

Abstract
Excitation–contraction coupling is the bridge between cardiac electrical activation and mechanical contraction. It is driven by the influx of Ca2+ across the sarcolemma triggering Ca2+ release from the sarcoplasmic reticulum (SR) – a process termed Ca2+‐induced Ca2+ release (CICR) – followed by re‐sequestration of Ca2+ into the SR. The Na+/Ca2+ exchanger inextricably couples the cycling of Ca2+ and Na+ in cardiac myocytes. Thus, influx of Na+ via voltage‐gated Na+ channels (NaV) has emerged as an important regulator of CICR both in health and in disease. Recent insights into the subcellular distribution of cardiac and neuronal NaV isoforms and their ultrastructural milieu have important implications for the roles of these channels in mediating Ca2+‐driven arrhythmias. This review will discuss functional insights into the role of neuronal NaV isoforms vis‐à‐vis cardiac NaVs in triggering such arrhythmias and their potential as therapeutic targets in the context of the aforementioned structural observations.
Keywords: arrhythmia, calcium, heart, neuronal sodium channels
Abbreviations
- BIN1
bridging integrator 1
- CaMKII
Ca2+/calmodulin‐dependent protein kinase II
- CICR
Ca2+‐induced Ca2+ release
- cNaV
cardiac sodium channel
- CPVT
catecholaminergic polymorphic ventricular tachycardia
- ECC
excitation‐contraction coupling
- HF
heart failure
- LCC
L‐type Ca2+ channel
- NaV
voltage‐gated Na+ channel
- NCX
sodium–calcium exchanger
- nNaV
neuronal sodium channel
- RyR
ryanodine receptor
- SR
sarcoplasmic reticulum
- t‐tubule
transverse tubule
- TTX
tetrodotoxin
- WT
wild‐type
Cardiac calcium (Ca2+) cycling involves the release of Ca2+ from intracellular stores, i.e. the sarcoplasmic reticulum (SR), prompted by Ca2+ influx across the sarcolemma – a process termed Ca2+‐induced Ca2+ release (CICR), and its re‐sequestration into the SR. The interaction of the Ca2+ released from the SR with contractile proteins links the heart's electrical activity with mechanical contraction and is termed excitation–contraction coupling (ECC); as such, it is a vital process for cardiac function. Abnormalities in ECC underlie life‐threatening arrhythmias in several pathologies ranging from catecholaminergic polymorphic ventricular tachycardia (CPVT) to heart failure (HF) (Belevych et al. 2013; Radwanski et al. 2013a). That Ca2+ cycling within cardiac myocytes is inextricably intertwined with the regulation of intracellular sodium (Na+) is well established (Bers et al. 2003; Murphy & Eisner, 2009). Voltage‐gated Na+ channels (NaVs) permit Na+ into the cell, resulting in electrical excitation, thereby initiating ECC. The Na+–Ca2+ exchanger (NCX), on the other hand, electrogenically exchanges 1 Ca2+ ion for 3 Na+ ions (Blaustein & Lederer, 1999; Philipson & Nicoll, 2000), creating a direct link between Na+ influx into the myocytes and Ca2+ cycling. This review will focus on the roles of NaV isoforms, the principal pathways for Na+ influx into cardiomyocytes, in modulating cardiac Ca2+ cycling in health and in disease, with particular emphasis on the spatial integration of NaVs with Ca2+ cycling proteins to form a larger macromolecular machine.
The interplay between sodium and calcium
Classically, the NCX's role in the cardiac cycle was viewed as the extrusion, from the cardiac myocyte, of all Ca2+ entering the cell during ECC. However, since ion transport via NCX is governed by the concentration gradients of Na+ and Ca2+ as well as the membrane potential, it can operate in both forward (3 Na+ in: 1 Ca2+ out) and reverse modes (3 Na+ out: 1 Ca2+ in; reversal potential according to the Nernst equation). As early as 1990, Leblanc and Hume demonstrated that, in the absence of Ca2+ entry via voltage‐dependent Ca2+ channels, Ca2+ entry via reverse mode NCX could elicit Ca2+ release from the SR (Leblanc & Hume, 1990). Using tetrodotoxin (TTX; 5 μm) to block NaVs, they further demonstrated that this Ca2+ entry via reverse mode NCX was dependent upon Na+ entry via NaVs: Na+ entry through NaVs elevates subsarcolemmal Na+ levels, causing NCX to reverse (3 Na+ out: 1 Ca2+ in) and bring in Ca2+, eliciting Ca2+ release from the SR. Shortly thereafter, Lipp and Niggli demonstrated Na+ current (I Na)‐induced Ca2+ transients in guinea pig ventricular myocytes, which were mediated by reverse mode NCX (Lipp & Niggli, 1994). These investigators concluded that Na+ influx via NaVs resulted in a rapid rise in subsarcolemmal [Na+], causing NCX to operate in reverse mode.
Although these studies demonstrated a link between Na+ influx via NaVs and SR Ca2+ release, the question remained as to the physiological role of I Na‐induced Ca2+ release. Larbig et al. (2010) determined that blockade of I Na with 10 μm TTX decreased the influx of trigger Ca2+ resulting in a lower rate of Ca2+ release from the SR and a reduced Ca2+ transient amplitude in myocytes isolated from wild‐type (WT) mice but not in those isolated from NCX knockout mice. These results suggested that Ca2+ entry via reverse mode NCX secondary to Na+ entry via NaVs does indeed contribute to activation of SR Ca2+ release.
