also possible that hypertrophy of the heart requires collagen crosslink breakup to accommodate angiogenesis (Yabluchanskiy et al. 2013). Then, when the myocyte performance matches the functional overload, the myocyte‐specific inhibition of structural integrity may stop and the other arm of this system, fibroblasts, may promote stiffening in order to maintain ECM integrity. At the same time, when the functional overload is severe and the Frank–Starling mechanism combined with hypertrophy does not compensate for the increased load, the collagen breakup signalling from myocytes may lead to critical destabilization of ECM, leading to ventricular dilatation and heart failure. This could, together with the collagen‐depositing signalling of fibroblasts under stretch overload, cause the established phenotype of systolic heart failure, where collagen production is high, but the crosslinking is diminished (Gunja‐Smith et al. 1996; Woodiwiss et al. 2001).
From fibrosis to sympathetic activation
Diffuse fibrosis occurs as a consequence of pressure overload as described above, but it is also associated with normal ageing, contributing to progressive cardiac stiffening even in the absence of an obvious cardiovascular disease (Biernacka & Frangogiannis, 2011). Any stiffening might be expected to be associated with a loss of the ability both to contract and to relax due to a part of the work performed by myocytes being absorbed by the ECM (Brilla et al. 1996). However, in ageing humans, the contractile capabilities of a heart are often preserved even when the ability to relax is significantly impaired (Chen, 2009; Santulli & Iaccarino, 2016). This suggests the existence of a compensatory mechanism that allows the heart to increase its contractility so that cardiac output can be maintained.
One such compensatory mechanism could be the activity of the cardiac sympathetic nervous system, which is known to be pro‐inotropic, i.e. increasing contractile strength (Goldberg et al. 1960). Like fibrosis, increased activity of the sympathetic system is often considered pathological, as it is found in dilatation‐induced heart failure (Kyuma et al. 2004), and is a contributing factor in the incidence of ventricular fibrillation, especially after a myocardial infarction (Shen & Zipes, 2014).
Despite these observations, we believe that the studies below show that CSNS is a crucial feature allowing stiff hearts to maintain sufficient cardiac output and that in certain well‐defined circumstances, excessive inhibition of the sympathetic system may be dangerous, rather than beneficial.
An example of a medical condition that could be related to CSNS inhibition is sudden cardiac death (SCD) due to pulseless electrical activity (PEA), the incidence of which is increasing, while effective treatment strategies are lacking (Hallstrom et al. 2009). In PEA, the electrical signal is propagated normally, but no contraction occurs. A second medical condition is asystole, where both electrical conduction and contraction are absent. We note that both these conditions are likely to improve with bolstering cardiac sympathetic nervous activation (which is among the recommended therapies): in PEA, the inotropic effect of sympathetic stimulation is expected to improve the issue of insufficient contractility. In asystole, sympathetic stimulation is expected to improve conduction of electrical signals from pacemaking cells, as it makes myocytes more excitable overall via activation of fast sodium channels (Baba et al. 2004) and the L‐type calcium current (Herzig et al. 1993).
Systems‐level evidence
An excellent human model of CSNS inhibition is heart transplantation, where the heart's natural innervation is disrupted (Vaseghi et al. 2009); here, denervation results in low levels of myocardial catecholamines, which persist for several years after transplantation (Regitz et al. 1990). In the normal population, the main cause of SCD is ventricular fibrillation (VF); however, in heart transplanted patients, VF is rare and the main causes of SCD are PEA and asystole (Vaseghi et al. 2009). This suggests that the denervation decreases the likelihood of a VF by negating the pro‐fibrillatory effect of CSNS, but the benefit of increased inotropy and facilitation of conduction is also lost, increasing the likelihood of PEA and asystole. It should be noted that transplanted hearts may be reinnervated, but this process is quite slow; as shown by Überfuhr et al. (2000), even after 4.6 years, 40% of transplant patients were classified as ‘not reinnervated’. The mean time between transplant and death in the study by Vaseghi was 3.5 years in non‐sudden deaths (and 1.3 years in sudden deaths), suggesting that these deaths could have happened before reinnervation took place and that the hearts can be indeed considered at least partially denervated.
