

Abstract
Key points
Spontaneous sarcoplasmic reticulum (SR) Ca2+ release events increased in fructose‐rich diet mouse (FRD) myocytes vs. control diet (CD) mice, in the absence of significant changes in SR Ca2+ load.
In HEK293 cells, hyperglycaemia significantly enhanced [3H]ryanodine binding and Ca2+/calmodulin‐dependent protein kinase II (CaMKII) phosphorylation of RyR2‐S2814 residue vs. normoglycaemia. These increases were prevented by CaMKII inhibition.
FRD significantly augmented cardiac apoptosis in WT vs. CD‐WT mice, which was prevented by co‐treatment with the reactive oxygen species scavenger Tempol. Oxidative stress was also increased in FRD‐SR‐autocamide inhibitory peptide (AIP) mice, expressing the SR‐targeted CaMKII inhibitor AIP, without any significant enhancement of apoptosis vs. CD‐SR‐AIP mice.
FRD produced mitochondrial swelling and membrane depolarization in FRD‐WT mice but not in FRD‐S2814A mice, in which the CaMKII site on ryanodine receptor 2 was ablated.
FRD decreased mitochondrial area, mean Feret diameter and the mean distance between SR and the outer mitochondrial membrane vs. CD hearts. This remodelling was prevented in AC3I mice, with cardiac‐targeted CaMKII inhibition.
Abstract
The impact of cardiac apoptosis in pre‐diabetic stages of diabetic cardiomyopathy is unknown. We show that myocytes from fructose‐rich diet (FRD) animals exhibit arrhythmias produced by exacerbated Ca2+/calmodulin‐protein kinase (CaMKII) activity, ryanodine receptor 2 (RyR2) phosphorylation and sarcoplasmic reticulum (SR) Ca2+ leak. We tested the hypothesis that this mechanism also underlies cardiac apoptosis in pre‐diabetes. We generated a pre‐diabetic model in FRD mice. FRD mice showed an increase in oxidative stress, hypertrophy and systolic dysfunction. FRD myocytes exhibited enhanced SR Ca2+ spontaneous events in the absence of SR Ca2+ load alterations vs. control‐diet (CD) myocytes. In HEK293 cells, hyperglycaemia significantly enhanced [3H]ryanodine binding and CaMKII phosphorylation of RyR2‐S2814 residue vs. normoglycaemia. CaMKII inhibition prevented hyperglycaemia‐induced alterations. FRD also evoked cardiac apoptosis in WT mice vs. CD‐WT mice. Co‐treatment with the reactive oxygen species scavenger Tempol prevented FRD‐induced apoptosis in WT mice. In contrast, FRD enhanced oxidative stress but not apoptosis in FRD‐SR‐AIP mice, in which a CaMKII inhibitor is targeted to the SR. FRD produced mitochondrial membrane depolarization in WT mice but not in S2814A mice, in which the CaMKII phosphorylation site on RyR2 was ablated. Furthermore, FRD decreased mitochondrial area, mean Feret diameter and mean SR–mitochondrial distance vs. CD‐WT hearts. This remodelling was prevented in AC3I mice, with cardiac‐targeted CaMKII inhibition. CaMKII phosphorylation of RyR2, SR Ca2+ leak and mitochondrial membrane depolarization are critically involved in the apoptotic pathway of the pre‐diabetic heart. The FRD‐induced decrease in SR–mitochondrial distance is likely to additionally favour Ca2+ transit between the two organelles.
Keywords: apoptosis, CaMKII, diabetes, mitochondria, sarcoplasmic reticulum
Key points
Spontaneous sarcoplasmic reticulum (SR) Ca2+ release events increased in fructose‐rich diet mouse (FRD) myocytes vs. control diet (CD) mice, in the absence of significant changes in SR Ca2+ load.
In HEK293 cells, hyperglycaemia significantly enhanced [3H]ryanodine binding and Ca2+/calmodulin‐dependent protein kinase II (CaMKII) phosphorylation of RyR2‐S2814 residue vs. normoglycaemia. These increases were prevented by CaMKII inhibition.
FRD significantly augmented cardiac apoptosis in WT vs. CD‐WT mice, which was prevented by co‐treatment with the reactive oxygen species scavenger Tempol. Oxidative stress was also increased in FRD‐SR‐autocamide inhibitory peptide (AIP) mice, expressing the SR‐targeted CaMKII inhibitor AIP, without any significant enhancement of apoptosis vs. CD‐SR‐AIP mice.
FRD produced mitochondrial swelling and membrane depolarization in FRD‐WT mice but not in FRD‐S2814A mice, in which the CaMKII site on ryanodine receptor 2 was ablated.
FRD decreased mitochondrial area, mean Feret diameter and the mean distance between SR and the outer mitochondrial membrane vs. CD hearts. This remodelling was prevented in AC3I mice, with cardiac‐targeted CaMKII inhibition.
Abbreviations
- AUC
area under the curve
- Caff
caffeine
- CaMKII
Ca2+/calmodulin‐dependent protein kinase II
- CD
control diet
- DAPI
4′,6‐diamino−2‐fenilindol
- DCM
diabetic cardiomyopathy
- Δψ m
mitochondrial membrane potential
- FRD
fructose‐rich diet
- FS
fractional shortening
- HEK293
human embryonic kidney cells 293
- IFG
impaired fasting glucose
- IGT
impaired glucose tolerance
- IpTGT
intraperitoneal test for glucose tolerance
- LVDD
left ventricular diastolic diameter
- LVFS
left ventricular fractional shortening
- LVMI
left ventricular mass index
- LVSD
left ventricular systolic diameter
- Mfn2
mitofusin 2
- mPTP
membrane permeability transition pore
- NCX
Na+/Ca2+ exchanger
- ROS
reactive oxygen species
- RyR2
ryanodine receptor 2
- SERCA2a
sarcoplasmic/endoplasmic reticulum calcium ATPase
- S2814A
knock‐in mice with substitution of S2814 residue of RyR2 by Ala
- SR
sarcoplasmic reticulum
- TBARS
thiobarbituric acid reactive substances
- T2DM
type 2 diabetes
- TdT
terminal deoxynucleotidyltransferase
- TEM
transmission electron microscopy
- TUNEL
terminal deoxynucleotidyltransferase dUTP nick end labelling
Introduction
Obesity and type 2 diabetes (T2DM) are occurring at epidemic rates worldwide, including developing countries (Pan et al. 1997; Mokdad et al. 2001). This increasing incidence is a result of changes in human behaviour, available nutrition and the adoption of more sedentary lifestyles. The main driving forces for the increased prevalence of insulin resistance are modern westernized diets and patterns of eating associated with the dramatic rises in obesity.