The identity of NaVs responsible for the enhanced Ca2+ release in the heart is the subject of ongoing research. The predominant NaV isoform identified in the heart is Nav1.5 (Chen‐Izu et al. 2015), which is sensitive to micromolar concentrations of TTX and is therefore, categorized as a TTX‐resistant cardiac‐type NaV (cNav) (Satin et al. 1992). Recent work in the heart (Dhar Malhotra et al. 2001; Maier et al. 2002, 2004; Westenbroek et al. 2013; Radwanski et al. 2015), however, has identified the presence of neuronal NaV isoforms (nNavs), so called because they were first identified in neurons. Unlike cNaVs (Nav1.5), nNavs (Nav1.1, 1.3, 1.6) are sensitive to nanomolar concentrations of TTX (Ritchie & Rogart, 1977; Renaud et al. 1983). In subsequent work, the groups of Goldhaber and Bridge demonstrated that 100 nm TTX suppressed SR Ca2+ release flux and Ca2+ transient amplitude in rabbit ventricular myocytes, pointing to a role for TTX‐sensitive nNaVs in triggering SR Ca2+ release (Torres et al. 2010). In this vein, our group recently obtained similar findings with 100 nm TTX using optical mapping in intact guinea pig ventricles (Radwanski et al. 2013b). These results underscore the importance of NaVs as modulators of cardiac Ca2+ cycling and complement the structural observations that nNavs in cardiac myocytes are localized to invaginations of the surface membrane known as the transverse tubules (t‐tubules; Fig. 1; Dhar Malhotra et al. 2001; Maier et al. 2002, 2004; Westenbroek et al. 2013; Radwanski et al. 2015) – where Ca2+ cycling proteins are also localized. Thus, it has been postulated that, at the beginning of an action potential, they admit Na+ into areas of closest proximity between the t‐tubule sarcolemma and SR. This Na+ is then extruded out of the cell by the NCX in exchange for Ca2+, ‘priming’ this nanodomain with Ca2+. This, in conjunction with Ca2+ that enters the cell through the L‐type Ca2+ channels (LCCs), facilities opening of SR Ca2+ release channels, ryanodine receptors (RyRs), resulting in a robust Ca2+ release and contraction.
Figure 1. Schematic diagram showing the protein machinery of cardiac Na+–Ca2+ cycling.
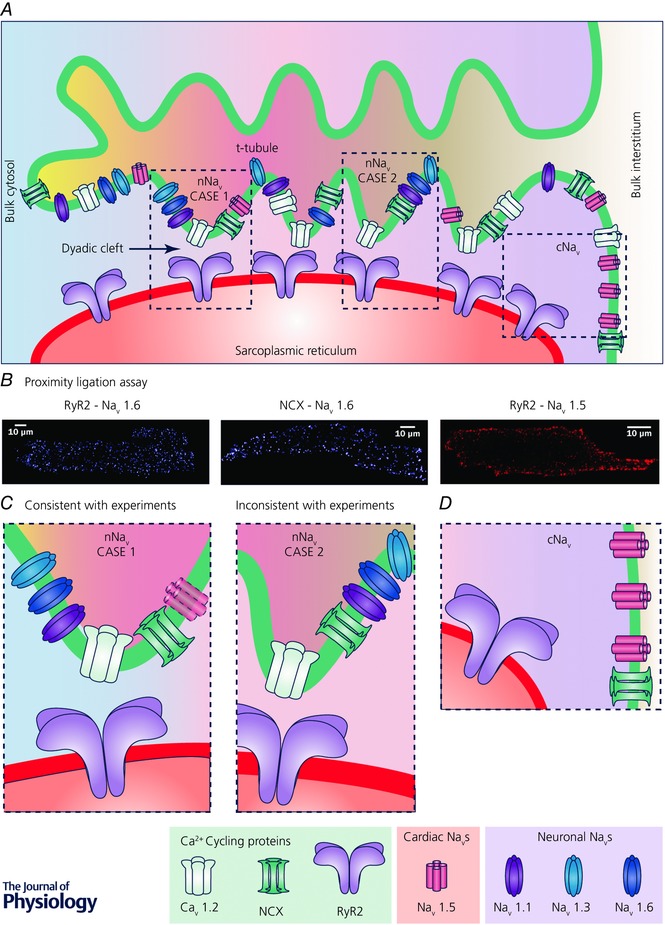
A, schematic diagram of a t‐tubule and associated junctional SR. Microfolds in t‐tubule are depicted based on recent findings (Hong et al. 2014; Lavorato et al. 2015). Different arrangements of Ca2+ cycling proteins and sodium channels are depicted along the t‐tubule. Regions highlighted by the dashed boxes are presented at higher magnification in C and D. Note that differential shading of the interstitial space within the t‐tubule and the cytoplasm within the dyadic cleft indicates local differences in ionic concentrations within these spaces due to their diffusional isolation from the bulk interstitial space and cytoplasm, respectively. B, results from Duolink proximity ligation assays (PLAs) show close association of nNaV isoform NaV1.6 with both RyR2 and NCX throughout murine myocytes, consistent with enrichment of nNaVs in t‐tubules. In contrast, PLA signal corresponding to association between cNaV (NaV1.5) and RyR2 is only observed at the periphery of the cell, consistent with cNaV localization at the lateral membrane. Adapted from Radwański et al. (2016). C, higher magnification views of regions from A showing two possible scenarios of nNaV localization within t‐tubules. Left, case 1, very close association between nNaVs and RyRs, which is consistent with PLA results. A cNaV is depicted faded since experimental results including PLA results argue against cNaV enrichment in t‐tubules. Right, case 2, nNaVs localized to t‐tubules but not very closely associated with RyRs, which is not consistent with PLA results. D, higher magnification view of region from A showing cNaV (NaV1.5) localization at the lateral membrane.
Compartmentation of cardiac sodium–calcium cycling: neuronal Na+ channels in the t‐tubules
In both skeletal and cardiac muscle, the dynamics of Ca2+ cycling are heavily influenced by the spatial organization of the RyRs, relative to the primary source of trigger Ca2+, LCCs (Fabiato, 1983). Whereas in skeletal muscle, sarcolemmal Ca2+ channels (CaV1.1) are mechanically linked to RyRs (RyR1), in cardiac muscle, LCCs (CaV1.2) and RyRs (RyR2) are closely associated in space without any known direct mechanical linkage. Therefore, the 10–12 nm wide dyadic cleft separating the SR membrane containing the RyRs and the sarcolemma at the t‐tubule, containing the LCCs, is of critical importance in determining the functional properties of cardiac Ca2+ cycling (Greenstein et al. 2006; Koh et al. 2006; Cannell et al. 2013). In consequence, the spatial organization of Navs and NCX relative to RyRs and LCCs is likely to be a critical modulator of Na+–Ca2+ cycling. Further underscoring the importance of the spatial organization of Na+ and Ca2+ cycling proteins relative to each other, Sobie and colleagues demonstrated that LCCs allosterically activate NCX in rabbit cardiomyocytes at positive membrane potentials, thereby enabling further augmentation of trigger Ca2+ for SR Ca2+ release by Na+ influx (Sobie et al. 2008).