A different model of CSNS inhibition is drug treatment, such as beta‐blockade. This is a much more complex model, however, since beta‐blockers are far wider‐acting than selective cardiac denervation. While beta‐blockade is very likely to decrease the intrinsic contractility of the heart by blocking pro‐inotropic catecholamine activity, it also decreases heart rate (thus improving diastolic filling) and usually decreases systemic blood pressure, decreasing overall cardiac workload (Frishman & Saunders, 2011). Therefore, beta‐blockade may not invariably introduce problems with contractility. It has been shown by Hall et al. (1995) that in cases of dilated cardiomyopathy (and thus systolic heart failure), beta‐blocker administration causes initial and transient decrease of cardiac output (proposed to be due to decreased inotropy), but the cardiac output then improves above the original value (proposed to be due to decreased cardiac workload). To our knowledge, there is unfortunately no such study for diastolic heart failure. However, the study by Youngquist et al. (2008) is perhaps indicative of the effect of beta‐blockers on patients with stiff hearts. The authors of this study have performed a study in aged patients (median 70 years, interquartile range 57–79 years) who are likely to have increased myocardial stiffness, demonstrating that within the observed group, beta‐blocker use was associated with a significant and large increase of likelihood of PEA over VF compared to patients without beta‐blockers. We note, however, that this study was an observational one and not randomized, i.e. the patients administered beta‐blockers might have had an a priori high likelihood of PEA. Nevertheless, as the authors state, this study suggests that widespread use of beta‐blockers might be the key factor in changing epidemiology of VF and PEA between the 1980s and now. While the incidence of VF as the initial rhythm in out‐of‐hospital cardiac arrest has decreased from 61–65% to 35–48%, the incidence of PEA grew to the current state of 22–30% (Saarinen et al. 2012).
Indirect evidence of the compensatory role of CSNS is also seen in the study by Grassi et al. (2009), which found significantly increased muscle sympathetic activity in hypertensive patients with diastolic dysfunction, compared both to hypertensive patients with normal diastolic function and to a control group of non‐hypertensive patients. At the same time, systolic function did not differ significantly between the three observed groups, as evaluated by ejection fraction and fractional shortening. If we assume that the diastolic dysfunction in the observed group was caused by increased cardiac stiffness, sustained systolic function is indicative of a compensatory mechanism that increases contraction strength: CSNS with its known pro‐inotropic effect, and being so clearly elevated in this study, seems a very likely candidate. One caveat is that this study measured striated muscle sympathetic activity, but it is known to be correlated to cardiac sympathetic activation (Lambert et al. 2011).
Animal studies also suggest the importance of the CSNS for maintaining sufficient contractility. One such example is the study by Albrecht et al. (1975), where sympathetic deactivation via pithing caused an almost 50% decrease in cardiac output, which could be only partially explained by decreased heart rate.
In summary, the existing literature suggests that in the presence of myocardial stiffness, increasing CSNS reduces the risk of PEA due to improved contractility, but at the same time, increases the risk of VF. This perspective may lead to a framework that can guide therapy: e.g. when a patient is treated for fibrillation with sympathoinhibitory drugs, the dose might be adjusted according to how stiff the patient's heart is.
Myocardial infarction and pressure overload
In this section, we briefly comment on why the stretch‐sensing mechanisms described above are relevant also to the heart with healed myocardial infarction. After myocardial infarction, a stiff scar is eventually formed in place of the infarcted tissue (Czubryt, 2012). Being composed mainly of collagen, the scar is far less contractile than the surrounding myocardium (Fomovsky & Holmes, 2010); when it is not sufficiently firm, the infarcted tissue is prone to dilatation and/or rupture (Noppe et al. 2014).
When a segment of the myocardial wall becomes stiff, assuming constant internal volume, the remaining myocardium is stretched with greater force during diastole. In addition, in order to maintain cardiac output, the remaining healthy tissue must generate more force to compensate for the scar. This makes the presence of stiff scars in myocardium a sort of analogue of elevated systolic and diastolic pressure. Indeed, certain processes we described earlier as being linked to hypertension have been observed after myocardial infarction, i.e. increased angiotensin‐II (Ang‐II) secretion (Sutton & Sharpe, 2000) and subsequent fibrosis in non‐infarcted myocardium (Volders, 1993); the hyperinnervation of non‐infarcted myocardium has also been described (Zhou et al. 2004). It is possible that these changes are mediated largely by similar stretch‐sensing mechanisms as the ones discussed earlier in this text, with the same consequences of their imbalance. To our knowledge, there is no study researching the role of post‐infarction interstitial hyperinnervation in maintaining contractility, but the role of matrix metalloproteinases and their inhibition after MI has been studied in animal models. The results are largely consistent with the role of collagen dynamics in hypertension, where inhibition of degradation via MMP inhibition attenuates left ventricular dilatation (Rohde et al. 1999; Lindsey et al. 2002); this effect is not entirely due to the acute healing phase, but has a role in the late healing phase as well (Mukherjee et al. 2003).