Diabetic cardiomyopathy (DCM) is defined as a disease with structural and functional changes in the myocardium, independent of hypertension, chronic artery disease or any other known cardiac illness. It is caused by metabolic and cellular abnormalities induced by diabetes mellitus, ultimately resulting in heart failure (Lebeche et al. 2008; Dobrin & Lebeche, 2010).
The transition from the early metabolic abnormalities that precede diabetes to overt diabetes, characterized by impaired fasting glucose (IFG) and impaired glucose tolerance (IGT), may take decades. However, current estimates indicate that most individuals with these pre‐diabetic states eventually develop DCM (Nguyen et al. 2010; Wang et al. 2010; Colagiuri, 2011). DCM may be subclinical for a long time, a state that is difficult to diagnose yet may cause future complications. Moreover, during the pre‐diabetic state, the risk of cardiovascular events is already increased and myocardial abnormalities might appear prior to the diagnosis of T2DM. Thus, the earlier identification of cardiac changes in pre‐diabetic/insulin‐resistant patients, the better the results of the strategy used to prevent the evolution to most serious stages of the disease.
Experimental evidence indicates that a critical factor in the transition from compensated to non‐compensated cardiac hypertrophy is myocyte cell loss by apoptosis and necrosis (Whelan et al. 2010). Several reports have focused on the role of myocardial cell death because of the increasing number of studies showing high levels of apoptotic and necrotic cardiomyocytes in experimental models of diabetes (Fiordaliso et al. 2004; Ghosh & Rodrigues, 2006; Ares‐Carrasco et al. 2009) and in cardiac tissue from diabetic patients (Frustaci et al. 2000; Chowdhry et al. 2007; Kuethe et al. 2007). Signals initiating myocardial cell death originate from intrinsic (e.g. mitochondria) and extrinsic (e.g. neurohumoral factors) sources. It is not fully known whether and to what extent one pathway predominates over the other in DCM, although studies using experimental models have recently shown that the mitochondrial‐dependent intrinsic pathway may play a large role (Ghosh et al. 2005; Williamson et al. 2010). Despite the vast bibliography on overt diabetes, apoptosis, its intracellular pathways and its impact in the pre‐diabetic heart are an unexplored field.
Recent findings from our group described that hearts from fructose‐rich diet (FRD) animals develop remarkable cardiac remodeling (Sommese et al. 2016). In addition, ventricular myocytes from FRD animals exhibit cardiac arrhythmogenic events. Our results indicate that Ca2+‐calmodulin protein kinase II (CaMKII)‐dependent phosphorylation of ryanodine receptor 2 (RyR2) is a mechanism underlying the pro‐arrhythmogenic pattern observed at the cellular level in FRD‐treated rats and mice. Moreover, the enhanced activity of CaMKII observed in FRD animals is mainly reliant on an increase in oxidative stress. Both, increased CaMKII activity and oxidative stress have been related to apoptotic and necrotic cell death in different diseases (Yang et al. 2006; Velez Rueda et al. 2012). Indeed, one underlying mechanism proposed for the link between DCM and heart failure is activation of CaMKII (Luo et al. 2013).
We hypothesized that ventricular dysfunction in the FRD model, which is associated with minor metabolic abnormalities and mimics the human pre‐diabetic state of insulin resistance (Sommese et al. 2016), is due at least in part, to the activation of apoptosis via mitochondrial damage initiated by sarcoplasmic reticulum (SR) Ca2+ leak promoted by CaMKII‐dependent phosphorylation of RyR2. This enhanced Ca2+ loss is proposed to drain to the mitochondria via the Ca2+ uniporter, burdening the organelle and igniting the apoptotic pathway. Knowledge of the cellular and molecular aspects underlying the metabolic disturbances on cardiomyocytes in the pre‐diabetic state should be useful in predicting and developing strategies to prevent, avoid or even reverse the structural and functional cardiac consequences of the overt disease.
Methods
Ethical approval
All experiments involving mice were performed according to institutional guidelines and appropriate laws, and were approved by the Faculty of Medicine, University of La Plata Institutional Animal Care and Use Committee (CICUAL no. T‐03‐01‐14). The authors have read and understood the policies and regulations of The Journal of Physiology as outlined by Grundy (2015) and ensured that all experiments complied with these regulations.
Cellular model
Cell culture and transfection
Stable, inducible HEK293 cells expressing mouse RyR2 (Helms et al. 2016) were grown in low glucose Dulbecco's modified Eagle medium (100 mg dl−1; Life Technologies, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum, 100 U ml−1 penicillin and 100 μg ml−1 streptomycin on 100‐mm tissue culture dishes. RyR2 expression was induced by adding 1 μg ml−1 tetracycline to the culture medium. At this point, cells were maintained either in low glucose or high glucose medium (450 mg dl−1; Life Technologies); 1 μm KN92 (Calbiochem, Billerica, MA, USA) or KN93 (Enzo Life Sciences, Plymouth Meeting, PA, USA) was also added to the culture medium as indicated. Forty‐eight hours after induction, cells were washed twice with PBS, then scraped from the dish and solubilized in 200 μL of lysis buffer, containing 25 mm Tris (pH 7.4), 137 mm NaCl, 1% CHAPS, 0.5% egg yolk phosphatidylcholine (Sigma, St Louis, MO, USA), 2.5 mm dithiothreitol and protease inhibitors (2 μm leupeptin, 100 μm phenylmethylsulphonyl fluoride, 500 μm benzamidine, 100 nm aprotinin). The cells were incubated at 4°C for 1 h with rotation. Finally, the lysates were centrifuged at 14 000 r.p.m. for 10 min and the supernatants were collected. Protein concentrations were determined with the Bradford method (Bio‐Rad, Hercules, CA, USA).
Animal model and protocols
Male C57bl/6 mice (wild type, WT) were divided into two groups: a control group, fed with a standard commercial diet, control diet (CD); and a fructose group, which received the same diet plus 10% (w/v) fructose in the drinking water for 3 weeks, fructose‐rich diet (FRD). This protocol model has been proved to generate a useful pre‐diabetic model (Alzugaray et al. 2009; Felice et al. 2014; Sommese et al. 2016).
Transgenic mice with cardiomyocite‐delimited transgenic expression of SR‐targeted CaMKII autocamide inhibitory peptide, AIP (SR‐AIP) (Ji et al. 2003) were divided into three groups: (1) a control group (CD) fed with a standard commercial diet; (2) a fructose group (FRD) which received the same diet plus 10% (w/v) fructose in the drinking water; and (3) 10% (w/v) fructose plus 0.8 mm 4‐hydroxy‐2,2,6,6‐tetramethylpiperidin−1‐oxyl (Tempol, used as an antioxidant) (FRD+T). In addition, knock‐in male mice in which the S2814 site of RyR2 was replaced by alanine (S2814A mice), to genetically inhibit RyR2 phosphorylation by CaMKII (Chelu et al. 2009), were divided in two groups, CD and FRD. Moreover, transgenic mice with cardiomyocyte‐delimited transgenic expression of a CaMKII inhibitory peptide (AC3I) (Zhang et al. 2005) were also divided and treated as the S2814A mice. These mice, SR‐AIP, S2814A and AC3I, were generously supplied by Drs Marcia Kaetzel and John Dedman (Cincinnati, OH, USA), Dr Xander Wehrens (Houston, TX, USA) and Dr Mark Anderson (Baltimore, MD, USA), respectively, and reproduced and genotyped in our laboratory. All treatments were performed for 3 weeks and the animals were 4 months old at the time of the experiments.
