

Abstract
Contraction and relaxation of the heart consume large amounts of energy that need to be replenished by oxidative phosphorylation in mitochondria, and matching energy supply to demand involves the complimentary control of respiration through ADP and Ca2+. In heart failure, an imbalance between ADP and Ca2+ leads to oxidation of mitochondrial pyridine nucleotides, where NADH oxidation may limit ATP production and contractile function, while NADPH oxidation can induce oxidative stress with consecutive maladaptive remodelling. Understanding the complex mechanisms that disturb this finely tuned equilibrium may aid the development of drugs that could ameliorate the progression of heart failure beyond the classical neuroendocrine inhibition.
Keywords: calcium mitochondria, heart failure, mitochondria, reactive oxygen species, redox
Abbreviations
- β‐AR
β‐adrenergic receptor
- DN
dominant negative
- EC
excitation–contraction
- ETC
electron transport chain
- H2O2
hydrogen peroxide
- KO
knock‐out
- MCU
mitochondrial Ca2+ uniporter
- MitoQ
mitoquinone
- NNT
nicotinamide nucleotide transhydrogenase
- ROS
reactive oxygen species
- SR
sarcoplasmic reticulum
Introduction
Chronic heart failure is the most common cause of hospital admissions in Western countries, and its prevalence is expected to increase further. The current medical treatments target in particular the excessive activation of the sympathetic nervous system and the renin–angiotensin–aldosterone system. Beyond these neuroendocrine interventions, few novel targets to treat heart failure have been identified. Over recent years, metabolic aspects to heart failure have gained increasing interest, since in patients with heart failure, the energy stores of the heart are depleted, with decreased ratios of phosphocreatine to adenosine triphosphate (ATP) predicting an adverse outcome (Neubauer, 2007). More recent work suggests that while depletion of phosphocreatine per se may limit maximal exercise capacity, maladaptive remodelling – typically closely associated with the prognosis – is not affected by such an energetic deficit (Lygate et al. 2013). Instead, the accumulation of metabolic intermediates of glycolysis and fatty acid oxidation as well as oxidative stress, which can all induce maladaptive signalling in their own right, may be the more relevant triggers for disease progression than the mere depletion of ATP (or phosphocreatine) stores (Chatham & Young, 2012; Nickel et al. 2013).
In the plasma and hearts of patients with heart failure, the levels of oxidative stress are increased and correlate with left ventricular dysfunction (Belch et al. 1991; Maack et al. 2003). Oxidative stress is an imbalance between the production and detoxification of reactive oxygen species (ROS), and their major sources in the heart are NADPH oxidases, uncoupled nitric oxide synthases and mitochondria (Burgoyne et al. 2012; Nickel et al. 2014). Mitochondria are considered to be the main source of ROS in most cell types and in particular in cardiac myocytes by most (Turrens, 2003; Adam‐Vizi, 2005; Balaban et al. 2005; Hool et al. 2005; Murphy, 2009) but not all authors (Brown & Borutaite, 2012). Oxidative stress impairs excitation–contraction (EC) coupling in cardiac myocytes, causes arrhythmias (Wagner et al. 2013; Yang et al. 2015), activates pro‐hypertrophic signalling (Ago et al. 2008; Erickson et al. 2008) and induces apoptotic and/or necrotic cell death through activation of the permeability transition pore (Halestrap, 2005). In particular, mitochondrial ROS play a causal role in the development and progression of heart failure in response to various pathological stimuli, such as ischaemia–reperfusion, pressure overload and angiotensin II (Matsushima et al. 2006; Dai et al. 2011, 2012). Moreover, mitochondria appear to amplify ROS production from other ROS sources, such as NADPH oxidase, by a mechanism termed ROS‐induced ROS release (Zorov et al. 2000; Aon et al. 2003; Brandes, 2005; Kimura et al. 2005; Dai et al. 2011; Maack & Böhm, 2011). Therefore, understanding the regulation of mitochondrial ROS is key to eventually developing treatments that may prevent or ameliorate heart failure development beyond the inhibition of neuroendocrine activation.
In the past 10 years, we (Nickel et al. 2014, 2015) and others (Aon et al. 2010; Gauthier et al. 2013; Kembro et al. 2013) have developed a concept in which ROS emission from cardiac mitochondria is dynamically regulated by Ca2+ and ADP, with the redox state of mitochondrial pyridine nucleotides as a central level of control. According to this concept, a mismatch between decreased mitochondrial Ca2+ uptake and increased cardiac workload induces net oxidation of NADH, which also oxidizes NADPH and thereby depletes the anti‐oxidative capacity of the matrix, giving rise to excessive emission of H2O2 (Fig. 1). In the following, we will discuss the pathophysiological basis of this concept and address current controversies, future directions of research and potential points of intervention.
Figure 1. Regulation of mitochondrial respiration and redox state by Ca2+ and ADP.
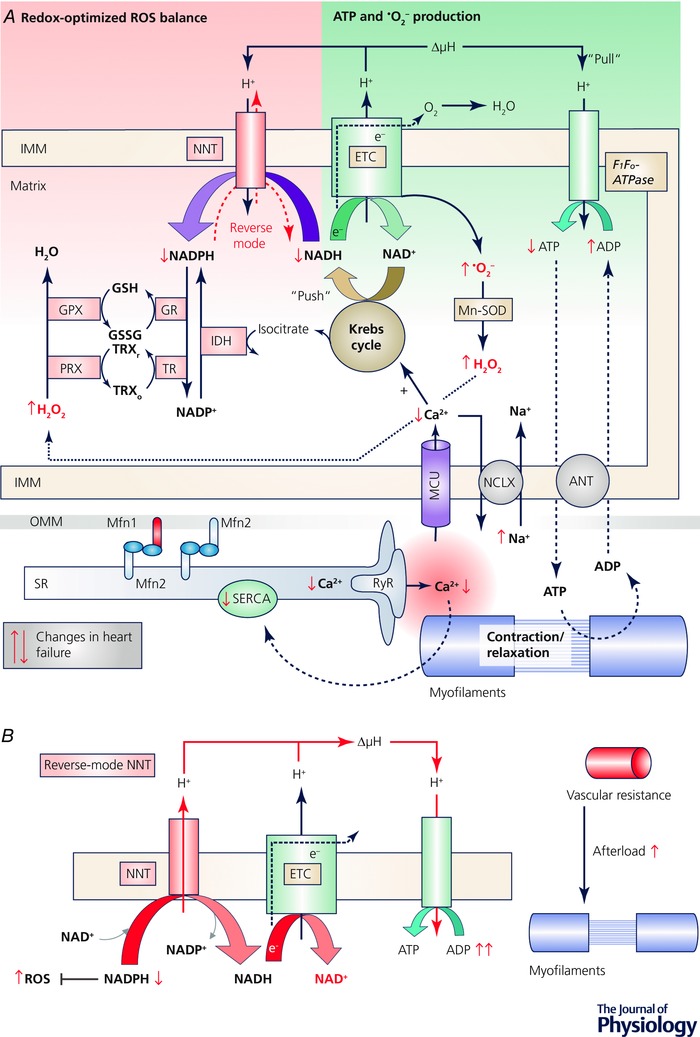
A, integration of mitochondrial ROS production with ROS elimination and the control through ion handling. Abbreviations: ΔμH, proton motive force; ANT, adenine nucleotide translocator; GPX, glutathione peroxidase; GR, glutathione reductase; GSH/GSSG, reduced/oxidized glutathione; IDH, isocitrate dehydrogenase; IMM, inner mitochondrial membrane; MCU, mitochondrial Ca2+ uniporter; Mfn, mitofusin; Mn‐SOD, Mn2+‐dependent superoxide dismutase; NCLX, mitochondrial Na+/Ca2+ (and Li+) exchanger; NNT, nicotinamide nucleotide transhydrogenase; OMM; outer mitochondrial membrane; PRX, peroxiredoxin; TR, thioredoxin reductase; TRXr/o, reduced/oxidized thioredoxin; RyR, ryanodine receptor; SERCA, SR Ca2+ ATPase; SR, sarcoplasmic reticulum. B, reverse‐mode of the nicotinamide nucleotide transhydrogenase (NNT). Through an increase in cardiac afterload, NADH is consumed by the ETC, and the NNT reverses, oxidizing NADPH to regenerate NADH and ATP, but at the cost of the antioxidative capacity, giving rise to ROS emission.