Summary and discussion
Putting together the links between pressure overload and fibrosis and between fibrosis and CSNS, a simplified model of disease based on pressure overload can be synthesized, as shown in Fig. 2. This model provides additional insight to the observation that fibrosis is associated with occurrence of arrhythmias (Shiozaki et al. 2013). In addition to the role of increased conduction heterogeneity in a fibrotic environment, the model explains why fibrosis might be indirectly associated with an increase of cardiac sympathetic activity, promoting arrhythmias by a mechanism not directly linked to fibrotic remodelling.
Figure 2. A model of disease development according to presence of remodelling steps.
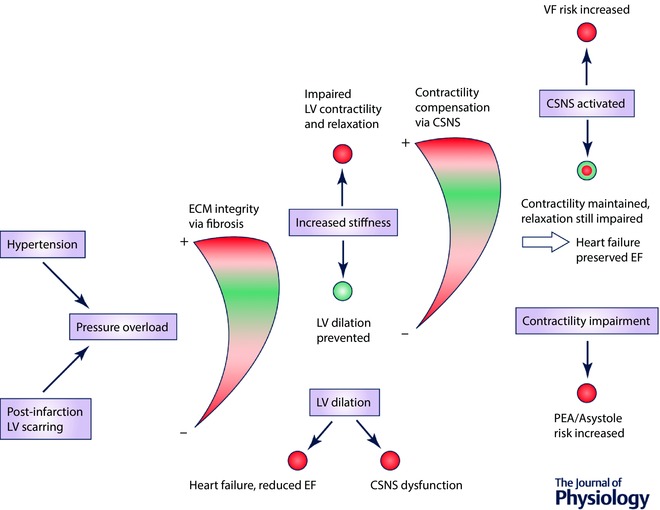
Blue nodes represent concepts of heart disease, with some of their consequences given as coloured circles: red, detrimental; green, beneficial. The gauges represent a spectrum of their respective feature (how much ECM integrity is increased, or how much CSNS is activated), with the red representing detrimental extremes and green representing a ‘sweet spot’, where total risk of both extremes is minimized. EF, ejection fraction; LV, left ventricular.
We consider the model to be a useful tool in evaluating existing and developed therapies. For example, anti‐fibrotic therapy has been proposed to ameliorate fibrosis‐associated arrhythmias (Morita et al. 2014), which is indeed expected to decrease the occurrence of arrhythmias. However, our model suggests that if pressure overload is also present and is not targeted by the therapy, the outcome can be detrimental to patients’ health by a disproportionate increase of risk of myocardial dilatation. An example of a controversial therapy would be the potentiation of MMPs, which is expected to ameliorate fibrosis, but might diminish the structural integrity of myocardium. On the other hand, angiotensin converting enzyme (ACE) inhibitors or angiotensin receptor antagonists seem to be a safer way to treat fibrosis, as they not only act anti‐fibrotically (Ciulla et al. 2009), but also decrease blood pressure, which in turn reduces the stress on myocardial structure and thus the underlying pro‐fibrotic signalling. It is yet to be determined how much the anti‐fibrotic effect of ACE inhibitors is due to a direct effect and how much is due to prevention of stretch‐induced pro‐fibrotic remodelling via decreased blood pressure.
The model might also be relevant to the understanding of the two flavours of heart failure: systolic and diastolic. These are conditions that are frequently researched together (‘heart failure’) in clinical studies, but which are largely different diseases with different underlying causes and thus requiring an entirely different therapy (this topic is discussed in greater depth in Iwano & Little (2013)). For example, there are therapies shown to be efficient in systolic heart failure, but a successful therapy for diastolic heart failure is lacking (Udelson, 2011). According to the studies discussed in this article, both types of heart failure may be viewed as two sides of a single coin: ECM integrity.
An initial loss of ECM integrity, caused by a breakdown of collagen or reduced crosslinking, may result in dilatation and systolic heart failure. Myocardial stretch promotes CSNS activity, which initially improves contractility, but this can fail due to excessive CSNS overactivation and neurotransmitter depletion on β‐receptor downregulation (Port & Bristow, 2001), possibly via β‐arrestins (Bathgate‐Siryk et al. 2014). Interestingly, the benefit of beta‐blockers in systolic heart failure is partly due to restoration of beta‐receptor density, which then paradoxically potentiates CSNS (López‐Sendón et al. 2004). It remains an open question of how the loss of collagen integrity may be prevented in the first place: MMP inhibition has been proposed (King et al. 2003), but human mortality studies of MMP inhibitor therapy are lacking.