After the treatment, animals were weighed, systolic blood pressure was measured by the tail‐cuff method, and echocardiography and glucose determination were performed. Animals were then anaesthetized with an intraperitoneal injection of ketamine/diazepam (100 and 5 mg kg−1, respectively). Central thoracotomy and heart excision were performed immediately after phase III anaesthesia was reached, verified by the loss of pedal withdrawal reflex. At this moment, tibia length was measured and the hearts were assigned for biochemical studies, immunohistochemical staining, electron microscopy, reactive oxygen species (ROS) production determinations, or myocyte isolation for cytosolic Ca2+ and mitochondrial membrane potential measurements. Some hearts were submitted to mitochondrial isolation for swelling experiments.
Glucose determinations
Before the animals were killed, blood samples were drawn from the tail vein to measure plasma glucose levels (One Touch Ultra, Johnson & Johnson, USA) and an intraperitoneal test for glucose tolerance, IpTGT, expressed as area under the curve (AUC) was performed; both determinations were performed after a 12 h fasting period, as previously described (Sommese et al. 2016). A significant difference on AUC between CD and FRD animals was considered an index of insulin deficiency inherent of impaired glucose tolerance status.
Transthoracic echocardiography
Echocardiographic examinations were performed using a 14 MHz linear transducer (Toshiba Nemio XG, Tokyo, Japan). After bidimensional short‐axis images of the left ventricle were obtained, M‐mode tracings were recorded (Lang et al. 2005). Measurements from three consecutive cardiac cycles were made by a trained operator blinded to the genotype of the animals.
[3H]Ryanodine binding assays
Binding assays were carried out following a modified version of a protocol previously described (Helms et al. 2016). Binding mixtures were prepared containing 50 μg of protein from cell lysates, 0.2 m KCl, 20 mm Na‐Hepes (pH 7.4), 6.5 nm [3H]ryanodine (Perkin Elmer, Boston, MA, USA) and enough CaCl2 to set free [Ca2+] between 100 nm and 100 μm. EGTA (1 mm) was used to buffer Ca2+. The Ca2+/EGTA ratio for these solutions was determined using MaxChelator (WEBMAXCLITE v1.15, http://maxchelator.stanford.edu/webmaxc/webmaxclite115.htm). The binding reactions were incubated for 2 h at 37°C, then filtered through Whatman GF/B filters presoaked with 5% polyethilenimine and washed three times with 5 ml of distilled water in a Brandel M24‐R Harvester. Non‐specific binding was determined in the presence of 20 μm unlabelled ryanodine (MP Biomedicals). [3H]Ryanodine binding was determined by liquid scintillation. Hill's equation was used to determine the maximum [3H]ryanodine binding and the EC50 in Origin 9 (Origin Lab, Northampton, MA, USA).
Myocyte isolation
Mice were anaesthetized (100 mg kg−1 of ketamine) and submitted to analgesia (5 mg kg−1 of diazepam) before being killed. Immediately after plane three of phase III of anaesthesia was verified by loss of corneal reflex and the appearance of slow deep diaphragmatic breathing, central thoracotomy and heart excision were performed. Myocytes were isolated by enzymatic digestion as previously described (Palomeque et al. 2009). Briefly, the hearts were attached via aorta to a cannula, excised and mounted in a Langendorff apparatus. Hearts were retrogradely perfused at 37°C at a constant coronary flow with a Krebs–Henseleit solution of the following composition (mm): 146.2 NaCl, 1.0 CaCl2, 10.0 Hepes, 0.35 NaH2PO4, 1.05 MgSO4, 10.0 glucose (pH adjusted to 7.4 with NaOH). The solution was continuously bubbled with 100% O2. After a stabilization period of 5 min, perfusion was switched to a nominally Ca2+‐free Krebs–Henseleit solution with EGTA for 4 min. Hearts were then perfused with collagenase (118 u ml−1), 0.1 mg ml−1 proteinase and 1% BSA, in Krebs–Henseleit solution containing 50 μm CaCl2. Perfusion continued until hearts became flaccid (15–20 min).
Ca2+ i measurements
Mouse cardiac isolated myocytes were loaded with Fura‐2/AM (10 μmol l−1 for 15 min). Ca2+ i fluorescence was measured with an epifluorescence system (Ion Optix, Milton, MA, USA), as described (Palomeque et al. 2009). Briefly, dye‐loaded cells were placed in a chamber on the stage of an inverted microscope (Nikon TE 2000‐U) and continuously perfused with a Hepes‐buffered solution at a constant flow of 1 ml min−1 at room temperature (20–22°C). Myocytes were stimulated via two platinum electrodes on either side of the bath at 0.5 Hz. Fura‐2 fluorescence was taken as an index of Ca2+ i.
Myocytes were stimulated at 0.5 Hz until stabilization. A caffeine pulse (15 mm) was rapidly applied after stabilization in the absence of field electrical stimulation, to assess SR Ca2+ load. The caffeine contracture decay rate constant (k Caff) was used to estimate the velocity of Ca2+ extrusion by the Na+/Ca2+ exchanger (NCX). SERCA2a activity (K SERCA2a) was estimated by subtracting k caff (1/τ of the caffeine transient decay) from kt (1/τ) of the systolic Ca2+ transient decay (Diaz et al. 2004). These estimations assume that decay of the systolic Ca2+ transient is contributed by a combination of SR and surface membrane fluxes, whereas the SR does not contribute to the decay of the caffeine response. Fluorescence data were stored for an off‐line analysis (ION WIZARD fluorescence analysis software).
Confocal imaging
Confocal images of Ca2+ sparks, waves and spontaneous contractile activity were taken in line scan mode. Cells were loaded with 10 μm Fluo‐3 AM for 20 min at room temperature excited with a 488 nm (argon laser) and fluorescence was collected at >515 nm (Mazzocchi et al. 2016). Each image consisted of 512 line scans obtained at 4 ms intervals.
Data were visualized using Leica Application Suite and Ca2+ sparks were measured using the ‘Sparkmaster’ plugin for ImageJ. Sparks and waves were obtained in quiescent cells. Spark frequency was expressed in units (100 μm)−1 s−1. Spark‐mediated Ca2+ leak from the SR was defined as spark frequency × spark mass. Spark mass, was calculated as spark amplitude × full width half maximum × full duration half maximum (Biesmans et al. 2011).