Energy supply and demand matching
During EC coupling, Ca2+ influx via L‐type Ca2+ channels triggers an even greater release of Ca2+ from the sarcoplasmic reticulum (SR), and this Ca2+ binds to troponin C and thereby induces contraction. During diastole, Ca2+ is taken back up into the SR by the SR Ca2+‐ATPase or exported across the cell membrane via the Na+/Ca2+ exchanger (Bers, 2006). A physiological increase in workload is triggered by β‐adrenergic stimulation, elevating the rate and amplitude of cytosolic Ca2+ transients and thereby increasing force generation at the myofilaments. Through this increase of work, ATP is hydrolysed to adenosine diphosphate (ADP), and after shuttling into mitochondria via the adenine nucleotide transporter, ADP activates the F1Fo‐ATPase to regenerate ATP (Fig. 1 A). This hastens electron flux along the electron transport chain (ETC), oxidizing NADH to NAD+ (so‐called ‘pull’ condition; Fig. 1 A). At the same time, Ca2+ is taken up into mitochondria via the mitochondrial Ca2+ uniporter (MCU), where it activates Krebs cycle dehydrogenases to adapt NADH regeneration to ETC‐induced oxidation (‘push’ condition). This dual role of Ca2+, i.e. increasing both energy consumption (‘pull’ on electrons along the ETC) and regeneration (‘push’ of electrons from the Krebs cycle into the ETC), is termed ‘parallel activation’ (Fig. 1 A) (Balaban, 2002; Cortassa et al. 2006).
The discovery of the molecular identity of the MCU (Baughman et al. 2011; De Stefani et al. 2011) has led to the consecutive identification of its various regulatory proteins, such as MICU1, MICU2, MCUR and EMRE, together forming the mitochondrial Ca2+ ‘uniplex’ (Finkel et al. 2015; Kamer & Mootha, 2015) (Kwong (2017), in this issue). Since the affinity of the MCU for Ca2+ is rather low (i.e. in the micromolar range), the close association of mitochondria to the SR creates a Ca2+ microdomain between these two organelles that allows efficient mitochondrial Ca2+ uptake and privileges this pathway over trans‐sarcolemmal Ca2+ influx (Kohlhaas & Maack, 2010, 2013). This microdomain is governed by physical tethers that link both organelles, one of these being mitofusin 2 (Mfn‐2) (de Brito & Scorrano, 2008; Chen et al. 2012; Naon et al. 2016) (Fig. 1 A). Ca2+ is exported from mitochondria by a Na+/Ca2+ (Li+) exchanger (Palty et al. 2010), whose kinetics are much slower than those of uptake via the MCU and therefore Ca2+ accumulates in mitochondria when amplitude and/or rate of cytosolic Ca2+ transients increase (Di Lisa et al. 1993; Maack et al. 2006).
The absolute amounts of Ca2+ taken up by mitochondrial, and whether these are consequential for cytosolic Ca2+ handling, is currently not fully resolved yet. While in other, non‐cardiomyocyte cell types, mitochondrial Ca2+ uptake commonly buffers cytosolic Ca2+ (Rizzuto & Pozzan, 2006), this has been suggested to be the case in cardiac myocytes as well in some models, but refuted by others, as previously reviewed (O'Rourke & Blatter, 2009; Kohlhaas & Maack, 2013; Williams et al. 2013). These controversies are related to technical challenges, but also potential species differences. In this context, it should be considered that in humans, cardiac output during exertion can increase 4‐ to 6‐fold (Chapman et al. 1960; Grimby et al. 1966), while in the mouse, baseline heart rate is already ∼600 beats per minute and increases by a factor of only ∼1.5 during maximal exertion with no increase in blood pressure (Desai et al. 1997; Georgakopoulos & Kass, 2001), indicating that workload variance requiring energetic adaptations is clearly much smaller in mice than in humans. Since mitochondrial Ca2+ uptake is required to match energy supply to demand, particularly during β‐adrenergic stimulation, it is likely that in humans, mitochondrial Ca2+ uptake may play a more important role than in the mouse. In fact, when comparing MCU current density in various organs within the mouse, it is lowest in the heart (Fieni et al. 2012). However, no study has systematically compared cardiac MCU current densities between different species so far. Therefore, the interesting results obtained in mice with genetic inactivation of the MCU (Pan et al. 2013; Kwong et al. 2015; Luongo et al. 2015; Rasmussen et al. 2015; Wu et al. 2015), which will be discussed in more detail further below, have to be extrapolated with some care and may to some extent underestimate the importance of mitochondrial Ca2+ uptake in the human situation.
Regulation of mitochondrial ROS emission
During respiration, the superoxide anion radical (.O2 −) is generated at complexes I and III of the ETC, and is dismutated to hydrogen peroxide (H2O2) by the Mn2+‐dependent superoxide dismutase (Balaban et al. 2005; Murphy, 2009; Chen & Zweier, 2014; Zorov et al. 2014; Murphy et al. 2016). H2O2 is then detoxified by glutathione peroxidase (GPX) and the thioredoxin/peroxiredoxin systems that all require reduced NADPH (Fig. 1 A). The regeneration of NADPH is governed by enzymes that derive their substrates from the Krebs cycle, in particular, isocitrate dehydrogenase and nicotinamide nucleotide transhydrogenase (NNT) (Ying, 2008; Nickel et al. 2015) (Fig. 1 A). Therefore, Ca2+‐induced stimulation of the Krebs cycle not only matches energy supply to demand, but also regenerates the antioxidative capacity of the matrix to prevent the emission of H2O2 during transitions of workload (Kohlhaas et al. 2010). Together with studies from isolated mitochondria, this helped to establish the concept of a ‘redox‐optimized ROS balance’ (Aon et al. 2010), in which the physiological steady state in cardiac mitochondria is tuned to an intermediate redox state that on the one hand prevents excessive formation of ROS at the ETC under highly reduced conditions (Starkov & Fiskum, 2003; Balaban et al. 2005), and on the other prevents depletion of the antioxidative capacity under highly oxidized conditions (Aon et al. 2010; Kohlhaas et al. 2010; Gauthier et al. 2013; Kembro et al. 2013).