On the other hand, diastolic heart failure is caused by an increase in ECM integrity in our model, manifesting as increased myocardial stiffness (which might be either a physiological compensation of pressure overload or a pathological dysregulation). In this context, CSNS is elevated to maintain sufficient contractility, but there is a lack of evidence for activation and desensitization to the same degree as is seen in systolic heart failure, which could underlie the observation that beta‐blockers are not nearly as efficient in treating diastolic heart failure (Paulus & van Ballegoij, 2010). Our review suggests that the lack of overt CSNS dysregulation in diastolic heart failure could be due to myocardial stiffness limiting myocardial stretch, preventing excessive pro‐CSNS signalling based on stretch sensing. Several studies cited in this review indirectly suggest that when anti‐inotropic drugs are administered to patients with diastolic heart failure, the degree of cardiac stiffness should be taken into account. Among the recommended treatment of diastolic heart failure are symptom relief and an aggressive treatment of hypertension (Rose‐Jones et al. 2014), which is again an interesting link to the model synthesized in this review, where diastolic heart failure is predicted to happen as a consequence of hypertension. From the point of future therapy, a possible research direction is a cardiac‐specific activation of certain MMPs with the motivation of reduction in cardiac stiffness. However, this would require a careful balancing of breakup of collagen and its crosslinks while maintaining the necessary structural integrity of ECM to prevent dilatation and/or rupture.
Hypertension and heart failure are complex systemic diseases with a vast range of possible interactions. In the current review we focus on the relationship between ECM integrity and CSNS, but many other factors play significant roles. For example, sustained β‐adrenergic stimulation is known to induce hypertrophy via several mechanisms (e.g. Sorriento et al. 2010) and activates G‐protein receptor kinases that have diverse effects (e.g. induction of insulin resistance, Santulli et al. 2013), which limits the ability of CSNS to compensate for the effects of heart failure. In addition, cell coupling modulators (Egan Benova et al. 2016) and immune regulators (González et al. 2016) have recently been identified as promising therapeutic targets that leverage different pathways from those discussed here. More research is needed to determine the relative importance of these interactions in disease progression. Nevertheless, we hope that this review provides insights and may eventually lead to new strategies for disease management and treatment.
Additional information
Competing interests
None declared.
Author contributions
Both authors have approved the final version of the manuscript and agree to be accountable for all aspects of the work. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Acknowledgements
The authors thank Professor Blanca Rodriguez, Oxford, who provided valuable feedback and editing for this review. G.B. acknowledges support from the Medical Research Council and J.T. acknowledges support from the Bakala Foundation.
Biographies
Jakub Tomek received his Master's degree in Computer science from Charles University in Prague, and continues his graduate studies in the Life Sciences Interface Doctoral Training Centre and Department of Physiology, Anatomy, and Genetics. He uses a combination of experimental approaches and computational modelling to understand cardiac physiology on the systemic level, particularly the role of sympathetic nervous system in heart disease. Further interests include image processing, machine learning, and data‐mining.

Gil Bub investigates the interplay of activity between neuron and cardiac cells in cell culture and in intact hearts, as well as new technologies for imaging and controlling excitable cells. He did his graduate work at McGill University, Canada on the dynamics of cardiac monolayers, spent several years as a University Research Lecturer in Oxford's Department of Physiology Anatomy and Genetics, and recently moved back to an academic position in McGill University's Physiology department.
[The copyright line for this article was changed on 5th May 2017 after original online publication]
Contributor Information
Jakub Tomek, Email: jakub.tomek@stkatz.ox.ac.uk.
Gil Bub, Email: gil.bub@mcgill.ca.