ROS determinations: lipid peroxidation
Lipid peroxidation was determined by measuring the rate of production of thiobarbituric acid reactive substances (TBARS), expressed as nmol mg−1 protein (Sommese et al. 2016). Heart homogenates were centrifuged at 2000 g for 10 min. Supernatants (0.5 ml) were mixed with 1.5 ml trichloroacetic acid (30%, w/v) and 0.5 ml water, followed by boiling for 15 min. After cooling, absorbance was determined spectrophotometrically at 535 nm, using an ε value of 1.56 × 105 (mmol l−1)−1 cm−1.
Western blotting
Hearts were freeze‐clamped, pulverized and processed as previously described (Sommese et al. 2016). Briefly, 0.1 g of tissue was homogenized in four volumes of lysis buffer (in mmol l−1: 20 sodium glycerolphosphate, 20 NaF, 1 EGTA, 2 EDTA, 0.2 Na2VO4, 2 dithiothreitol, 10 benzamide, 1 phenylmethylsulfonyl fluoride, 0.001 pepstatin, 1% Igepal, 0.01% Triton and 0.048 mg ml−1 leupeptin). Protein was measured by the Bradford method using BSA as standard. Lysates (∼90 μg of total protein) were separated per gel line in 10% SDS polyacrylamide gel (Mundina‐Weilenmann et al. 1996) and transferred to polyvinylidene difluoride (PVDF) membranes. Blots were probed overnight with the following primary antibodies: Bax (1:2000, Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA) and Bcl‐2 (1:2000, Santa Cruz). GAPDH signals were used to normalize the signal intensity of the different proteins. Secondary antibodies were used as appropriate, goat anti‐mouse‐HRP (1:15 000, Santa Cruz) or goat anti‐rabbit‐HRP (1:15 000, Santa Cruz).
Isolated mitochondria and fractions were processed as described above after homogenization. As primary antibody, COX1 (1:1000, Santa Cruz) was used to evaluate the purity of the isolation. Immunoreactivity was visualized by a peroxidase‐based chemiluminescence detection kit (Millipore, Billerica, MA, USA) using a Chemidoc Imaging system (Bio‐Rad). The signal intensity of the bands in the immunoblots was quantified by densitometry, using Image J software (NIH, Bethesda, MD, USA).
In total, 50 μg of protein from HEK293 cell lysates was suspended in Laemmli buffer and separated by SDS‐PAGE in 4–20% TGX precast gels (Bio‐Rad). Proteins were then transferred to PVDF membranes using the iblot2 transfer system (Thermo Fisher Scientific Inc., Waltham, MA, USA). Membranes were probed using the ibind flex system (ThermoFisher) with the following primary antibodies: anti‐RyR (1:2000, ThermoFisher), pS2808‐RyR (1:5000, Badrilla, Leeds, UK) and pS2814‐RyR (1:5,000, Badrilla). Secondary antibodies, used as appropriate, were goat anti‐mouse‐HRP (1:1000, ThermoFisher) or goat anti‐rabbit‐HRP (1:2000, ThermoFisher). Membranes were developed using SuperSignal Femto ECL reagent (ThermoFisher) and imaged with a ChemiDoc MP apparatus (Bio‐Rad). Band intensity was quantified with ImageLab (Bio‐Rad).
Isolation of mouse heart mitochondria
Mitochondria from four to six mouse hearts were isolated by differential centrifugation, according to a modified method described previously (Pardo et al. 2015). Briefly, hearts were rapidly excised from pentobarbital‐anaesthetized rats and placed in an ice‐cold isolation buffer containing (in mm): 75 sucrose, 225 mannitol and 0.01 EGTA, pH 7.4. After both atria and right ventricle were removed, the remaining left ventricle was homogenized manually with a Dounce homogenizer (∼20 strokes) in the presence of proteinase (0.8 mg in 5 ml of isolation buffer, Sigma). Homogenized tissue was centrifuged for 5 min at 480 g (4°C), and the pellet containing unbroken cells and nuclei was discarded. The resulting supernatant containing the mitochondrial fraction was further centrifuged at 7700 g (three times for 5 min), and the final pellet was resuspended in isolation buffer with no EGTA and further centrifuged at 7700 g for 5 min. Protein concentration of the mitochondrial suspension was determined as described above.
Mitochondrial swelling determination
Mitochondrial swelling was measured as a decrease in the 90 degrees light‐scattering signal induced by the addition of either 20, 100 or 200 μm CaCl2, which promotes the influx of solutes through the opened mitochondrial membrane permeability transition pore (mPTP) and decreases light scattering (Pardo et al. 2015). After 5 min of preincubation at 37°C in a medium containing (in mmol l−1) 120 KCl, 20 MOPS, 10 Tris‐HCl and 5 KH2PO4, pH 7.4, the different CaCl2 concentrations were added to induce mPTP opening. The decrease in light scattering was detected with a temperature‐controlled Aminco Bowman Series 2 spectrofluorometer operating with continuous stirring at excitation and emission wavelengths of 520 nm. Light‐scattering decrease was calculated for each sample as the difference between the values before and after the addition of CaCl2. Each determination was made in triplicate.
Tissue preparation and electron microscopy
Isolated mitochondria and samples of 1 mm2 excised from the left ventricles were harvested in 0.1 m PBS (pH 7.4) and fixed with 5% glutaraldehyde at 4°C (Pelco International, CA, USA) in 0.1 m sodium cacodylate buffer at 10°C. Each set was washed in the same buffer, postfixed in 1% OsO4 for 1 h at room temperature, dehydrated in a graded acetone series and embedded in low‐viscosity epoxy resin (Pelco International) as described previously (Spurr, 1969). Polymerization was performed for 48 h at 70°C. Ultrathin sections with interference colour grey were cut by an ultramicrotome (Ultracut R; Leica, Vienna, Austria), mounted on grids, and stained with uranyl acetate and lead citrate (Reynolds, 1963). Grids were examined by transmission electron microscopy (TEM) (model 900; Zeiss, Jena, Germany) with a Gatan digital camera (model Orius SC 1000).
TUNEL technique
The terminal deoxynucleotidyltransferase dUTP nick end labelling (TUNEL) technique was used to study apoptosis, using the Tunel In situ Cell Death Detection Kit, TMR red (Roche, Indianapolis, IN, USA), as previously described (Salas et al. 2010). Briefly, sections were deparaffinized, dehydrated and incubated with proteinase K for antigen retrieval. After washing with PBS containing 0.5% Tween 20 (Merck, Schuchardt OHG, Hohenbrunn, Germany), slides were incubated with the reaction mixture containing modified nucleotides (TMR‐dUTP) and the enzyme terminal deoxynucleotidyltransferase (TdT) that catalyses the template‐independent polymerization of deoxyribonucleotides to the 3′‐end of single‐ and double‐stranded DNA. After washing, nuclei were counterstained with 5 μg ml−1 of DAPI (6‐diamidino‐2‐phenylindole; Invitrogen Life Technologies, Eugene, OR, USA) following the manufacturer's protocol. Coverslips were mounted on slides using the aqueous medium Reagent FluoroSave (Calbiochem, La Jolla, CA, USA) and then examined under confocal microscopy (FV1000, Olympus Co., Tokyo, Japan). The TUNEL reaction mixture replaced by the label solution of the kit was used as a negative control.