While the presence of NaVs within t‐tubules is widely accepted, there remains some debate regarding which isoforms are localized there. This question has important implications given the biophysical differences between cNaVs and nNaVs: the TTX‐sensitive nNaVs exhibit more positive voltage dependence of gating, i.e. greater channel availability at positive potentials, as well as more rapid activation and inactivation compared to the TTX‐resistant cNaVs (Fozzard & Hanck, 1996; Maier et al. 2004). In a 2000 study, Moore and colleagues, using wide‐field microscopy, observed no close association between cNaVs and RyRs (Scriven et al. 2000). In subsequent work using confocal microscopy, cNaVs (NaV1.5) were found to be enriched at the intercalated disk whereas nNaVs (NaV1.1, NaV1.3 and NaV1.6) and skeletal muscle NaVs (NaV1.4) were enriched within t‐tubules (Dhar Malhotra et al. 2001; Maier et al. 2002, 2004; Westenbroek et al. 2013; Radwanski et al. 2015). In this context, the aforementioned results highlighting the role of TTX‐sensitive Na+ current in modulating Ca2+ release (Larbig et al. 2010; Torres et al. 2010; Gershome et al. 2011; Radwanski et al. 2013b) argue in favour of nNaVs rather than cNaVs being a part of the Na+–Ca2+ cycling machinery. However, when interpreting these findings, it is important to bear in mind that the resolution of these microscopy techniques is limited by diffraction to 200–300 nm resolution. Some functional validation was provided by measurements made by Brette and Orchard – these investigators examined Na+ current densities in both intact and detubulated rat ventricular myocytes and determined that TTX‐resistant cNaVs (NaV1.5) are predominantly located at the lateral sarcolemma whereas TTX‐sensitive nNaVs are preferentially localized to the t‐tubules (Brette & Orchard, 2006). Furthermore, Lin et al. (2011) using cell‐attached macropatch demonstrated that cNaVs were distributed between the intercalated disk and the midsection of myocytes whereas nNaVs were only observed at the latter location. Interestingly, these investigators also observed functional differences in cNaVs based on their subcellular location highlighting the role of subcellular location in modulating ion channel function: TTX‐resistant Navs (likely to be NaV1.5) along the lateral sarcolemma activated at more positive potentials than those at the intercalated disk suggesting that cellular excitability at physiological resting potentials is largely governed by NaV1.5 at the intercalated disk. A related approach to this problem was reported by Bhargava et al. (2013) who combined scanning ion conductance microscopy and cell‐attached patch clamp to record currents from LCCs as well as clusters of NaVs located within t‐tubules. However, these investigators used sensitivity to 30 μm TTX to confirm the identity of NaVs in their experiments; thus, they did not discriminate between cardiac and neuronal isoforms. It is important to note here that RyRs in cardiac muscle are distributed between the junctional and corbular SR, organized into clusters with uneven gaps (Cabra et al. 2016; Franzini‐Armstrong, 2016). Thus, it is possible that neuronal and/or cardiac NaVs may be present in sufficient density at or near the dyadic cleft interspersed with RyRs to significantly influence Ca2+ cycling.
In the context of characterizing t‐tubular NaVs, novel experimental techniques have proved to be of great value. One such technique is the proximity ligation assay (PLA), which identifies proteins located within 40 nm of each other with extremely high sensitivity (Gullberg & Andersson, 2010). Using PLA in conjunction with confocal immunofluorescence, we recently demonstrated that primarily nNaVs (NaV1.1, NaV1.3 and NaV1.6) and, to a lesser extent, cNaVs (NaV1.5) are closely associated with RyR2 and NCX in t‐tubules in ventricular myocytes isolated from murine hearts (Fig. 1; Radwański et al. 2016). Notably, PLA signal is only generated when colabelled proteins are within ∼40 nm of each other (Gullberg & Andersson, 2010) suggesting very close association between nNaVs and Ca2+ cycling proteins (Fig. 1 C – case 1). And, among the nNaVs, NaV1.6 demonstrated the greatest degree of association with Ca2+ handling proteins. Overall, these results highlight nNaVs and NaV1.6 as potentially important components of nanodomains which comprise the machinery of ECC. However, data from animal models with cardiac‐specific gene‐targeted deletion of nNaVs is necessary to confirm this hypothesis.
Physiological modulation of neuronal Na+ channels
Like cNaVs, nNaVs possess consensus Ca2+/calmodulin‐dependent protein kinase II (CaMKII) phosphorylation sites that correspond to DI–II linker (Marionneau et al. 2012). Therefore, phosphorylation of nNav by CaMKII may account for synergistic interaction between reverse mode NCX and LCC at positive membrane potentials during β‐adrenergic stimulation (Viatchenko‐Karpinski et al. 2005). In fact, recently, we demonstrated CaMKII augmentation of TTX‐sensitive nNaVs during β‐adrenergic stimulation result in an increased intracellular Na+ influx (Fig. 2 A, bottom panel) (Radwański et al. 2016). The notion of CaMKII regulation of Na+ influx is consistent with work in genetic models where CaMKII is either rendered constitutively active (Wagner et al. 2006) or knocked out (Dybkova et al. 2014). Further, there is evidence that augmentation of Na+ influx through nNaV in WT murine hearts may be potentially inotropic by increasing Ca2+ transient amplitude (Kirchhof et al. 2015; Radwanski et al. 2015). However, in a setting of altered RyR2 function, augmentation of nNaV activity may in fact be proarrhythmic (Radwański et al. 2016).
Figure 2. Schematic diagram showing normal and aberrant Na+–Ca2+ cycling.

A, top, during normal Na+–Ca2+ cycling, Na+ influx via nNaVs early in the action potential leads to Ca2+ influx via reverse mode NCX, which in turn, in conjunction with Ca2+ influx via LCCs, leads to SR Ca2+ release via RyRs. Inset shows normal electrocardiogram resulting from normal Na+–Ca2+ cycling. Bottom, during β‐adrenergic stimulation, Na+ influx is enhanced secondary to CaMKII‐mediated phosphorylation of nNaVs. This leads to augmented Ca2+ influx via reverse mode NCX, which in turn leads to enhanced SR Ca2+ release via RyRs. C, pathological elevated Na+ influx via nNaVs results in a larger Ca2+ influx via reverse mode NCX. This, particularly in the presence of elevated diastolic Ca2+ levels or RyR leak, can trigger arrhythmogenic diastolic Ca2+ release. Inset electrocardiogram shows premature beats and arrhythmias triggered by diastolic Ca2+ releases.