Pathological alterations in heart failure
In systolic heart failure, contractile dysfunction is the result of decreased systolic Ca2+ transients, and this is primarily related to decreased SR Ca2+ load secondary to lowered SR Ca2+‐ATPase activity and leaky ryanodine receptors (Bers, 2006) (Fig. 1 A). On the other hand, the cytosolic Na+ concentration is elevated, which activates the reverse‐mode of the sarcolemmal Na+/Ca2+ exchanger to contribute to cytosolic Ca2+ influx during the action potential (Armoundas et al. 2003; Weber et al. 2003) (Fig. 1 A). While this partly compensates for decreased SR Ca2+ release (Weisser‐Thomas et al. 2003), the rather slow trans‐sarcolemmal Na+/Ca2+ exchanger‐mediated Ca2+ influx (Sipido et al. 1997) is less efficient for mitochondrial Ca2+ uptake (Kohlhaas & Maack, 2010). Furthermore, elevated cytosolic Na+ accelerates mitochondrial Ca2+ efflux via the mitochondrial Na+/Ca2+ (Li+) exchanger (Maack et al. 2006; Liu & O'Rourke, 2008; Kohlhaas et al. 2010) (Fig. 1 A). Finally, in human cardiac mitochondria from patients with heart failure, the open probability of the MCU is decreased (Michels et al. 2009). Therefore, deterioration of EC coupling in heart failure compromises the well‐tuned mitochondrial Ca2+ uptake machinery. This has consequences for both energy supply‐and‐demand matching as well as the anti‐oxidative capacity: in isolated cardiac myocytes from a guinea‐pig model of systolic heart failure, NADH and NADPH oxidized, decreasing the amount of reducing equivalents for ATP production at the ETC and provoking the emission of ROS (Fig. 1 A) (Liu & O'Rourke, 2008; Kohlhaas et al. 2010). Since the inhibition of the mitochondrial Na+/Ca2+ (Li+) exchanger prevented NAD(P)H oxidation in myocytes (Liu & O'Rourke, 2008) and the development of heart failure in vivo (Liu et al. 2014), the Na+‐induced redox and energetic mismatch may play a causal role for heart failure progression and possibly represent a potential novel therapeutic target for patients with heart failure. However, this approach has not been followed up by large animal or clinical studies yet.
Mitochondrial transhydrogenase: the yin and yang of antioxidative capacity
Besides contractile dysfunction of the heart, cardiac haemodynamics are further compromised by an elevation of cardiac afterload due to increased systemic vascular resistance (Mason et al. 1964) as a result of the neuroendocrine activation that triggers vasoconstriction (Francis et al. 1984). We recently identified a mechanism in which the mere increase in cardiac afterload provokes mitochondrial ROS emission (Nickel et al. 2015). The NADH and NADPH pools are directly linked by NNT, catalysing the reaction NADH + NADP+ ↔ NADPH + NAD+, which is coupled to the protonmotive force (ΔμH; Fig. 1 A). In energized mitochondria, the forward NNT reaction towards NADPH regeneration is strongly favoured and therefore NNT is considered a key anti‐oxidative enzyme (Rydstrom, 2006). Intriguingly, the most commonly used mouse strain, C57BL/6J, but not C57BL/6N, has a loss‐of‐function mutation in the gene encoding NNT (Nnt), which causes oxidative stress and impairs ATP production in pancreatic islet cells, leading to glucose intolerance in this strain (Toye et al. 2005; Freeman et al. 2006). Furthermore, the Nnt mutation sensitized BL/6J hearts to develop cardiomyopathy upon deletion of Mn2+‐dependent superoxide dismutase (Huang et al. 2006; Kim et al. 2010). In contrast to this anti‐oxidative role, we observed that in response to an increase in cardiac afterload, NNT can reverse its direction when oxidation of NADH at the ETC outweighs its Krebs cycle‐mediated regeneration, consuming NADPH towards NADH and ATP regeneration, but at the cost of the anti‐oxidative capacity (Nickel et al. 2015) (Fig. 1 B). The ensuing oxidative stress accounts for necrosis, left ventricular dysfunction and death during pressure overload, which was prevented in BL/6J mice (with inactivated NNT) or when BL/6N mice (with intact NNT) were treated with SS‐31 (Nickel et al. 2015), a tetrapeptide that accumulates ∼1000‐fold in mitochondria and ameliorates mitochondrial ROS production (Szeto, 2014). While initial experiments suggested that SS‐31 was a ROS scavenger (Zhao et al. 2004), more recent work suggests that it does not have direct anti‐oxidative effects (Brown et al. 2014), but binds to cardiolipin, an essential phospholipid of the inner mitochondrial membrane (Szeto, 2014). This interaction with SS‐31 protects cardiolipin from oxidation and dysfunction, preventing disassembly of the ETC supercomplexes and thereby energetic deficit and mitochondrial ROS production (Szeto, 2014). SS‐31 also improved left ventricular function in the short and long term in a dog model of heart failure (Sabbah et al. 2016) and is currently being tested in phase II clinical trials on patients with systolic (NCT02788747, NCT02914665) and diastolic heart failure (NCT02814097).
Although the reverse‐mode of NNT mediates oxidative stress during pathological cardiac workload, targeting NNT itself may not be a valuable pharmacological concept since the forward mode of NNT is required in most cells and conditions for anti‐oxidative capacity (Rydstrom, 2006), and C57BL/6J mice, which lack a functional NNT, have impaired glucose tolerance and thereby are more prone to develop diabetes. Furthermore, mutations of Nnt in humans are associated with familial glucocorticoid deficiency (Meimaridou et al. 2012). Therefore, inhibiting NNT in humans globally may cause more harm than benefit.
An alternative therapeutic approach to reduce mitochondrial ROS is targeting the antioxidant electron acceptor ubiquinone to mitochondria using lipophilic cations, as with mitoquinone (MitoQ) (Murphy, 2016). MitoQ reduced infarct size after cardiac ischaemia–reperfusion (Adlam et al. 2005) and ameliorated cardiac remodelling in hypertensive rats (Graham et al. 2009). In clinical trials, MitoQ ameliorated hepatic necroinflammation in patients with hepatitis C (Gane et al. 2010), but had neutral results in patients with Parkinson's disease (Snow et al. 2010). MitoQ has not yet been tested in patients with cardiovascular diseases.