Collagen determination
Collagen evaluation was done in sections stained with the Picrosirius Red technique and viewed with polarized light (Montes, 1996). For this, sections were deparaffinized, hydrated through graded ethanol and stained for 1 h in a 0.1% solution of Sirius Red (Direct Red 80, Aldrich, Milwaukee, WI, USA) dissolved in aqueous saturated picric acid. Sections were then rapidly washed in running tap water and counterstained with Harris haematoxylin (Montes, 1996). A conventional optical microscope (BX53 Olympus microscope) with a strong light source (halogen lamp), an analyser (U‐ANT Olympus) and a polarizer (U‐POT Olympus) were used to study the birefringence of the stained collagen.
Morphometry analysis
For analysing TUNEL and Picrosirius Red‐stained slides, cellSens Dimension software (v1.7 Olympus) was used. For morphometric determinations of TEM images, ImagePro Plus software (v6.3 Media Cybernetics, Rockville, MD, USA) was used. Confocal images of slides stained with the TUNEL technique were analysed per fluorophore channel (Texas Red, red channel; DAPI, blue channel). The manual threshold option of the Count and Measure menu was used to select the range of both colours. Once selected, identified cells were counted per image. At least 10 images representing each about 4 × 105 μm2 of ventricle tissue were analysed per sample. Results were expressed as the ratio between apoptotic and normal cells. Samples stained with the Picrosirus Red technique were analysed in the same way but using a unique image to select both types of collagen, I and III, selecting the range yellow‐to‐red for the former and a range of greens for the latter. The area occupied by both structures was measured and their ratio expressed.
TEM images were used to measure different morphometric parameters. Low magnification images (12 000×) were used to measure sarcomere length, myofibril width and mitochondrial characteristics [area and mean Ferret diameter (which reports the shortest caliper – Feret – length). Higher magnification images (85 000×) were used to establish the separation between mitochondrial membrane and SR. For this, skeletonization of the images allowed us to determine the central line of both membrane structures. Lines perpendicular to both membranes were then drawn and their length was measured. At least one line every 20 nm of membrane length was drawn. Measurements of all lines were then averaged.
Statistics
Continuous variables were expressed as mean ± SEM and were evaluated with either unpaired Student t test or ANOVA followed by Tukey's post hoc test, when comparison among different groups was performed. A P value <0.05 was considered significant.
Results
Impaired glucose tolerance induced by fructose‐rich diet
FRD‐fed WT mice showed a significant increase in the area under the curve of serum glucose after intraperitoneal glucose administration, without significant alterations in fasting glycaemia, as well as body weight, tibia length and blood pressure when compared to CD mice (Table 1A). These findings are consistent with a pre‐diabetic state rather than an overt T2DM, and allowed for excluding effects at the cellular and organ level other than those produced by impaired glucose tolerance. In agreement with previously reported results (Sommese et al. 2016), mice treated with FRD showed a significant increase in ROS, indicated by the increase in TBARS. Results from echocardiography revealed hypertrophy and systolic dysfunction in FRD vs. DC mice, respectively (Table 1B).
Table 1A.
General characterization of the fructose‐rich diet model
| Treatment | ||
|---|---|---|
| Parameter | Control diet | Fructose‐rich diet |
| n | 9–23 | 9–17 |
| Body weight (g) | 31.4 ± 1.2 | 29.5 ± 1.1 |
| Tibia length (mm) | 17.7 ± 0.5 | 17.9 ± 0.0 |
| Blood pressure (mmHg) | 122.9 ± 1.5 | 122.7 ± 1.7 |
| FG (mg dl−1) | 132.9 ± 5.2 | 139.9 ± 2.9 |
| AUC [mmol glucosa (l.min)−1] | 970.3 ± 9 | 1112.6 ± 57* |
| Calorie intake (kcal day−1) | 9.0 ± 0.2 | 12.3 ± 0.4** |
| TBARS (%) | 100.0 ± 27.4 | 188.2 ± 13.2 |
Fructose‐rich diet (FRD) induces a significant increment in the area under the curve (AUC) of plasma glucose levels after an intraperitoneal load of glucose, in calorie intake and in the oxidative stress (measured by thiobarbituric acid reactive substances, TBARS) of the treated animals with respect to control diet (CD) animals. Other parameters, i.e. body weight, tibia length and fasting glucose (FG), were not modified by the FRD, indicating a pre‐diabetic state rather than an overt diabetes. Data are mean ± SEM. * P < 0.05, ** P < 0.01 vs. CD.
Table 1B.
Echocardiographic parameters from control and fructose‐treated mice
| CD | FRD | |
|---|---|---|
| n | 9 | 13 |
| LVDD (mm) | 3.40 ± 0.11 | 3.77 ± 0.20 |
| LVSD (mm) | 2.40 ± 0.10 | 2.87 ± 0.14* |
| FS (%) | 29.80 ± 2.35 | 23.93 ± 1.58* |
| LVMI (mg g−1) | 7.90 ± 0.66 | 12.19 ± 1.19* |
| LVMI (mg mm−1) | 14.10 ± 1.16 | 21.00 ± 2.43* |
Diastolic left ventricular diameter (LVDD) was not different between the fructose‐rich diet (FRD) and control diet (CD) mice. However, left ventricular systolic diameter (LVSD) and the hypertrophy indices [LVMI: left ventricular mass index, normalized by body weight (mg g−1) or by tibia length (mg mm−1)] were significantly increased in FRD with respect to CD animals. These altered parameters, in association with the fractional shortening (FS: endocardial fractional shortening) being significantly decreased in FRD vs. CD mice, support a cardiac dysfunction produced by the diet. Values are mean ± SEM. * P < 0.05 vs. control mice.
FRD increases spontaneous Ca2+ release events
Figure 1 A shows typical recordings and averaged results obtained using confocal microscopy of mouse myocytes isolated from WT mice on CD or FRD, showing Ca2+ sparks, waves and spontaneous contractions. Myocytes from FRD mice exhibited a significantly increased frequency of each of these spontaneous events compared to myocytes from CD mice. The calculated spark‐mediated SR Ca2+ leak was also significantly increased in FRD vs. CD WT myocytes.
Figure 1. Fructose rich diet induces SR Ca2+ leak without changes in SR Ca2+ content.