Role of neuronal Na+ channels in cardiac arrhythmias
Abnormal NaV function underlies arrhythmias in several pathologies (Ruan et al. 2009). Over‐active NaVs in neurons precipitate seizures, whereas in the heart, they can precipitate triggered arrhythmias. As noted above, nNaV activity is increased during β‐adrenergic stimulation in a CaMKII‐dependent manner (Radwański et al. 2016). Pathologically elevated Na+ influx into the confined space of the t‐tubular junction during late phase 3 of an action potential or during rest (diastole) can precipitate arrhythmias by modulating RyR function via Na+‐dependent signalling mechanisms. For instance, in inherited ryanopathies such as CPVT where RyRs are ‘leaky’ and the junctional cleft is primed with Ca2+, physiological enhancement of nNaV activity by isoproterenol precipitated aberrant diastolic Ca2+ releases and consequent arrhythmias in vivo via an NCX‐mediated mechanism (Radwanski et al. 2015, 2016). In silico studies suggest that pathological accumulation of cytosolic Na+ and Ca2+ facilitate NCX reversal (Armoundas et al. 2003; Radwanski & Poelzing, 2011), thereby facilitating aberrant SR Ca2+ releases. Such Na+‐mediated signalling increases Ca2+ spark frequency through sensitized RyR2 (Radwanski et al. 2015). Likewise, in CPVT cardiomyocytes with enhanced SR Ca2+ load (via acute, conditional overexpression of SERCA2a), selectively slowing nNaV inactivation with β‐pompilidotoxin increased Na+ influx, which in turn, through NCX, triggered aberrant Ca2+ releases and arrhythmias in vivo (Radwanski et al. 2015, 2016). Overall, these results point to nNaVs as important regulators of aberrant Ca2+ release events in such disease states.
Several further examples exist of the arrhythmogenic impact of pathologically increased Na+ influx via nNaVs. In mice lacking the Na+ channel auxiliary subunit β1, a compensatory upregulation of a nNav isoform, NaV1.3, results in higher rates of aberrant diastolic Ca2+ releases in β1‐knockout mice than in their WT littermates (Lin et al. 2014). Likewise, a rat pilocarpine‐induced epilepsy model evidenced increased persistent Na+ current secondary to an upregulation of NaV1.1 (Biet et al. 2015). This enhanced persistent Na+ current, in turn, precipitated cardiac arrhythmias in vivo. In yet another model of epileptic encephalopathy, gain of function mutation in Nav1.6 resulted in frequent aberrant diastolic Ca2+ releases and ventricular arrhythmias upon catecholamine challenge (Frasier et al. 2016). Taken together, these results strongly support a role of enhanced persistent Na+ influx via nNaVs, particularly within Na+–Ca2+ cycling nanodomains in precipitating aberrant diastolic Ca2+ releases and thereby cardiac arrhythmias (Fig. 2 B) in multiple pathologies where intracellular (and particularly dyadic) Na+ and Ca2+ is elevated.
Role of neuronal Na+ channels in the failing heart
Enhanced persistent Na+ influx is widely acknowledged as a contributor to arrhythmogenesis in acquired forms of ryanopathy, such as heart failure (Valdivia et al. 2005; Undrovinas & Maltsev, 2008; Undrovinas et al. 2010; Sossalla & Maier, 2012; Antzelevitch et al. 2014; Makielski, 2016). Undrovinas and colleagues identified NaV1.1 as a significant contributor to persistent Na+ influx in a coronary artery embolization‐mediated canine model of HF (Mishra et al. 2014), highlighting the role of nNaVs in this phenomenon. Likewise, NCX function is pathologically enhanced in failing hearts (Pogwizd & Bers, 2002). Taken together, these findings suggest that arrhythmogenic SR Ca2+ releases in failing hearts may be triggered by Na+‐driven Ca2+ entry. Interestingly, despite the enhanced Na+ influx and NCX function, failing rat hearts do not demonstrate a synergistic interaction between NCX and LCCs to increase ECC as part of CICR during β‐adrenergic stimulation (Viatchenko‐Karpinski et al. 2005). This may reflect the severe disruption of t‐tubules in failing hearts (Li et al. 2015), which contributes to compromised mechanical function and reduced functional reserve. Despite this disruption of the t‐tubular network in failing hearts, the remaining t‐tubules host abnormal functional nanodomains composed of nNavs, NCX and hypersensitive RyRs that facilitate temporal synchronization of aberrant Ca2+ release (Belevych et al. 2012), thereby precipitating cardiac arrhythmias.
An important debate in this context has been about whether the interplay between Na+ and Ca2+ cycling in the failing heart is mediated by Na+‐driven Ca2+ entry via NCX directly triggering SR Ca2+ release or by means of this enhanced Ca2+ influx being taken up into the SR, thereby enhancing SR Ca2+ load. SR Ca2+ load in the failing heart is reduced compared to non‐failing hearts. This is in line with observations that SR Ca2+ load was lowered even when Na+ entry was enhanced through cardiac glycoside treatment (Ho et al. 2011), pointing to Na+‐driven Ca2+ entry via NCX directly triggering SR Ca2+ release as the likely arrhythmia mechanism. However, this does not exclude a role for elevated SR Ca2+ load under other conditions. Thus, structural characterization of Na+–Ca2+ cycling nanodomains in this complex disease state will be crucial to fully understanding this arrhythmia mechanism. Equally, the contribution of nNaVs to arrhythmias needs to be investigated in mathematical models incorporating nano‐scale structural organization as well as in experimental heart failure models.
Neuronal Na+ channels as targets for anti‐arrhythmic therapy
Given that aberrant diastolic Ca2+ releases resulting from altered RyR2 function underlie arrhythmias in a wide range of pathologies, including CPVT(Radwanski et al. 2015, 2016) and ischaemic and non‐ischaemic cardiomyopathy (Belevych et al. 2013), nNaV inhibition may have wide‐ranging therapeutic applications. Such a strategy would also avoid a major pitfall of non‐isoform‐selective NaV inhibition: although initially beneficial in managing Ca2+‐mediated arrhythmias following myocardial infarction (The CAPS investigators, 1986), the loss of excitability resulting from blunting of peak Na+ current proved pro‐arrhythmic, increasing the incidence of sudden arrhythmic death in patients with structural heart disease (Echt et al. 1991; Starmer et al. 1991). Since TTX‐resistant cardiac NaVs (NaV1.5) are primarily responsible for cardiac excitability, selectively targeting nNaV could beneficially lower pathological persistent Na+ influx without any detrimental effects on excitability. Indeed, in mice with CPVT, selective inhibition of nNaVs with riluzole or 4,9‐anhydro‐TTX, a TTX analogue, desynchronized pathological diastolic Ca2+ releases in both isolated myocytes and in intact tissue, and proved potently antiarrhythmic in vivo (Radwanski et al. 2015, 2016). Last but not least, selective silencing of NaV1.6 recapitulated this antiarrhythmic effect in vivo, suggesting that the therapeutic strategy of inhibiting nNaVs could even be further refined to target specific Na+ channel isoforms.