Lessons learned from novel MCU‐deficient mouse models for bioenergetic feedback coupling
After the molecular identity of the MCU was resolved, three different mouse models with either constitutive global (Pan et al. 2013; Holmström et al. 2015) or conditional cardiac myocyte‐specific MCU knock‐out (KO) (Kwong et al. 2015; Luongo et al. 2015) or overexpression of a dominant negative (DN‐) MCU (Rasmussen et al. 2015; Wu et al. 2015) were generated (reviewed by Kwong (2017) in this issue). They all have in common that although mitochondria isolated from hearts are unable to rapidly accumulate Ca2+, their cardiac and/or cardiomyocyte function is normal in the absence of β‐adrenergic receptor (β‐AR) stimulation, indicating that MCU‐mediated mitochondrial Ca2+ uptake is required during physiological increases of workload, but not to sustain cardiac function at rest. Interestingly, mice with cardiomyocyte‐specific MCU‐KO had limited capacity to sprint at high speed (Kwong et al. 2015), which was attributed to a delay and/or blunting of the positive inotropic, but not chronotropic, effect of catecholamines in vivo (Kwong et al. 2015; Luongo et al. 2015). In DN‐MCU mice, both the positive inotropic and the chronotropic responses to β‐AR stimulation were blunted (Rasmussen et al. 2015; Wu et al. 2015). In both models, cytosolic Ca2+ concentrations in isolated cardiac myocytes were unaffected at baseline, but increased during systole in response to β‐AR stimulation in the absence of an MCU (Luongo et al. 2015; Rasmussen et al. 2015), in agreement with a study on neonatal cardiac myocytes with siRNA‐induced silencing of the MCU. This may suggest that, at least to some extent, mitochondrial Ca2+ uptake may contribute to cytosolic Ca2+ buffering, which, however, is a subject of continual debate (O'Rourke & Blatter, 2009; Williams et al. 2013). Rasmussen et al. (2015) argue that this increase in cytosolic Ca2+ may increase ATP demand through the activation of EC coupling.
As a metabolic consequence, and in agreement with our previous results on guinea pig cardiac myocytes, in which we acutely inhibited the MCU with Ru360 (Kohlhaas et al. 2010), the β‐AR‐induced stimulation of Krebs cycle‐mediated NAD(P)H production is blunted also by genetic MCU inactivation (Luongo et al. 2015; Wu et al. 2015). In quiescent cells, in which little ATP is consumed and thereby only a small ADP‐induced ‘pull’ on electrons along the ETC occurs, β‐AR stimulation increases NAD(P)H, while MCU inactivation blunts this increase (Fig. 2 B) (Luongo et al. 2015; Wu et al. 2015). In contrast, in working hearts of wild‐type animals, the redox states of NADH/NAD+ and NADPH/NADP+ remained balanced after β‐AR stimulation (Luongo et al. 2015), in agreement with the concept of the ‘parallel activation’ of ‘push’ and ‘pull’ conditions (Fig. 2 A). In MCU‐KO hearts, NADH/NAD+ and NADPH/NADP+ oxidized after β‐AR stimulation (Luongo et al. 2015), indicating that ‘pull’ (through ATP consumption) now outweighs the ‘push’ (in the absence of Ca2+‐induced and Krebs cycle‐mediated NADH production; Figs 1 A and 2 B). These data are in agreement with our previous results on beating guinea pig cardiac myocytes (Kohlhaas et al. 2010).
Figure 2. The impact of deficient mitochondrial Ca2+ uptake on mitochondrial redox state and energetics.
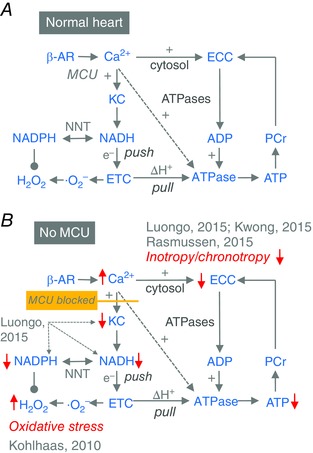
A, control of respiration from upstream by Ca2+, which increases the availability of NADH through the Krebs cycle (KC) to ‘push’ electrons into the electron transport chain (ETC), and from downstream by ADP, which ‘pulls’ electrons (e−) down the ETC to regenerate the proton gradient (ΔH+) that fuels the F1Fo‐ATPase. B, changes that occur in the control of respiration and redox state, according to experimental results by Luongo et al. 2015, Kwong et al. 2015, Rasmussen et al. 2015 and Kohlhaas et al. 2010. Abbreviations: ATPases, energy consumption by myosin ATPase, SR Ca2+‐ATPase, Na+/K+‐ATPase and other ATP‐consuming enzymes; ECC, excitation–contraction coupling; NNT, nicotinamide nucleotide transhydrogenase; PCr, phosphocreatinine. Red arrows indicate increases or decreases.
In experiments on the MCU‐KO model, Ca2+ activated respiration and ATP production in wild‐type mitochondria that respired maximally in the presence of ADP (‘state 3’ respiration) (Kwong et al. 2015; Luongo et al. 2015). However, computational modelling predicted that during state 3 respiration, when respiratory control is dominated by the ADP‐induced acceleration of electron flux along the ETC (‘pull’), Ca2+‐induced activation of the Krebs cycle with increased NADH generation would only increase ATP production by 5% (Cortassa et al. 2003). In addition to this Ca2+‐mediated ‘push’ effect on NADH, however, Ca2+ can also directly activate the F1Fo‐ATPase by ∼2‐fold (Territo et al. 2000). Therefore, the observed Ca2+‐induced (and MCU‐sensitive) acceleration of O2 consumption rates (Kwong et al. 2015; Luongo et al. 2015) are therefore presumably related to both Ca2+‐induced activation the Krebs cycle and of the F1Fo‐ATPase (arrow in Fig. 2 A).
One conflicting observation from studies on genetically modified mice deserves further considerations. While in both the conditional MCU‐KO and the DN‐MCU model, MCU inactivation increased cytosolic Ca2+ transients in unloaded cardiac myocytes, it decreased (and/or delayed) the inotropic response to β‐AR stimulation or an increase in stimulation rate in whole hearts (Kwong et al. 2015; Luongo et al. 2015; Rasmussen et al. 2015). It is unlikely that a decrease of myofilament Ca2+ affinity accounts for this effect, since under baseline conditions, contractility was similar in wild‐type and MCU‐deficient mouse hearts. It may therefore be assumed that the more cardiac workload increases, NADH oxidation may actually limit electron supply of the ETC and thereby ATP production. This would further demand that decreases in ATP affect contraction and relaxation. In fact, the SR Ca2+‐ATPase is the enzyme that is most sensitive to a decrease in the free energy of ATP (Tian & Ingwall, 1996). Furthermore, the parallel oxidation of NADPH/NADP+ may provoke excessive emission of H2O2 from mitochondria (Kohlhaas et al. 2010; Nickel et al. 2015), which may additionally hamper EC coupling (Wagner et al. 2013; Yang et al. 2015). This, however, remains to be addressed by future studies.