References
- Albrecht I, Hallback M, Julius S, Lundgren Y, Stage L, Weiss L & Folkow B (1975). Arterial pressure, cardiac output and systemic resistance before and after pithing in normotensive and spontaneously hypertensive rats. Acta Physiol Scand, 94, 378–385. [DOI] [PubMed] [Google Scholar]
- Baba S, Dun W & Boyden PA (2004). Can PKA activators rescue Na+ channel function in epicardial border zone cells that survive in the infarcted canine heart? Cardiovasc Res 64, 260–267. [DOI] [PubMed] [Google Scholar]
- Badenhorst D, Maseko M, Tsotetsi OJ, Naidoo A, Brooksbank R, Norton GR & Woodiwiss AJ (2003). Cross‐linking influences the impact of quantitative changes in myocardial collagen on cardiac stiffness and remodelling in hypertension in rats. Cardiovasc Res 57, 632–641. [DOI] [PubMed] [Google Scholar]
- Bathgate‐Siryk A, Dabul S, Pandya K, Walklett K, Rengo G, Cannavo A, De Lucia C, Liccardo D, Gao E, Leosco D, Koch WJ & Lymperopoulos A (2014). Negative impact of β‐arrestin‐1 on post‐myocardial infarction heart failure via cardiac and adrenal‐dependent neurohormonal mechanisms. Hypertension 63, 404–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biernacka A & Frangogiannis NG (2011). Aging and cardiac fibrosis. Aging Dis 2, 158–173. [PMC free article] [PubMed] [Google Scholar]
- Bishop JE & Laurent GJ (1995). Collagen turnover and its regulation in the normal and hypertrophying heart. Eur Heart J 16 Suppl C, 38–44. [DOI] [PubMed] [Google Scholar]
- Brilla CG, Matsubara L & Weber KT (1996). Advanced hypertensive heart disease in spontaneously hypertensive rats. Lisinopril‐mediated regression of myocardial fibrosis. Hypertension 28, 269–275. [DOI] [PubMed] [Google Scholar]
- Chen MA (2009). Heart failure with preserved ejection fraction in older adults. Am J Med 122, 713–723. [DOI] [PubMed] [Google Scholar]
- Ciulla M, Paliotti R, Esposito A, Cuspidi C, Muiesan M, Rosei E, Magrini F & Zanchetti A (2009). Effects of antihypertensive treatment on ultrasound measures of myocardial fibrosis in hypertensive patients with left ventricular hypertrophy: results of a randomized trial comparing the angiotensin receptor antagonist, candesartan and the angiotensin‐converting enzyme inhibitor, enalapril. J Hypertens 27, 626–632. [DOI] [PubMed] [Google Scholar]
- Creemers EE & Pinto YM (2011). Molecular mechanisms that control interstitial fibrosis in the pressure‐overloaded heart. Cardiovasc Res 89, 265–272. [DOI] [PubMed] [Google Scholar]
- Czubryt MP (2012). Common threads in cardiac fibrosis, infarct scar formation, and wound healing. Fibrogenes Tissue Repair 5, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drazner MH (2011). The progression of hypertensive heart disease. Circulation 123, 327–334. [DOI] [PubMed] [Google Scholar]
- Egan Benova T, Szeiffova Bacova B, Viczenczova C, Diez E, Barancik M & Tribulova N (2016). Protection of cardiac cell‐to‐cell coupling attenuate myocardial remodeling and proarrhythmia induced by hypertension. Physiol Res 65 Suppl 1, S29–S42. [DOI] [PubMed] [Google Scholar]
- Engebretsen K & Tønnessen T (2014). Attenuated development of cardiac fibrosis in left ventricular pressure overload by SM16, an orally active inhibitor of ALK5. J Mol Cell Cardiol 76, 148–157. [DOI] [PubMed] [Google Scholar]
- Fan D, Takawale A, Lee J & Kassiri Z (2012). Cardiac fibroblasts, fibrosis and extracellular matrix remodeling in heart disease. Fibrogenesis Tissue Repair 5, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fomovsky GM & Holmes JW (2010). Evolution of scar structure, mechanics, and ventricular function after myocardial infarction in the rat. Am J Physiol Heart Circ Physiol 298, H221–H228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frey N & Olson EN (2003). Cardiac hypertrophy: the good, the bad, and the ugly. Annu Rev Physiol 65, 45–79. [DOI] [PubMed] [Google Scholar]
- Frishman WH & Saunders E (2011). β‐Adrenergic blockers. J Clin Hypertens (Greenwich) 13, 649–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg LI, Bloodwell RD, Braunwald E & Morrow AG (1960). The direct effects of norepinephrine, epinephrine, and methoxamine on myocardial contractile force in man. Circulation 22, 1125–1132. [DOI] [PubMed] [Google Scholar]