Future directions: the subcellular milieu of Na+ channels
The development of novel, selective therapeutic strategies to prevent Ca2+‐mediated arrhythmias by targeting nNaVs depends on understanding the relationship between structure – the subcellular organization of different nNaV isoforms and their milieu at the nano‐scale, and function – the interplay between Na+ and Ca2+ cycling in the heart. In recent years, we have learned a great deal about the subcellular localization of cNaVs. In an elegant 2011 study, Petitprez and colleagues identified two distinct pools of NaV1.5 within cardiac myocytes, one located at the intercalated disk scaffolded by SAP97, and the other located at the lateral sarcolemma scaffolded by the syntrophin–dystrophin complex (Petitprez et al. 2011). More recently, we used super‐resolution microscopy techniques to identify a subpopulation of intercalated disk‐localized NaV1.5 located within 200 nm of connexin43 gap junctions (Veeraraghavan et al. 2015; Veeraraghavan & Gourdie, 2016). Importantly, the ultrastructural properties of this juxta‐gap junctional membrane region, where the extracellular cleft is just 5–10 nm wide, were shown to be an important modulator of cardiac conduction dependence on the sodium current (Veeraraghavan et al. 2015). Likewise, Delmar and colleagues have utilized super‐resolution microscopy correlated with electron microscopy to identify yet another subpopulation of intercalated disk‐localized NaV1.5 localized to N‐cadherin‐rich sites, where the membranes of adjacent cells are 50–75 nm apart (Agullo‐Pascual et al. 2014; Leo‐Macias et al. 2016). The techniques developed by these investigators could prove very useful in future investigation of the ultrastructural milieu of nNaVs in cardiac myocytes. Perhaps more importantly, the identification of these NaV1.5‐rich nano‐domains suggests that nNaVs may also be similarly organized into nano‐domains with distinct ultrastructural properties and therefore behave in a location‐dependent manner. The hypothesis that the nano‐scale milieu of t‐tubule‐localized NaVs may modulate their function is supported by the work of Hong and colleagues: having previously identified a role for the t‐tubule protein bridging integrator 1 (BIN1) in Ca2+ channel trafficking and clustering at the t‐tubule surface (Hong et al. 2010, 2012), these investigators recently demonstrated that BIN1 promotes micro‐folding of the t‐tubular membrane (Hong et al. 2014; Fu & Hong, 2016). These microfolds in the t‐tubular membrane can trap extracellular ions, thus diffusionally isolating the t‐tubular extracellular clefts from bulk extracellular space (Forssmann & Girardier, 1970; Hong et al. 2014; Lavorato et al. 2015). Importantly, loss of these t‐tubular microfolds in heart failure is associated with impaired contractile function secondary to desynchronized ECC as well as elevated risk of ventricular arrhythmias. Not only will any nNaVs co‐distributed with Ca2+ handling proteins be subject to intra‐ and extra‐cellular ionic concentrations modulated by local ultrastructure, but studies have also demonstrated that NaVs are directly regulated in a Ca2+‐dependent manner (Van Petegem et al. 2012; Wang et al. 2014; Gabelli et al. 2016). Therefore, studies of nNaV localization relative to Ca2+‐handling proteins and their local ultrastructural milieu will be critical in understanding the interplay between Na+ and Ca2+ cycling in health and in disease. Further, both the nNaVs themselves and the structural proteins that generate their ultrastructural niches could represent valuable therapeutic targets in a wide array of disease states.
Conclusions
In summary, the available evidence suggests that Na+ influx through Na+ channels, in particular neuronal Na+ channels (nNavs), contributes both to the triggering of CICR in normal physiology and to arrhythmogenic diastolic Ca2+ release during disease states as diverse as CPVT and heart failure. Importantly, selective inhibition of nNavs shows promise as a therapeutic strategy in such pathologies, further underscoring the importance of the interplay between cardiac Na+ and Ca2+ cycling. Going forward, understanding the subcellular organization of nNavs, particularly in relation to Ca2+ handling proteins, will be vital to elucidating the mechanisms underlying this phenomenon as well as to the development of effective therapies against Ca2+‐induced arrhythmias.
Additional information
Competing interests
None.
Author contributions
R.V., S.G. and P.B.R. contributed equally to the conceptualization of this article. The text was written by R.V. and P.B.R., and revised and edited by all three authors. All authors have approved the final version of the manuscript and agree to be accountable for all aspects of the work. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
This work was supported by National Institutes of Health grants R01‐HL074045 and R01‐HL063043 (to S.G.); K99‐HL127299 (to P.B.R.) and by an American Heart Association Scientist Development Grant 16SDG29870007 (to R.V.).
Biographies
Rengasayee (Sai) Veeraraghavan earned his PhD from the University of Utah's Department of Bioengineering and completed postdoctoral training at the University of Utah's Department of Mathematics and at the Virginia Tech Carilion Research Institute. He is currently a Research Assistant Professor at the Virginia Tech Carilion Research Institute investigating the structural underpinnings of cardiac arrhythmia mechanisms.

Sandor Gyorke earned his PhD from the Sechenov Institute of Evolutionary Physiology and Biochemistry and completed postdoctoral training at the University of Texas Medical Branch. He is a Professor at the Ohio State University and his group investigates cardiac excitation–contraction coupling in health and in disease, particularly mechanisms that govern the gating activity of the cardiac ryanodine receptor channels and control the release of calcium from the sarcoplasmic reticulum.
Przemysław Radwański earned his PharmD from the University of Illinois (Chicago) and his PhD from the University of Utah and completed postdoctoral training at the Ohio State University. He is currently a Research Assistant Professor at the Ohio State University studying the mechanisms by which sodium channels contribute to arrhythmogenic defects in Ca2+ cycling in the heart and developing novel anti‐arrhythmic therapies.
Contributor Information
Rengasayee Veeraraghavan, Email: saiv@vt.edu.
Przemysław B. Radwański, Email: przemyslaw.radwanski@osumc.edu.