Conclusions
There is now ample evidence for the tight and bidirectional interplay between cytosolic ion handling, mitochondrial redox regulation and ATP production in cardiac myocytes. Mitochondrial Ca2+ uptake and its impact on the redox state of pyridine nucleotides is a central element of this interplay (Fig. 1 A), and disease conditions that interfere at this level are likely to induce catastrophic events that induce contractile dysfunction, arrhythmias and maladaptive remodelling through oxidative stress and energetic deficit. The data of the novel mouse models have aided in further characterization of this interplay, but since the dynamic range of cardiac workloads is much smaller in the mouse than in humans, and mitochondrial Ca2+ uptake is required for matching ATP supply to demand particularly during these variations, these results may still underestimate the true effects that may occur in humans when mitochondrial Ca2+ uptake is impaired. Our growing understanding of the pathophysiology of these processes in heart failure may aid the development of novel mitochondria‐directed treatment options to ameliorate disease progression in these patients.
Additional information
Competing interests
None.
Author contributions
All authors have written the manuscript and are responsible for the content. All authors approved the final version of the manuscript and all persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
C.M. is supported by the Deutsche Forschungsgemeinschaft (DFG; Heisenberg Professur; SFB 894; Ma 2528/7‐1), Corona Stiftung and Margret Elisabeth Strauß‐Projektförderung of the Deutsche Herzstiftung.
Biographies
Michael Kohlhaas received his PhD at the Georg August University of Göttingen (Germany) in 2007 with focus on the impact of CaMKIIδ overexpression on the Ca2+ cycling in the cardiomyocyte. In May 2007, he joined the group of Professor Maack at the University of the Saarland in Homburg, Germany and works as a postdoctoral research fellow. His work focuses on pathophysiology of chronic heart failure, Ca2+‐cycling, excitation‐contraction coupling, mitochondrial energetics and oxidative stress in cardiomyocytes.
Alexander Georg Nickel received his PhD at the Technical University of Munich (Germany) in 2009 with focus on amino acid transport in kidney epithelial cells. In October 2009, he joined the group of Professor Maack at the University of the Saarland in Homburg, Germany and works as a postdoctoral research fellow. His work focuses on pathophysiology of chronic heart failure, mitochondrial energetics and oxidative stress in cardiomyocytes.
Christoph Maack received his MD at the University of Cologne (Germany) in 2000. After a post‐doctoral research fellowship with Brian O'Rourke at Johns Hopkins University (2002–2005) he established his working group in the Clinic for Cardiology at the University of the Saarland in Homburg, Germany, where since 2012 he is a Senior Physician and holds a Heisenberg Professorship for Cardiovascular Physiology and Bioenergetics. His work focuses on cellular defects in heart failure, with a special emphasis on the mechanisms of mitochondrial production of reactive oxygen species and the interplay between excitation‐contraction coupling and mitochondrial energetics.

References
- Adam‐Vizi V (2005). Production of reactive oxygen species in brain mitochondria: contribution by electron transport chain and non‐electron transport chain sources. Antioxid Redox Signal 7, 1140–1149. [DOI] [PubMed] [Google Scholar]
- Adlam VJ, Harrison JC, Porteous CM, James AM, Smith RAJ, Murphy MP & Sammut IA (2005). Targeting an antioxidant to mitochondria decreases cardiac ischemia‐reperfusion injury. FASEB J 19, 1088–1095. [DOI] [PubMed] [Google Scholar]
- Ago T, Liu T, Zhai P, Chen W, Li H, Molkentin JD, Vatner SF & Sadoshima J (2008). A redox‐dependent pathway for regulating class II HDACs and cardiac hypertrophy. Cell 133, 978–993. [DOI] [PubMed] [Google Scholar]
- Aon MA, Cortassa S, Marban E & O'Rourke B (2003). Synchronized whole cell oscillations in mitochondrial metabolism triggered by a local release of reactive oxygen species in cardiac myocytes. J Biol Chem 278, 44735–44744. [DOI] [PubMed] [Google Scholar]
- Aon MA, Cortassa S & O'Rourke B (2010). Redox‐optimized ROS balance: a unifying hypothesis. Biochim Biophys Acta 1797, 865–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armoundas AA, Hobai IA, Tomaselli GF, Winslow RL & O'Rourke B (2003). Role of sodium‐calcium exchanger in modulating the action potential of ventricular myocytes from normal and failing hearts. Circ Res 93, 46–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balaban RS (2002). Cardiac energy metabolism homeostasis: role of cytosolic calcium. J Mol Cell Cardiol 34, 1259–1271. [DOI] [PubMed] [Google Scholar]
- Balaban RS, Nemoto S & Finkel T (2005). Mitochondria, oxidants, and aging. Cell 120, 483–495. [DOI] [PubMed] [Google Scholar]
- Baughman JM, Perocchi F, Girgis HS, Plovanich M, Belcher‐Timme CA, Sancak Y, Bao XR, Strittmatter L, Goldberger O, Bogorad RL, Koteliansky V & Mootha VK (2011). Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature 476, 341–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belch JJ, Bridges AB, Scott N & Chopra M (1991). Oxygen free radicals and congestive heart failure. Br Heart J 65, 245–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bers DM (2006). Altered cardiac myocyte Ca regulation in heart failure. Physiology (Bethesda) 21, 380–387. [DOI] [PubMed] [Google Scholar]
- Brandes RP (2005). Triggering mitochondrial radical release: a new function for NADPH oxidases. Hypertension 45, 847–848. [DOI] [PubMed] [Google Scholar]
- Brown DA, Hale SL, Baines CP, del Rio CL, Hamlin RL, Yueyama Y, Kijtawornrat A, Yeh ST, Frasier CR, Stewart LM, Moukdar F, Shaikh SR, Fisher‐Wellman KH, Neufer PD & Kloner RA (2014). Reduction of early reperfusion injury with the mitochondria‐targeting peptide bendavia. J Cardiovasc Pharmacol Ther 19, 121–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown GC & Borutaite V (2012). There is no evidence that mitochondria are the main source of reactive oxygen species in mammalian cells. Mitochondrion 12, 1–4. [DOI] [PubMed] [Google Scholar]
- Burgoyne JR, Mongue‐Din H, Eaton P & Shah AM (2012). Redox signaling in cardiac physiology and pathology. Circ Res 111, 1091–1106. [DOI] [PubMed] [Google Scholar]