- González A, López B, Querejeta R & Díez J (2002). Regulation of myocardial fibrillar collagen by angiotensin II. A role in hypertensive heart disease? J Mol Cell Cardiol 34, 1585–1593. [DOI] [PubMed] [Google Scholar]
- González GE, Rhaleb N‐E, D'Ambrosio MA, Nakagawa P, Liao T‐D, Peterson EL, Leung P, Dai X, Janic B, Liu Y‐H, Yang X‐P & Carretero OA (2016). Cardiac‐deleterious role of galectin‐3 in chronic angiotensin II‐induced hypertension. Am J Physiol Hear Circ Physiol 311, H1287–H1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grassi G, Seravalle G, Quarti‐Trevano F, Dell'Oro R, Arenare F, Spaziani D & Mancia G (2009). Sympathetic and baroreflex cardiovascular control in hypertension‐related left ventricular dysfunction. Hypertension 53, 205–209. [DOI] [PubMed] [Google Scholar]
- Gunja‐Smith Z, Morales AR & Romanelli R (1996). Remodeling of human myocardial collagen in idiopathic dilated cardiomyopathy role of metalloproteinases and pyridinoline cross‐links. Am J Pathol 148, 1639–1648. [PMC free article] [PubMed] [Google Scholar]
- Hall SA, Cigarroa CG, Marcoux L, Risser RC, Grayburn PA & Eichhorn EJ (1995). Time course of improvement in left ventricular function, mass and geometry in patients with congestive heart failure treated with beta‐adrenergic blockade. J Am Coll Cardiol 25, 1154–1161. [DOI] [PubMed] [Google Scholar]
- Hallstrom A, Herlitz J, Kajino K & Olasveengen TM (2009). Treatment of asystole and PEA. Resuscitation 80, 975–976. [DOI] [PubMed] [Google Scholar]
- Herzig S, Patil P, Neumann J, Staschen CM & Yue DT (1993). Mechanisms of beta‐adrenergic stimulation of cardiac Ca2+ channels revealed by discrete‐time Markov analysis of slow gating. Biophys J 65, 1599–1612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwano H & Little WC (2013). Heart failure: What does ejection fraction have to do with it? J Cardiol 62, 1–3. [DOI] [PubMed] [Google Scholar]
- Janicki JS, Brower GL, Gardner JD, Forman MF, Stewart JA, Murray DB & Chancey AL (2006). Cardiac mast cell regulation of matrix metalloproteinase‐related ventricular remodeling in chronic pressure or volume overload. Cardiovasc Res 69, 657–665. [DOI] [PubMed] [Google Scholar]
- Kim HE, Dalal SS, Young E, Legato MJ, Weisfeldt ML & D'Armiento J (2000). Disruption of the myocardial extracellular matrix leads to cardiac dysfunction. J Clin Invest 106, 857–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King MK, Coker ML, Goldberg A, McElmurray JH 3rd, Gunasinghe HR, Mukherjee R, Zile MR, O'Neill TP & Spinale FG (2003). Selective matrix metalloproteinase inhibition with developing heart failure: effects on left ventricular function and structure. Circ Res 92, 177–185. [DOI] [PubMed] [Google Scholar]
- Koitabashi N, Danner T, Zaiman AL, Pinto YM, Rowell J, Mankowski J, Zhang D, Nakamura T, Takimoto E & Kass DA (2011). Pivotal role of cardiomyocyte TGF‐β signaling in the murine pathological response to sustained pressure overload. J Clin Invest 121, 2301–2312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyuma M, Nakata T, Hashimoto A & Nagao K (2004). Incremental prognostic implications of brain natriuretic peptide, cardiac sympathetic nerve innervation, and noncardiac disorders in patients with heart failure. J Nucl Med 45, 155–164. [PubMed] [Google Scholar]
- Lambert EA, Schlaich MP, Dawood T, Sari C, Chopra R, Barton DA, Kaye DM, Elam M, Esler MD & Lambert GW (2011). Single‐unit muscle sympathetic nervous activity and its relation to cardiac noradrenaline spillover. J Physiol 589, 2597–2605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leask A & Abraham DJ (2004). TGF‐β signaling and the fibrotic response. FASEB J 18, 816–827. [DOI] [PubMed] [Google Scholar]
- Lindsey ML, Gannon J, Aikawa M, Schoen FJ, Rabkin E, Lopresti‐Morrow L, Crawford J, Black S, Libby P, Mitchell PG & Lee RT (2002). Selective matrix metalloproteinase inhibition reduces left ventricular remodeling but does not inhibit angiogenesis after myocardial infarction. Circulation 105, 753–758. [DOI] [PubMed] [Google Scholar]