References
- Agullo‐Pascual E, Lin X, Leo‐Macias A, Zhang M, Liang FX, Li Z, Pfenniger A, Lubkemeier I, Keegan S, Fenyo D, Willecke K, Rothenberg E & Delmar M (2014). Super‐resolution imaging reveals that loss of the C‐terminus of connexin43 limits microtubule plus‐end capture and NaV1.5 localization at the intercalated disc. Cardiovasc Res 104, 371–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antzelevitch C, Nesterenko V, Shryock JC, Rajamani S, Song Y & Belardinelli L (2014). The role of late I Na in development of cardiac arrhythmias. Handb Exp Pharmacol 221, 137–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armoundas AA, Hobai IA, Tomaselli GF, Winslow RL & O'Rourke B (2003). Role of sodium‐calcium exchanger in modulating the action potential of ventricular myocytes from normal and failing hearts. Circ Res 93, 46–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belevych AE, Radwanski PB, Carnes CA & Gyorke S (2013). ‘Ryanopathy’: causes and manifestations of RyR2 dysfunction in heart failure. Cardiovasc Res 98, 240–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belevych AE, Terentyev D, Terentyeva R, Ho HT, Gyorke I, Bonilla IM, Carnes CA, Billman GE & Gyorke S (2012). Shortened Ca2+ signaling refractoriness underlies cellular arrhythmogenesis in a postinfarction model of sudden cardiac death. Circ Res 110, 569–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bers DM, Barry WH & Despa S (2003). Intracellular Na+ regulation in cardiac myocytes. Cardiovasc Res 57, 897–912. [DOI] [PubMed] [Google Scholar]
- Bhargava A, Lin X, Novak P, Mehta K, Korchev Y, Delmar M & Gorelik J (2013). Super‐resolution scanning patch clamp reveals clustering of functional ion channels in adult ventricular myocyte. Circ Res 112, 1112–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biet M, Morin N, Lessard‐Beaudoin M, Graham RK, Duss S, Gagne J, Sanon NT, Carmant L & Dumaine R (2015). Prolongation of action potential duration and QT interval during epilepsy linked to increased contribution of neuronal sodium channels to cardiac late Na+ current: potential mechanism for sudden death in epilepsy. Circ Arrhythm Electrophysiol 8, 912–920. [DOI] [PubMed] [Google Scholar]
- Blaustein MP & Lederer WJ (1999). Sodium/calcium exchange: its physiological implications. Physiol Rev 79, 763–854. [DOI] [PubMed] [Google Scholar]
- Brette F & Orchard CH (2006). Density and sub‐cellular distribution of cardiac and neuronal sodium channel isoforms in rat ventricular myocytes. Biochem Biophys Res Commun 348, 1163–1166. [DOI] [PubMed] [Google Scholar]
- Cabra V, Murayama T & Samso M (2016). Ultrastructural analysis of self‐associated RyR2s. Biophys J 110, 2651–2662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannell MB, Kong CH, Imtiaz MS & Laver DR (2013). Control of sarcoplasmic reticulum Ca2+ release by stochastic RyR gating within a 3D model of the cardiac dyad and importance of induction decay for CICR termination. Biophys J 104, 2149–2159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen‐Izu Y, Shaw RM, Pitt GS, Yarov‐Yarovoy V, Sack JT, Abriel H, Aldrich RW, Belardinelli L, Cannell MB, Catterall WA, Chazin WJ, Chiamvimonvat N, Deschenes I, Grandi E, Hund TJ, Izu LT, Maier LS, Maltsev VA, Marionneau C, Mohler PJ, Rajamani S, Rasmusson RL, Sobie EA, Clancy CE & Bers DM (2015). Na+ channel function, regulation, structure, trafficking and sequestration. J Physiol 593, 1347–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhar Malhotra J, Chen C, Rivolta I, Abriel H, Malhotra R, Mattei LN, Brosius FC, Kass RS & Isom LL (2001). Characterization of sodium channel α‐ and β‐subunits in rat and mouse cardiac myocytes. Circulation 103, 1303–1310. [DOI] [PubMed] [Google Scholar]
- Dybkova N, Wagner S, Backs J, Hund TJ, Mohler PJ, Sowa T, Nikolaev VO & Maier LS (2014). Tubulin polymerization disrupts cardiac beta‐adrenergic regulation of late INa. Cardiovasc Res 103, 168–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Echt DS, Liebson PR, Mitchell LB, Peters RW, Obias‐Manno D, Barker AH, Arensberg D, Baker A, Friedman L, Greene HL et al (1991). Mortality and morbidity in patients receiving encainide, flecainide, or placebo. The Cardiac Arrhythmia Suppression Trial. N Engl J Med 324, 781–788. [DOI] [PubMed] [Google Scholar]
- Fabiato A (1983). Calcium‐induced release of calcium from the cardiac sarcoplasmic reticulum. Am J Physiol Heart Circ Physiol 245, C1–C14. [DOI] [PubMed] [Google Scholar]
- Forssmann WG & Girardier L (1970). A study of the T system in rat heart. J Cell Biol 44, 1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fozzard HA & Hanck DA (1996). Structure and function of voltage‐dependent sodium channels: comparison of brain II and cardiac isoforms. Physiol Rev 76, 887–926. [DOI] [PubMed] [Google Scholar]
- Franzini‐Armstrong C (2016). Can the arrangement of RyR2 in cardiac muscle be predicted? Biophys J 110, 2563–2565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frasier CR, Wagnon JL, Bao YO, McVeigh LG, Lopez‐Santiago LF, Meisler MH & Isom LL (2016). Cardiac arrhythmia in a mouse model of sodium channel SCN8A epileptic encephalopathy. Proc Natl Acad Sci USA 45, 12838–12843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y & Hong T (2016). BIN1 regulates dynamic t‐tubule membrane. Biochim Biophys Acta 1863, 1839–1847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabelli SB, Yoder JB, Tomaselli GF & Amzel LM (2016). Calmodulin and Ca2+ control of voltage gated Na+ channels. Channels (Austin) 10, 45–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gershome C, Lin E, Kashihara H, Hove‐Madsen L & Tibbits GF (2011). Colocalization of voltage‐gated Na+ channels with the Na+/Ca2+ exchanger in rabbit cardiomyocytes during development. Am J Physiol Heart Circ Physiol 300, H300–H311. [DOI] [PubMed] [Google Scholar]
- Greenstein JL, Hinch R & Winslow RL (2006). Mechanisms of excitation‐contraction coupling in an integrative model of the cardiac ventricular myocyte. Biophys J 90, 77–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gullberg M & Andersson A‐C (2010). Visualization and quantification of protein‐protein interactions in cells and tissues. Nat Methods 7, 1608. [Google Scholar]