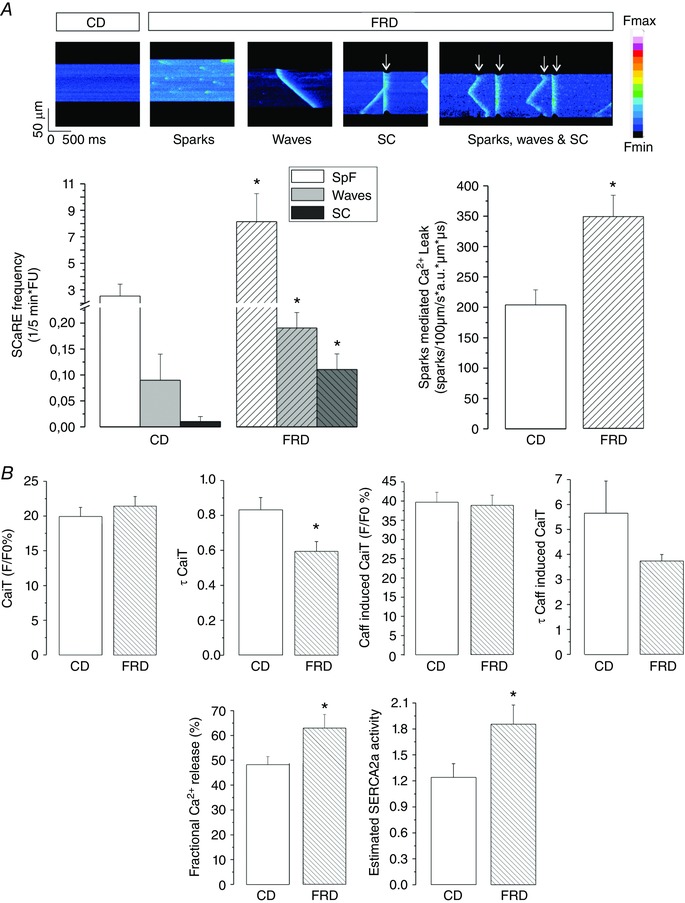
A, representative confocal images of isolated myocytes from control diet (CD) or fructose‐rich diet (FRD) mice. While CD myocytes barely show spontaneous Ca2+ release events (SCaRE), FRD myocytes showed sparks, waves and spontaneous contractions (SC and white arrows). Below, bar graphs showing average data of SCaRE frequency and spark‐mediated SR Ca2+ leak (see Methods for details). * P < 0.05 vs. CD myocytes, n = 9–13 myocytes from four mice per group. B, bar graphs showing Ca2+ transient amplitude (CaiT), the decay time of CaiT (τ CaiT), caffeine induced CaiT amplitude (Caff‐induced CaiT), the decay time of the Caff‐induced CaiT (τ Caff‐induced CaiT), fractional Ca2+ release and the estimated SERCA2a activity from CD and FRD myocytes. CaiT‐ and Caff‐induced CaiT are not different between the groups, but due to the slight increase in CaiT and decrease in Caff‐induced CaiT, fractional Ca2+ release is significantly increased in FRD compared to CD myocytes. Moreover, the SERCA2a estimated activity is also significantly increased in FRD myocytes. In this and the following figures the data are expressed as mean ± SEM. * P < 0.05, n = 35−50 myocytes from 3–4 mice per group.
An increase in SR Ca2+ leak may be produced by an increase in SR Ca2+ load, an alteration of RyR2 activity or a combination of both. Figure 1 B shows that SR Ca2+ content was similar in FRD vs. CD myocytes. These findings are consistent with previous reports showing that CaMKII‐dependent phosphorylation of RyR2 may evoke by itself an increase in diastolic SR Ca2+ leak in the absence of changes in SR Ca2+ content (van Oort et al. 2010; Gonano et al. 2011; Erickson et al. 2013; Sommese et al. 2016). Moreover, the lack of SR Ca2+ content decrease associated with an increase in SR Ca2+ leak can be explained only by an increase in SR Ca2+ uptake. Figure 1 B shows that the estimated SERCA2a activity increased significantly in FRD myocytes compared to CD cells. Furthermore, recent experiments from our laboratory showed an increase in CaMKII activity and phosphorylation of the Thr17 site on phospholamban (PLN) in FRD animals (Sommese et al. 2016), which may account for the increase in SERCA2a activity and the lack of change in SR Ca2+ content in the presence of a leaky RyR2. In these previous experiments, FRD also increased RyR2 S2814 phosphorylation (the CaMKII site), without significant changes in RyR2 expression levels in mouse myocytes. In contrast, phosphorylation at the protein kinase A site S2808 was not altered in pre‐diabetic compared with control hearts.
To further support these previous findings, we performed experiments in cultured HEK293 cells expressing wild type RyR2 exposed to normal glucose or high glucose media to simulate hyperglycaemia. Figure 2 A and B shows that high glucose significantly increases [3H]ryanodine binding. This increase was prevented by treatment of the cells with the CaMKII inhibitor KN93 but not with its inactive analogue KN92. Moreover, phosphorylation of the CaMKII site, S2814, was significantly increased, without significant changes in either RyR2 expression or the protein kinase A site, S2808 (Fig. 2 C and D). Similar to [3H]ryanodine binding, co‐treatment of the cells with KN93 prevented the increase of CaMKII phosphorylation of RyR2 produced by hyperglycaemia. Although the osmolality of the low and high glucose culture media is different (316 vs. 340 mosmol kg−1 respectively, Life Technologies), this difference seems not to affect the conclusions, since high glucose in the presence of KN93 reproduced the results of low glucose.
Figure 2. Recombinant RyR2 presents more activity in high glucose media due to CaMKII phosphorylation of the channel.