- Chapman CB, Fisher JN & Sproule BJ (1960). Behavior of stroke volume at rest and during exercise in human beings. J Clin Invest 39, 1208–1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatham JC & Young ME (2012). Metabolic remodeling in the hypertrophic heart: fuel for thought. Circ Res 111, 666–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y‐R & Zweier JL (2014). Cardiac mitochondria and reactive oxygen species generation. Circ Res 114, 524–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Csordas G, Jowdy C, Schneider TG, Csordas N, Wang W, Liu Y, Kohlhaas M, Meiser M, Bergem S, Nerbonne JM, Dorn GW 2nd & Maack C (2012). Mitofusin 2‐containing mitochondrial‐reticular microdomains direct rapid cardiomyocyte bioenergetic responses via interorganelle Ca2+ crosstalk. Circ Res 111, 863–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortassa S, Aon MA, Marban E, Winslow RL & O'Rourke B (2003). An integrated model of cardiac mitochondrial energy metabolism and calcium dynamics. Biophys J 84, 2734–2755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortassa S, Aon MA, O'Rourke B, Jacques R, Tseng HJ, Marban E & Winslow RL (2006). A computational model integrating electrophysiology, contraction, and mitochondrial bioenergetics in the ventricular myocyte. Biophys J 91, 1564–1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai DF, Hsieh EJ, Liu Y, Chen T, Beyer RP, Chin MT, MacCoss MJ & Rabinovitch PS (2012). Mitochondrial proteome remodelling in pressure overload‐induced heart failure: the role of mitochondrial oxidative stress. Cardiovasc Res 93, 79–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai DF, Johnson SC, Villarin JJ, Chin MT, Nieves‐Cintron M, Chen T, Marcinek DJ, Dorn GW 2nd, Kang YJ, Prolla TA, Santana LF & Rabinovitch PS (2011). Mitochondrial oxidative stress mediates angiotensin II‐induced cardiac hypertrophy and Gαq overexpression‐induced heart failure. Circ Res 108, 837–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Brito OM & Scorrano L (2008). Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature 456, 605–610. [DOI] [PubMed] [Google Scholar]
- Desai KH, Sato R, Schauble E, Barsh GS, Kobilka BK & Bernstein D (1997). Cardiovascular indexes in the mouse at rest and with exercise: new tools to study models of cardiac disease. Am J Physiol Heart Circ Physiol 272, H1053–H1061. [DOI] [PubMed] [Google Scholar]
- De Stefani D, Raffaello A, Teardo E, Szabo I & Rizzuto R (2011). A forty‐kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature 476, 336–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Lisa F, Gambassi G, Spurgeon H & Hansford RG (1993). Intramitochondrial free calcium in cardiac myocytes in relation to dehydrogenase activation. Cardiovasc Res 27, 1840–1844. [DOI] [PubMed] [Google Scholar]
- Erickson JR, Joiner ML, Guan X, Kutschke W, Yang J, Oddis CV, Bartlett RK, Lowe JS, O'Donnell SE, Aykin‐Burns N, Zimmerman MC, Zimmerman K, Ham AJ, Weiss RM, Spitz DR, Shea MA, Colbran RJ, Mohler PJ & Anderson ME (2008). A dynamic pathway for calcium‐independent activation of CaMKII by methionine oxidation. Cell 133, 462–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fieni F, Lee SB, Jan YN & Kirichok Y (2012). Activity of the mitochondrial calcium uniporter varies greatly between tissues. Nat Commun 3, 1317. doi: 10.1038/ncomms2325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finkel T, Menazza S, Holmstrom KM, Parks RJ, Liu J, Sun J, Liu J, Pan X & Murphy E (2015). The ins and outs of mitochondrial calcium. Circ Res 116, 1810–1819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis GS, Goldsmith SR, Levine T, Olivari M & Cohn JN (1984). The neurohumoral axis in congestive heart failure. Ann Intern Med 101, 370–377. [DOI] [PubMed] [Google Scholar]
- Freeman H, Shimomura K, Horner E, Cox RD & Ashcroft FM (2006). Nicotinamide nucleotide transhydrogenase: a key role in insulin secretion. Cell Metab 3, 35–45. [DOI] [PubMed] [Google Scholar]
- Gane EJ, Weilert F, Orr DW, Keogh GF, Gibson M, Lockhart MM, Frampton CM, Taylor KM, Smith RAJ & Murphy MP (2010). The mitochondria‐targeted anti‐oxidant mitoquinone decreases liver damage in a phase II study of hepatitis C patients. Liver Int 30, 1019–1026. [DOI] [PubMed] [Google Scholar]
- Gauthier LD, Greenstein JL, Cortassa S, O'Rourke B & Winslow RL (2013). A computational model of reactive oxygen species and redox balance in cardiac mitochondria. Biophys J 105, 1045–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgakopoulos D & Kass DA (2001). Minimal force‐frequency modulation of inotropy and relaxation of in situ murine heart. J Physiol 534, 535–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham D, Huynh NN, Hamilton CA, Beattie E, Smith RAJ, Cochemé HM, Murphy MP & Dominiczak AF (2009). Mitochondria‐targeted antioxidant MitoQ10 improves endothelial function and attenuates cardiac hypertrophy. Hypertension 54, 322–328. [DOI] [PubMed] [Google Scholar]
- Grimby G, Nilsson NJ & Saltin B (1966). Cardiac output during submaximal and maximal exercise in active middle‐aged athletes. J Appl Physiol 21, 1150–1156. [DOI] [PubMed] [Google Scholar]
- Halestrap A (2005). Biochemistry: a pore way to die. Nature 434, 578–579. [DOI] [PubMed] [Google Scholar]
- Holmström KM, Pan X, Liu JC, Menazza S, Liu J, Nguyen TT, Pan H, Parks RJ, Anderson S, Noguchi A, Springer D, Murphy E & Finkel T (2015). Assessment of cardiac function in mice lacking the mitochondrial calcium uniporter. J Mol Cell Cardiol 85, 178–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hool LC, Di Maria CA, Viola HM & Arthur PG (2005). Role of NAD(P)H oxidase in the regulation of cardiac L‐type Ca2+ channel function during acute hypoxia. Cardiovasc Res 67, 624–635. [DOI] [PubMed] [Google Scholar]
- Huang TT, Naeemuddin M, Elchuri S, Yamaguchi M, Kozy HM, Carlson EJ & Epstein CJ (2006). Genetic modifiers of the phenotype of mice deficient in mitochondrial superoxide dismutase. Hum Mol Genet 15, 1187–1194. [DOI] [PubMed] [Google Scholar]
- Kamer KJ & Mootha VK (2015). The molecular era of the mitochondrial calcium uniporter. Nat Rev Mol Cell Biol 16, 545–553. [DOI] [PubMed] [Google Scholar]