- López‐Sendón J, Swedberg K, McMurray J, Tamargo J, Maggioni A, Dargie H, Tendera M, Waagstein F, Kjeshus J, Lechat P & Torp‐Pedersen C (2004). Expert consensus document on β‐adrenergic receptor blockers. Eur Heart J 25, 1341–1362. [DOI] [PubMed] [Google Scholar]
- McCain ML & Parker KK (2011). Mechanotransduction: the role of mechanical stress, myocyte shape, and cytoskeletal architecture on cardiac function. Pflugers Arch 462, 89–104. [DOI] [PubMed] [Google Scholar]
- McCormick RJ & Thomas DP (1998). Collagen crosslinking in the heart: relationship to development and function. Basic Appl Myol 8, 143–150. [Google Scholar]
- McElmurray JH, Mukherjee R, New RB, Sampson AC, King MK, Hendrick JW, Goldberg A, Peterson TJ, Hallak H, Zile MR & Spinale FG (1999). Angiotensin‐converting enzyme and matrix metalloproteinase inhibition with developing heart failure: comparative effects on left ventricular function and geometry. J Pharmacol Exp Ther 291, 799–811. [PubMed] [Google Scholar]
- Morita N, Mandel WJ, Kobayashi Y & Karagueuzian HS (2014). Cardiac fibrosis as a determinant of ventricular tachyarrhythmias. J Arrhythmia 30, 389–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee R, Brinsa TA, Dowdy KB, Scott AA, Baskin JM, Deschamps AM, Lowry AS, Escobar GP, Lucas DG, Yarbrough WM, Zile MR & Spinale FG (2003). Myocardial infarct expansion and matrix metalloproteinase inhibition. Circulation 107, 618–625. [DOI] [PubMed] [Google Scholar]
- Noppe G, Dufeys C, Buchlin P, Marquet N, Castanares‐Zapatero D, Balteau M, Hermida N, Bouzin C, Esfahani H, Viollet B, Bertrand L, Balligand JL, Vanoverschelde JL, Beauloye C & Horman S (2014). Reduced scar maturation and contractility lead to exaggerated left ventricular dilation after myocardial infarction in mice lacking AMPKα1. J Mol Cell Cardiol 74, 32–43. [DOI] [PubMed] [Google Scholar]
- Paulus WJ & van Ballegoij JJM (2010). Treatment of heart failure with normal ejection fraction: an inconvenient truth! J Am Coll Cardiol 55, 526–537. [DOI] [PubMed] [Google Scholar]
- Peterson JT, Hallak H, Johnson L, Li H, O'Brien PM, Sliskovic DR, Bocan TMA., Coker ML, Etoh T & Spinale FG (2001). Matrix metalloproteinase inhibition attenuates left ventricular remodeling and dysfunction in a rat model of progressive heart failure. Circulation 103, 2303–2309. [DOI] [PubMed] [Google Scholar]
- Port JD & Bristow MR (2001). Altered beta‐adrenergic receptor gene regulation and signaling in chronic heart failure. J Mol Cell Cardiol 33, 887–905. [DOI] [PubMed] [Google Scholar]
- Rainer PP, Hao S, Vanhoutte D, Lee DI, Koitabashi N, Molkentin JD & Kass DA (2014). Cardiomyocyte‐specific transforming growth factor β suppression blocks neutrophil infiltration, augments multiple cytoprotective cascades, and reduces early mortality after myocardial infarction. Circ Res 114, 1246–1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regitz V, Bossaller C, Strasser R, Schüler S, Hetzer R & Fleck E (1990). Myocardial catecholamine content after heart transplantation. Circulation 82, 620–623. [DOI] [PubMed] [Google Scholar]
- Rohde LE, Ducharme A, Arroyo LH, Aikawa M, Sukhova GH, Lopez‐Anaya A, McClure KF, Mitchell PG, Libby P & Lee RT (1999). Matrix metalloproteinase inhibition attenuates early left ventricular enlargement after experimental myocardial infarction in mice. Circulation 99, 3063–3070. [DOI] [PubMed] [Google Scholar]
- Rose‐Jones LJ, Rommel JJ & Chang PP (2014). Heart failure with preserved ejection fraction. An ongoing enigma. Cardiol Clin 32, 151–161. [DOI] [PubMed] [Google Scholar]
- Ruwhof C & Laarse A Van Der (2000). Mechanical stress‐induced cardiac hypertrophy: mechanisms and signal transduction pathways. Cardiovasc Res 47, 23–37. [DOI] [PubMed] [Google Scholar]