- Ho HT, Stevens SC, Terentyeva R, Carnes CA, Terentyev D & Gyorke S (2011). Arrhythmogenic adverse effects of cardiac glycosides are mediated by redox modification of ryanodine receptors. J Physiol 589, 4697–4708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong TT, Smyth JW, Chu KY, Vogan JM, Fong TS, Jensen BC, Fang K, Halushka MK, Russell SD, Colecraft H, Hoopes CW, Ocorr K, Chi NC & Shaw RM (2012). BIN1 is reduced and Cav1.2 trafficking is impaired in human failing cardiomyocytes. Heart Rhythm 9, 812–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong TT, Smyth JW, Gao D, Chu KY, Vogan JM, Fong TS, Jensen BC, Colecraft HM & Shaw RM (2010). BIN1 localizes the L‐type calcium channel to cardiac T‐tubules. PLoS Biol 8, e1000312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong TT, Yang H, Zhang SS, Cho HC, Kalashnikova M, Sun B, Zhang H, Bhargava A, Grabe M, Olgin J, Gorelik J, Marban E, Jan LY & Shaw RM (2014). Cardiac BIN1 folds T‐tubule membrane, controlling ion flux and limiting arrhythmia. Nat Med 20, 624–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirchhof P, Tal T, Fabritz L, Klimas J, Nesher N, Schulte JS, Ehling P, Kanyshkova T, Budde T, Nikol S, Fortmueller L, Stallmeyer B, Muller FU, Schulze‐Bahr E, Schmitz W, Zlotkin E & Kirchhefer U (2015). First report on an inotropic peptide activating tetrodotoxin‐sensitive, “neuronal” sodium currents in the heart. Circ Heart Fail 8, 79–88. [DOI] [PubMed] [Google Scholar]
- Koh X, Srinivasan B, Ching HS & Levchenko A (2006). A 3D Monte Carlo analysis of the role of dyadic space geometry in spark generation. Biophys J 90, 1999–2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larbig R, Torres N, Bridge JH, Goldhaber JI & Philipson KD (2010). Activation of reverse Na+‐Ca2+ exchange by the Na+ current augments the cardiac Ca2+ transient: evidence from NCX knockout mice. J Physiol 588, 3267–3276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavorato M, Huang TQ, Iyer VR, Perni S, Meissner G & Franzini‐Armstrong C (2015). Dyad content is reduced in cardiac myocytes of mice with impaired calmodulin regulation of RyR2. J Muscle Res Cell Motil 36, 205–214. [DOI] [PubMed] [Google Scholar]
- Leblanc N & Hume JR (1990). Sodium current‐induced release of calcium from cardiac sarcoplasmic reticulum. Science 248, 372–376. [DOI] [PubMed] [Google Scholar]
- Leo‐Macias A, Agullo‐Pascual E, Sanchez‐Alonso JL, Keegan S, Lin X, Arcos T, Feng Xia L, Korchev YE, Gorelik J, Fenyo D, Rothenberg E & Delmar M (2016). Nanoscale visualization of functional adhesion/excitability nodes at the intercalated disc. Nat Commun 7, 10342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Lichter JG, Seidel T, Tomaselli GF, Bridge JH & Sachse FB (2015). Cardiac resynchronization therapy reduces subcellular heterogeneity of ryanodine receptors, T‐tubules, and Ca2+ sparks produced by dyssynchronous heart failure. Circ Heart Fail 8, 1105–1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin X, Liu N, Lu J, Zhang J, Anumonwo JM, Isom LL, Fishman GI & Delmar M (2011). Subcellular heterogeneity of sodium current properties in adult cardiac ventricular myocytes. Heart Rhythm 8, 1923–1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin X, O'Malley H, Chen C, Auerbach D, Foster M, Shekhar A, Zhang M, Coetzee W, Jalife J, Fishman GI, Isom L & Delmar M (2014). Scn1b deletion leads to increased tetrodotoxin‐sensitive sodium current, altered intracellular calcium homeostasis and arrhythmias in murine hearts. J Physiol 593, 1389–1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipp P & Niggli E (1994). Sodium current‐induced calcium signals in isolated guinea‐pig ventricular myocytes. J Physiol 474, 439–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier SK, Westenbroek RE, McCormick KA, Curtis R, Scheuer T & Catterall WA (2004). Distinct subcellular localization of different sodium channel α and β subunits in single ventricular myocytes from mouse heart. Circulation 109, 1421–1427. [DOI] [PubMed] [Google Scholar]
- Maier SK, Westenbroek RE, Schenkman KA, Feigl EO, Scheuer T & Catterall WA (2002). An unexpected role for brain‐type sodium channels in coupling of cell surface depolarization to contraction in the heart. Proc Natl Acad Sci USA 99, 4073–4078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makielski JC (2016). Late sodium current: A mechanism for angina, heart failure, and arrhythmia. Trends Cardiovasc Med 26, 115–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marionneau C, Lichti CF, Lindenbaum P, Charpentier F, Nerbonne JM, Townsend RR & Mérot J (2012). Mass spectrometry‐based identification of native cardiac Nav1.5 channel α subunit phosphorylation sites. J Proteome Res 11, 5994–6007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra S, Reznikov V, Maltsev VA, Undrovinas NA, Sabbah HN & Undrovinas A (2014). Contribution of sodium channel neuronal isoform Nav1.1 to late sodium current in ventricular myocytes from failing hearts. J Physiol 593, 1409–1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy E & Eisner DA (2009). Regulation of intracellular and mitochondrial sodium in health and disease. Circ Res 104, 292–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petitprez S, Zmoos AF, Ogrodnik J, Balse E, Raad N, El‐Haou S, Albesa M, Bittihn P, Luther S, Lehnart SE, Hatem SN, Coulombe A & Abriel H (2011). SAP97 and dystrophin macromolecular complexes determine two pools of cardiac sodium channels Nav1.5 in cardiomyocytes. Circ Res 108, 294–304. [DOI] [PubMed] [Google Scholar]
- Philipson KD & Nicoll DA (2000). Sodium‐calcium exchange: a molecular perspective. Annu Rev Physiol 62, 111–133. [DOI] [PubMed] [Google Scholar]
- Pogwizd SM & Bers DM (2002). Na/Ca exchange in heart failure: contractile dysfunction and arrhythmogenesis. Ann N Y Acad Sci 976, 454–465. [DOI] [PubMed] [Google Scholar]