- Kembro JM, Aon MA, Winslow RL, O'Rourke B & Cortassa S (2013). Integrating mitochondrial energetics, redox and ROS metabolic networks: a two‐compartment model. Biophys J 104, 332–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim A, Chen CH, Ursell P & Huang TT (2010). Genetic modifier of mitochondrial superoxide dismutase‐deficient mice delays heart failure and prolongs survival. Mamm Genome 21, 534–542. [DOI] [PubMed] [Google Scholar]
- Kimura S, Zhang G‐X, Nishiyama A, Shokoji T, Yao L, Fan Y‐Y, Rahman M, Suzuki T, Maeta H & Abe Y (2005). Role of NAD(P)H oxidase‐ and mitochondria‐derived reactive oxygen species in cardioprotection of ischemic reperfusion injury by angiotensin II. Hypertension 45, 860–866. [DOI] [PubMed] [Google Scholar]
- Kohlhaas M, Liu T, Knopp A, Zeller T, Ong MF, Böhm M, O'Rourke B & Maack C (2010). Elevated cytosolic Na+ increases mitochondrial formation of reactive oxygen species in failing cardiac myocytes. Circulation 121, 1606–1613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohlhaas M & Maack C (2010). Adverse bioenergetic consequences of Na+‐Ca2+ exchanger‐mediated Ca2+ influx in cardiac myocytes. Circulation 122, 2273–2280. [DOI] [PubMed] [Google Scholar]
- Kohlhaas M & Maack C (2013). Calcium release microdomains and mitochondria. Cardiovasc Res 98, 259–268. [DOI] [PubMed] [Google Scholar]
- Kwong JQ (2017). The mitochondrial calcium uniporter in the heart: energetics and beyond. J Physiol 595, 3743–3751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwong JQ, Lu X, Correll RN, Schwanekamp JA, Vagnozzi RJ, Sargent MA, York AJ, Zhang J, Bers DM & Molkentin JD (2015). The mitochondrial calcium uniporter selectively matches metabolic output to acute contractile stress in the heart. Cell Rep 12, 15–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu T & O'Rourke B (2008). Enhancing mitochondrial Ca2+ uptake in myocytes from failing hearts restores energy supply and demand matching. Circ Res 103, 279–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu T, Takimoto E, Dimaano VL, DeMazumder D, Kettlewell S, Smith G, Sidor A, Abraham TP & O'Rourke B (2014). Inhibiting mitochondrial Na+/Ca2+ exchange prevents sudden death in a guinea pig model of heart failure. Circ Res 115, 44–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luongo TS, Lambert JP, Yuan A, Zhang X, Gross P, Song J, Shanmughapriya S, Gao E, Jain M, Houser SR, Koch WJ, Cheung JY, Madesh M & Elrod JW (2015). The mitochondrial calcium uniporter matches energetic supply with cardiac workload during stress and modulates permeability transition. Cell Rep 12, 23–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lygate CA, Aksentijevic D, Dawson D, Ten Hove M, Phillips D, de Bono JP, Medway DJ, Sebag‐Montefiore LM, Hunyor I, Channon K, Clarke K, Zervou S, Watkins H, Balaban R & Neubauer S (2013). Living without creatine: unchanged exercise capacity and response to chronic myocardial infarction in creatine‐deficient mice. Circ Res 112, 945–955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maack C & Böhm M (2011). Targeting mitochondrial oxidative stress in heart failure: throttling the afterburner. J Am Coll Cardiol 58, 83–86. [DOI] [PubMed] [Google Scholar]
- Maack C, Cortassa S, Aon MA, Ganesan AN, Liu T & O'Rourke B (2006). Elevated cytosolic Na+ decreases mitochondrial Ca2+ uptake during excitation‐contraction coupling and impairs energetic adaptation in cardiac myocytes. Circ Res 99, 172–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maack C, Kartes T, Kilter H, Schafers HJ, Nickenig G, Bohm M & Laufs U (2003). Oxygen free radical release in human failing myocardium is associated with increased activity of rac1‐GTPase and represents a target for statin treatment. Circulation 108, 1567–1574. [DOI] [PubMed] [Google Scholar]
- Mason DT, Braunwald E, Karsh RB & Bullock FA (1964). Studies on Digitalis. X. Effects of ouabain on forearm vascular resistance and venous tone in normal subjects and in patients in heart failure. J Clin Invest 43, 532–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsushima S, Ide T, Yamato M, Matsusaka H, Hattori F, Ikeuchi M, Kubota T, Sunagawa K, Hasegawa Y, Kurihara T, Oikawa S, Kinugawa S & Tsutsui H (2006). Overexpression of mitochondrial peroxiredoxin‐3 prevents left ventricular remodeling and failure after myocardial infarction in mice. Circulation 113, 1779–1786. [DOI] [PubMed] [Google Scholar]
- Meimaridou E, Kowalczyk J, Guasti L, Hughes CR, Wagner F, Frommolt P, Nurnberg P, Mann NP, Banerjee R, Saka HN, Chapple JP, King PJ, Clark AJL & Metherell LA (2012). Mutations in NNT encoding nicotinamide nucleotide transhydrogenase cause familial glucocorticoid deficiency. Nat Genet 44, 740–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michels G, Khan IF, Endres‐Becker J, Rottlaender D, Herzig S, Ruhparwar A, Wahlers T & Hoppe UC (2009). Regulation of the human cardiac mitochondrial Ca2+ uptake by 2 different voltage‐gated Ca2+ channels. Circulation 119, 2435–2443. [DOI] [PubMed] [Google Scholar]
- Murphy E, Ardehali H, Balaban RS, DiLisa F, Dorn GW, Kitsis RN, Otsu K, Ping P, Rizzuto R, Sack MN, Wallace D & Youle RJ (2016). Mitochondrial function, biology, and role in disease. A Scientific Statement From the American Heart Association. Circ Res 118, 1960–1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy MP (2009). How mitochondria produce reactive oxygen species. Biochem J 417, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy MP (2016). Understanding and preventing mitochondrial oxidative damage. Biochem Soc Trans 44, 1219–1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naon D, Zaninello M, Giacomello M, Varanita T, Grespi F, Lakshminaranayan S, Serafini A, Semenzato M, Herkenne S, Hernandez‐Alvarez MI, Zorzano A, De Stefani D, Dorn GW 2nd & Scorrano L (2016). Critical reappraisal confirms that Mitofusin 2 is an endoplasmic reticulum‐mitochondria tether. Proc Natl Acad Sci USA 113, 11249–11254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neubauer S (2007). The failing heart–an engine out of fuel. N Engl J Med 356, 1140–1151. [DOI] [PubMed] [Google Scholar]
- Nickel A, Kohlhaas M & Maack C (2014). Mitochondrial reactive oxygen species production and elimination. J Mol Cell Cardiol 73C, 26–33. [DOI] [PubMed] [Google Scholar]
- Nickel A, Löffler J & Maack C (2013). Myocardial energetics in heart failure. Basic Res Cardiol 108, 358. [DOI] [PubMed] [Google Scholar]