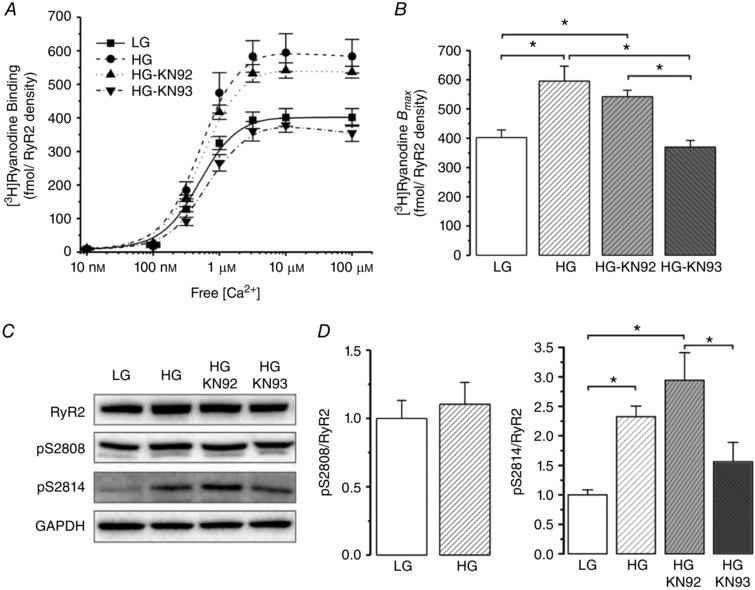
A, [3H]ryanodine binding normalized by RyR2 density. Since low glucose (LG) media alone did not show a difference in [3H]ryanodine binding with respect to LG + KN93 or KN92 (the CaMKII inhibitor or its inactive analogue of KN93, respectively), the LG groups were pooled (full line). High glucose (HG) significantly increased the [3H]ryanodine binding, as well as HG + KN92. However HG + KN93 completely prevented the increase in [3H]ryanodine binding. B, B max of the experiments shown in A. C, representative immunoblots of RyR2 expression, phosphorylation sites of RyR2, S2808 (pS2808) and S2814 (pS2814), and GAPDH as a loading control. D, average data of the experiments in C. HG media did not change the phosphorylation status at S2808 but increased the phosphorylation of RyR2 at S2814, the CaMKII site, which could be prevented by the CaMKII inhibitor, KN93. * P < 0.05.
Taken together, the results support the notion that the increase in SR Ca2+ leak in FRD myocytes is produced by CaMKII‐dependent phosphorylation of RyR2.
FRD‐induced increase in cardiac apoptosis is dependent on CaMKII activity
To explore the possibility that CaMKII‐induced diastolic Ca2+ mishandling may trigger apoptosis, detection of cardiac apoptosis was performed using the fluorescence TUNEL method that identifies broken DNA. Figure 3 A shows typical photographs of specimens from mice fed with CD, FRD and FRD co‐treated with Tempol, an intracellular ROS scavenger. Previous studies showed that Tempol per se does not affect CaMKII‐dependent RyR2 phosphorylation (Sommese et al. 2016). Figure 3 B shows that treatment with FRD dramatically increased the number of apoptotic cells, and that this effect was prevented by co‐treatment with Tempol. The apoptotic effect of FRD was also supported by a significant increase in the Bax/Bcl‐2 ratio (Fig. 3 C). These results indicate that apoptosis is associated with an increase in oxidative stress in FRD animals. Notably, the increase in apoptosis was not associated with an increase in collagen, either type I or III, in mice treated with either FRD or FRD + Tempol, which indicates that apoptosis is an early sign in the evolution of the disease that precedes fibrosis.
Figure 3. Fructose rich diet induces ROS dependent apoptosis.
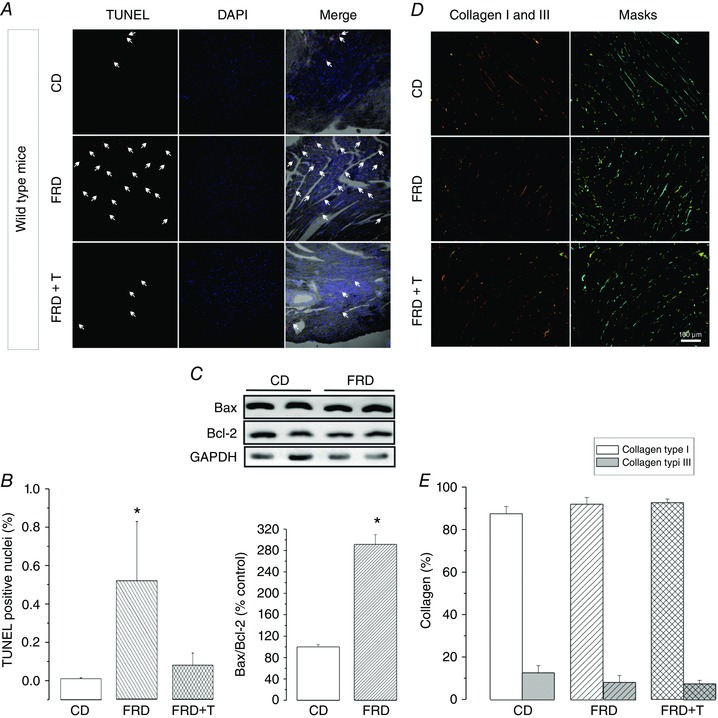
A, representative images of TUNEL (red dots) to denote the apoptotic cells, DAPI (blue dots) to mark the nuclei and the merging of both in a bright field image. White arrows highlight some of the TUNEL‐positive nuclei. The specimens correspond to control diet (CD), fructose‐rich diet (FRD) and FRD + Tempol (FRD+T), an intracellular ROS scavenger, in WT mice. B, average data of the experiments in A, indicating a significant increase in the apoptotic nuclei in the FRD group, which is prevented when animals are treated with the ROS scavenger. C, typical immunoblots of the proapoptotic protein Bax, and the anti‐apoptotic protein Bcl‐2, and GAPDH as a loading control. Below the blots, the bar graph shows that the apoptotic index (Bax/Bcl‐2) is significantly increased in FRD with respect to CD samples. D, collagen types I and III were detected in the cardiac tissue after staining, using the Picrosirius Red technique. Polarized microscopy identified type I collagen fibres as red or yellow structures. Type III collagen fibres are seen as green filaments. To homogenize the range of colour that identifies each collagen type, a light‐blue mask for collagen I and a yellow mask for collagen III were applied on the original images. E, average data of the experiments in D, showing no difference in collagen types I and III between CD and FRD specimens. * P < 0.05 vs. CD, n = 3–5 animals per group.
To elucidate the role of CaMKII and CaMKII‐dependent phosphorylation of RyR2 on cell death produced by hyperglycaemia, we next treated SR‐AIP mice, which express the cardiomyocyte‐delimited transgenic SR‐targeted CaMKII inhibitor, AIP, with FRD. In these mice, CaMKII‐dependent phosphorylation of SR proteins is significantly reduced (Ji et al. 2003). FRD fed SR‐AIP mice did not show any significant increase in TUNEL‐positive nuclei in comparison with control diet‐matched samples. As expected, Tempol treatment did not modify these results. Similarly, the Bax/Bcl‐2 ratio did not change significantly between SR‐AIP CD and FRD mice (Fig. 4 A and B). Interestingly, and in agreement with our previous work (Sommese et al. 2016), although FRD increased oxidative stress, assessed by TBARS (Fig. 4 D), apoptosis was not increased in SR‐AIP mice. This finding indicates that ROS per se are not sufficient to cause apoptosis and that CaMKII‐dependent phosphorylation at the SR level may play a role in the apoptotic pathway in the FRD model.
Figure 4. Inhibition of CaMKII activity at the SR level prevents FRD‐induced apoptosis.