- Saarinen S, Kämäräinen A, Silfvast T, Yli‐Hankala A & Virkkunen I (2012). Pulseless electrical activity and successful out‐of‐hospital resuscitation – long‐term survival and quality of life: an observational cohort study. Scand J Trauma Resusc Emerg Med 20, 74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santulli G & Iaccarino G (2016). Adrenergic signaling in heart failure and cardiovascular aging. Maturitas 93, 65–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santulli G, Trimarco B & Iaccarino G (2013). G‐protein‐coupled receptor kinase 2 and hypertension. High Blood Press Cardiovasc Prev 20, 5–12. [DOI] [PubMed] [Google Scholar]
- Shen MJ & Zipes DP (2014). Role of the autonomic nervous system in modulating cardiac arrhythmias. Circ Res 114, 1004–1021. [DOI] [PubMed] [Google Scholar]
- Shiozaki AA, Senra T, Arteaga E, Martinelli Filho M, Pita CG, Ávila LFR, Parga Filho JR, Mady C, Kalil‐Filho R, Bluemke DA & Rochitte CE (2013). Myocardial fibrosis detected by cardiac CT predicts ventricular fibrillation/ventricular tachycardia events in patients with hypertrophic cardiomyopathy. J Cardiovasc Comput Tomogr 7, 173–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorriento D, Santulli G, Fusco A, Anastasio A, Trimarco B & Iaccarino G (2010). Intracardiac injection of AdGRK5‐NT reduces left ventricular hypertrophy by inhibiting NF‐κB‐dependent hypertrophic gene expression. Hypertension 56, 696–704. [DOI] [PubMed] [Google Scholar]
- Sutton MG & Sharpe N (2000). Left ventricular remodeling after myocardial infarction: pathophysiology and therapy. Circulation 101, 2981–2988. [DOI] [PubMed] [Google Scholar]
- Takeda N, Manabe I, Uchino Y, Eguchi K, Matsumoto S, Nishimura S, Shindo T, Sano M, Otsu K, Snider P, Conway SJ & Nagai R (2010). Cardiac fibroblasts are essential for the adaptive response of the murine heart to pressure overload. J Clin Invest 120, 254–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Überfuhr P, Frey AW, Ziegler S, Reichart B & Schwaiger M (2000). Sympathetic reinnervation of sinus node and left ventricle after heart transplantation in humans: Regional differences assessed by heart rate variability and positron emission tomography. J Hear Lung Transplant 19, 317–323. [DOI] [PubMed] [Google Scholar]
- Udelson J (2011). Heart failure with preserved ejection fraction. Circulation 124, e540–e543. [DOI] [PubMed] [Google Scholar]
- Vaseghi M, Lellouche N, Ritter H, Fonarow GC, Patel JK, Moriguchi J, Fishbein MC, Kobashigawa JA & Shivkumar K (2009). Mode and mechanisms of death after orthotopic heart transplantation. Hear Rhythm 6, 503–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volders P (1993). Interstitial collagen is increased in the non‐infarcted human myocardium after myocardial infarction. J Mol Cell Cardiol 25, 1317–1323. [DOI] [PubMed] [Google Scholar]
- Voorhees AP, DeLeon‐Pennell KY, Ma Y, Halade G V, Yabluchanskiy A, Iyer RP, Flynn E, Cates CA, Lindsey ML & Han H‐C (2015). Building a better infarct: Modulation of collagen cross‐linking to increase infarct stiffness and reduce left ventricular dilation post‐myocardial infarction. J Mol Cell Cardiol 85, 229–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson WRW, Anderton M, Choke EC, Dawson J, Loftus IM & Thompson MM (2008). Elevated plasma MMP1 and MMP9 are associated with abdominal aortic aneurysm rupture. Eur J Vasc Endovasc Surg 35, 580–584. [DOI] [PubMed] [Google Scholar]
- Woodiwiss AJ, Tsotetsi OJ, Sprott S, Lancaster EJ, Mela T, Chung ES, Meyer TE & Norton GR (2001). Reduction in myocardial collagen cross‐linking parallels left ventricular dilatation in rat models of systolic chamber dysfunction. Circulation 103, 155–160. [DOI] [PubMed] [Google Scholar]
- Yabluchanskiy A, Ma Y, Iyer RP, Hall ME & Lindsey ML (2013). Matrix metalloproteinase‐9: Many shades of function in cardiovascular disease. Physiology (Bethesda) 28, 391–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youngquist ST, Kaji AH & Niemann JT (2008). Beta‐blocker use and the changing epidemiology of out‐of‐hospital cardiac arrest rhythms. Resuscitation 76, 376–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou S, Chen LS, Miyauchi Y, Miyauchi M, Kar S, Kangavari S, Fishbein MC, Sharifi B & Chen P‐S (2004). Mechanisms of cardiac nerve sprouting after myocardial infarction in dogs. Circ Res 95, 76–83. [DOI] [PubMed] [Google Scholar]