- Nickel AG, von Hardenberg A, Hohl M, Löffler JR, Kohlhaas M, Becker J, Reil JC, Kazakov A, Bonnekoh J, Stadelmaier M, Puhl SL, Wagner M, Bogeski I, Cortassa S, Kappl R, Pasieka B, Lafontaine M, Lancaster CR, Blacker TS, Hall AR, Duchen MR, Kaestner L, Lipp P, Zeller T, Müller C, Knopp A, Laufs U, Böhm M, Hoth M & Maack C (2015). Reversal of mitochondrial transhydrogenase causes oxidative stress in heart failure. Cell Metab 22, 472–484. [DOI] [PubMed] [Google Scholar]
- O'Rourke B & Blatter LA (2009). Mitochondrial Ca2+ uptake: tortoise or hare? J Mol Cell Cardiol 46, 767–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palty R, Silverman WF, Hershfinkel M, Caporale T, Sensi SL, Parnis J, Nolte C, Fishman D, Shoshan‐Barmatz V, Herrmann S, Khananshvili D & Sekler I (2010). NCLX is an essential component of mitochondrial Na+/Ca2+ exchange. Proc Natl Acad Sci USA 107, 436–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan X, Liu J, Nguyen T, Liu C, Sun J, Teng Y, Fergusson MM, Rovira II, Allen M, Springer DA, Aponte AM, Gucek M, Balaban RS, Murphy E & Finkel T (2013). The physiological role of mitochondrial calcium revealed by mice lacking the mitochondrial calcium uniporter. Nat Cell Biol 15, 1464–1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen TP, Wu Y, Joiner M‐L, Koval OM, Wilson NR, Luczak ED, Wang Q, Chen B, Gao Z, Zhu Z, Wagner BA, Soto J, McCormick ML, Kutschke W, Weiss RM, Yu L, Boudreau RL, Abel ED, Zhan F, Spitz DR, Buettner GR, Song L‐S, Zingman LV & Anderson ME (2015). Inhibition of MCU forces extramitochondrial adaptations governing physiological and pathological stress responses in heart. Proc Natl Acad Sci USA 112, 9129–9134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzuto R & Pozzan T (2006). Microdomains of intracellular Ca2+: molecular determinants and functional consequences. Physiol Rev 86, 369–408. [DOI] [PubMed] [Google Scholar]
- Rydstrom J (2006). Mitochondrial NADPH, transhydrogenase and disease. Biochim Biophys Acta 1757, 721–726. [DOI] [PubMed] [Google Scholar]
- Sabbah HN, Gupta RC, Kohli S, Wang M, Hachem S & Zhang K (2016). Chronic therapy with elamipretide (MTP‐131), a novel mitochondria‐targeting peptide, improves left ventricular and mitochondrial function in dogs with advanced heart failure. Circ Heart Fail 9, e002206. doi: 10.1161/CIRCHEARTFAILURE.115.002206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sipido KR, Maes M & Van de Werf F (1997). Low efficiency of Ca2+ entry through the Na+‐Ca2+ exchanger as trigger for Ca2+ release from the sarcoplasmic reticulum. A comparison between L‐type Ca2+ current and reverse‐mode Na+‐Ca2+ exchange. Circ Res 81, 1034–1044. [DOI] [PubMed] [Google Scholar]
- Snow BJ, Rolfe FL, Lockhart MM, Frampton CM, O'Sullivan JD, Fung V, Smith RAJ, Murphy MP & Taylor KM (2010). A double‐blind, placebo‐controlled study to assess the mitochondria‐targeted antioxidant MitoQ as a disease‐modifying therapy in Parkinson's disease. Mov Disord 25, 1670–1674. [DOI] [PubMed] [Google Scholar]
- Starkov AA & Fiskum G (2003). Regulation of brain mitochondrial H2O2 production by membrane potential and NAD(P)H redox state. J Neurochem 86, 1101–1107. [DOI] [PubMed] [Google Scholar]
- Szeto HH (2014). First‐in‐class cardiolipin‐protective compound as a therapeutic agent to restore mitochondrial bioenergetics. Br J Pharmacol 171, 2029–2050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Territo PR, Mootha VK, French SA & Balaban RS (2000). Ca2+ activation of heart mitochondrial oxidative phosphorylation: role of the F0/F1‐ATPase. Am J Physiol Cell Physiol 278, C423–C435. [DOI] [PubMed] [Google Scholar]
- Tian R & Ingwall JS (1996). Energetic basis for reduced contractile reserve in isolated rat hearts. Am J Physiol Heart Circ Physiol 270, H1207–H1216. [DOI] [PubMed] [Google Scholar]
- Toye AA, Lippiat JD, Proks P, Shimomura K, Bentley L, Hugill A, Mijat V, Goldsworthy M, Moir L, Haynes A, Quarterman J, Freeman HC, Ashcroft FM & Cox RD (2005). A genetic and physiological study of impaired glucose homeostasis control in C57BL/6J mice. Diabetologia 48, 675–686. [DOI] [PubMed] [Google Scholar]
- Turrens JF (2003). Mitochondrial formation of reactive oxygen species. J Physiol 552, 335–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner S, Rokita AG, Anderson ME & Maier LS (2013). Redox regulation of sodium and calcium handling. Antioxid Redox Signal 18, 1063–1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber CR, Piacentino V 3rd, Houser SR & Bers DM (2003). Dynamic regulation of sodium/calcium exchange function in human heart failure. Circulation 108, 2224–2229. [DOI] [PubMed] [Google Scholar]
- Weisser‐Thomas J, Piacentino V 3rd, Gaughan JP, Margulies K & Houser SR (2003). Calcium entry via Na/Ca exchange during the action potential directly contributes to contraction of failing human ventricular myocytes. Cardiovasc Res 57, 974–985. [DOI] [PubMed] [Google Scholar]
- Williams GS, Boyman L, Chikando AC, Khairallah RJ & Lederer WJ (2013). Mitochondrial calcium uptake. Proc Natl Acad Sci USA 110, 10479–10486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Rasmussen TP, Koval OM, Joiner ML, Hall DD, Chen B, Luczak ED, Wang Q, Rokita AG, Wehrens XH, Song LS & Anderson ME (2015). The mitochondrial uniporter controls fight or flight heart rate increases. Nat Commun 6, 6081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang KC, Kyle JW, Makielski JC & Dudley SC Jr (2015). Mechanisms of sudden cardiac death: oxidants and metabolism. Circ Res 116, 1937–1955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying W (2008). NAD+/NADH and NADP+/NADPH in cellular functions and cell death: regulation and biological consequences. Antioxid Redox Signal 10, 179–206. [DOI] [PubMed] [Google Scholar]
- Zhao K, Zhao G‐M, Wu D, Soong Y, Birk AV, Schiller PW & Szeto HH (2004). Cell‐permeable peptide antioxidants targeted to inner mitochondrial membrane inhibit mitochondrial swelling, oxidative cell death, and reperfusion injury. J Biol Chem 279, 34682–34690. [DOI] [PubMed] [Google Scholar]
- Zorov DB, Filburn CR, Klotz L‐O, Zweier JL & Sollott SJ (2000). Reactive oxygen species (ROS)‐induced ROS release. A new phenomenon accompanying induction of the mitochondrial permeability transition in cardiac myocytes. J Exp Med 192, 1001–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zorov DB, Juhaszova M & Sollott SJ (2014). Mitochondrial reactive oxygen species (ROS) and ROS‐induced ROS release. Physiol Rev 94, 909–950. [DOI] [PMC free article] [PubMed] [Google Scholar]