- Radwanski PB, Belevych AE, Brunello L, Carnes CA & Gyorke S (2013a). Store‐dependent deactivation: cooling the chain‐reaction of myocardial calcium signaling. J Mol Cell Cardiol 58, 77–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radwanski PB, Brunello L, Veeraraghavan R, Ho HT, Lou Q, Makara MA, Belevych AE, Anghelescu M, Priori SG, Volpe P, Hund TJ, Janssen PM, Mohler PJ, Bridge JH, Poelzing S & Gyorke S (2015). Neuronal Na+ channel blockade suppresses arrhythmogenic diastolic Ca2+ release. Cardiovasc Res 106, 143–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radwanski PB, Greer‐Short A & Poelzing S (2013b). Inhibition of Na+ channels ameliorates arrhythmias in a drug‐induced model of Andersen‐Tawil syndrome. Heart Rhythm 10, 255–263. [DOI] [PubMed] [Google Scholar]
- Radwański PB, Ho H‐T, Veeraraghavan R, Brunello L, Liu B, Belevych AE, Unudurthi SD, Makara MA, Priori SG, Volpe P, Armoundas AA, Dillmann WH, Knollmann BC, Mohler PJ, Hund TJ & Györke S (2016). Neuronal Na+ channels are integral components of pro‐arrhythmic Na+/Ca2+ signaling nanodomain that promotes cardiac arrhythmias during β‐adrenergic stimulation. JACC Basic Transl Sci 1, 251–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radwanski PB & Poelzing S (2011). NCX is an important determinant for premature ventricular activity in a drug‐induced model of Andersen‐Tawil syndrome. Cardiovasc Res 92, 57–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renaud JF, Kazazoglou T, Lombet A, Chicheportiche R, Jaimovich E, Romey G & Lazdunski M (1983). The Na+ channel in mammalian cardiac cells. Two kinds of tetrodotoxin receptors in rat heart membranes. J Biol Chem 258, 8799–8805. [PubMed] [Google Scholar]
- Ritchie JM & Rogart RB (1977). The binding of saxitoxin and tetrodotoxin to excitable tissue. Rev Physiol Biochem Pharmacol 79, 1–50. [DOI] [PubMed] [Google Scholar]
- Ruan Y, Liu N & Priori SG (2009). Sodium channel mutations and arrhythmias. Nat Rev Cardiol 6, 337–348. [DOI] [PubMed] [Google Scholar]
- Satin J, Kyle JW, Chen M, Bell P, Cribbs LL, Fozzard HA & Rogart RB (1992). A mutant of TTX‐resistant cardiac sodium channels with TTX‐sensitive properties. Science 256, 1202–1205. [DOI] [PubMed] [Google Scholar]
- Scriven DR, Dan P & Moore ED (2000). Distribution of proteins implicated in excitation‐contraction coupling in rat ventricular myocytes. Biophys J 79, 2682–2691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobie EA, Cannell MB & Bridge JH (2008). Allosteric activation of Na+‐Ca2+ exchange by L‐type Ca2+ current augments the trigger flux for SR Ca2+ release in ventricular myocytes. Biophys J 94, L54–L56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sossalla S & Maier LS (2012). Role of ranolazine in angina, heart failure, arrhythmias, and diabetes. Pharmacol Ther 133, 311–323. [DOI] [PubMed] [Google Scholar]
- Starmer CF, Lastra AA, Nesterenko VV & Grant AO (1991). Proarrhythmic response to sodium channel blockade. Theoretical model and numerical experiments. Circulation 84, 1364–1377. [DOI] [PubMed] [Google Scholar]
- The CAPS investigators (1986). The Cardiac Arrhythmia Pilot Study. Am J Cardiol 57, 91–95. [DOI] [PubMed] [Google Scholar]
- Torres NS, Larbig R, Rock A, Goldhaber JI & Bridge JH (2010). Na+ currents are required for efficient excitation‐contraction coupling in rabbit ventricular myocytes: a possible contribution of neuronal Na+ channels. J Physiol 588, 4249–4260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Undrovinas A & Maltsev VA (2008). Late sodium current is a new therapeutic target to improve contractility and rhythm in failing heart. Cardiovasc Hematol Agents Med Chem 6, 348–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Undrovinas NA, Maltsev VA, Belardinelli L, Sabbah HN & Undrovinas A (2010). Late sodium current contributes to diastolic cell Ca2+ accumulation in chronic heart failure. J Physiol Sci 60, 245–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valdivia CR, Chu WW, Pu J, Foell JD, Haworth RA, Wolff MR, Kamp TJ & Makielski JC (2005). Increased late sodium current in myocytes from a canine heart failure model and from failing human heart. J Mol Cell Cardiol 38, 475–483. [DOI] [PubMed] [Google Scholar]
- Van Petegem F, Lobo PA & Ahern CA (2012). Seeing the forest through the trees: towards a unified view on physiological calcium regulation of voltage‐gated sodium channels. Biophys J 103, 2243–2251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veeraraghavan R & Gourdie R (2016). Stochastic optical reconstruction microscopy‐based relative localization analysis (STORM‐RLA) for quantitative nanoscale assessment of spatial protein organization. Mol Biol Cell 27, 3583–3590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veeraraghavan R, Lin J, Hoeker GS, Keener JP, Gourdie RG & Poelzing S (2015). Sodium channels in the Cx43 gap junction perinexus may constitute a cardiac ephapse: an experimental and modeling study. Pflugers Arch 467, 2093–2105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viatchenko‐Karpinski S, Terentyev D, Jenkins LA, Lutherer LO & Gyorke S (2005). Synergistic interactions between Ca2+ entries through L‐type Ca2+ channels and Na+‐Ca2+ exchanger in normal and failing rat heart. J Physiol 567, 493–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner S, Dybkova N, Rasenack EC, Jacobshagen C, Fabritz L, Kirchhof P, Maier SK, Zhang T, Hasenfuss G, Brown JH, Bers DM & Maier LS (2006). Ca2+/calmodulin‐dependent protein kinase II regulates cardiac Na+ channels. J Clin Invest 116, 3127–3138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Chung BC, Yan H, Wang HG, Lee SY & Pitt GS (2014). Structural analyses of Ca2+/CaM interaction with NaV channel C‐termini reveal mechanisms of calcium‐dependent regulation. Nat Commun 5, 4896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westenbroek RE, Bischoff S, Fu Y, Maier SK, Catterall WA & Scheuer T (2013). Localization of sodium channel subtypes in mouse ventricular myocytes using quantitative immunocytochemistry. J Mol Cell Cardiol 64, 69–78. [DOI] [PMC free article] [PubMed] [Google Scholar]