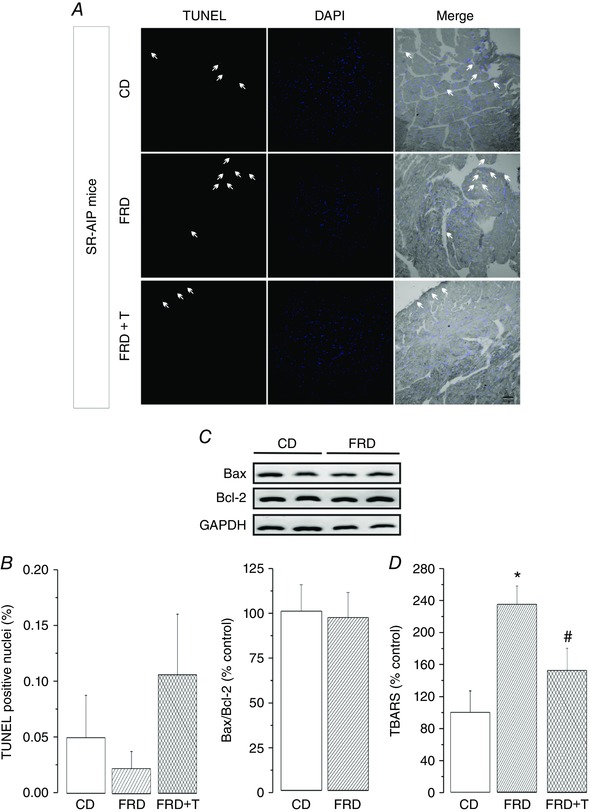
A, representative images of TUNEL to denote the apoptotic cells (red dots and arrows), DAPI (blue dots) to mark intact nuclei and the merging of both in a bright field image. The specimens correspond to control diet (CD), fructose‐rich diet (FRD) and FRD + Tempol (FRD+T), in SR‐AIP mice. These mice express an inhibitor peptide of CaMKII targeted to SR membranes. B, bar graph showing the average data of A, indicating no difference in the apoptotic nuclei among groups. C, typical immunoblots of the pro‐apoptotic protein Bax and the anti‐apoptotic protein Bcl‐2, and GAPDH as a loading control. Below the blots, the bar graph shows that the apoptotic index (Bax/Bcl‐2) is similar between CD and FRD in SR‐AIP mice; n = 3–4 mice per group. D, lipid peroxidation results, measured with the TBARS technique. FRD induces an increase in oxidative stress in FRD SR‐AIP mice. This increase is prevented by Tempol treatment. Taken together, these results indicate that ROS per se are not sufficient to induce apoptosis in the FRD model. Moreover, CaMKII‐dependent phosphorylation at the SR level is a necessary step in the FRD‐induced apoptosis. * P < 0.05 vs CD, # P < 0.05 vs FRD, n = 4–6 mice per group.
FRD induces mitochondrial damage through an increase in CaMKII‐induced SR Ca2+ leak
Apoptosis may occur by activation of the extrinsic and/or the intrinsic pathway. The intrinsic apoptotic pathway involves mitochondrial participation and is the most common intracellular cascade observed in the heart when apoptosis takes place (Ghosh et al. 2005; Williamson et al. 2010). Considering the SR Ca2+ leak observed in FRD mice, we hypothesized that at least some of the Ca2+ lost by the SR enters into the mitochondria inducing mitochondrial Ca2+ overload. This in turn would dissipate mitochondrial membrane potential (Δψ m) through opening of the mPTP, and favours the release of apoptotic factors (Olichon et al. 2003). If our hypothesis is correct, we should expect an increase in mitochondrial damage in these myocytes and that this damage could be avoided by preventing CaMKII phosphorylation of RyR2.
Figure 5 A and B shows representative transmission electron micrographs at two different magnifications (left) and Western blots (right), revealing the integrity and purity of the isolated mitochondrial fraction used. Loss of mitochondria function by mPTP opening can be measured in isolated mitochondria in response to supraphysiological mitochondrial Ca2+ increases. Fig. 5 C and D provdes typical examples and overall results showing that mitochondria from FRD‐fed mice were more susceptible to Ca2+‐triggered decreases in light scattering, which correlate with mPTP opening (Joiner et al. 2012), than mitochondria from CD mice. In contrast, FRD and CD myocytes from S2814A mice, in which the CaMKII site at RyR2 was replaced by Ala and therefore is not phosphorylatable, behaved similarly to CD mice (Fig. 5 E and F). Similar results were obtained with a second mitochondrial integrity assay that uses the mitochondrial membrane voltage‐sensitive fluorescent indicator, JC‐1. Mitochondria from FRD myocytes showed a significant loss of Δψ m compared to mitochondria from CD‐WT and FRD S2814A myocytes (Fig. 5 G). Taken together, these results indicate that CaMKII‐dependent phosphorylation of RyR2 and the consequent SR Ca2+ leak are involved in mitochondrial damage of FRD mice.
Figure 5. Phosphorylation of Ser2418 of RyR2 is related with mitochondria damage.
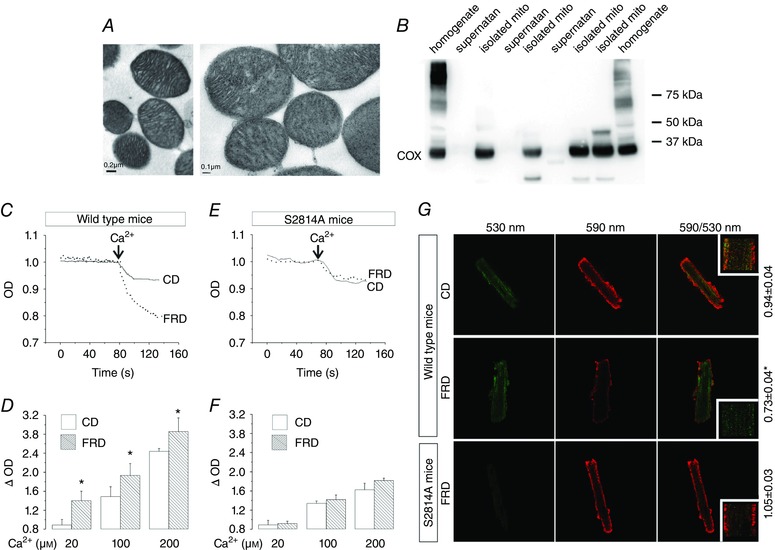
A, transmission electron micrographs of isolated mitochondria at two different magnifications. B, representative immunoblots of different fractions of the mitochondria isolation procedure; expression of the cyclooxygenase enzyme (COX, present only in mitochondria), in the final fraction (isolated mito), shows the preservation of the organelle during the protocol. A and B denote the purity of the isolated mitochondria preparations. C, representative trace of the optical density (OD) of isolated mitochondria from WT mice, showing a more pronounced light scattering in FRD than in CD mouse mitochondria after a Ca2+pulse. D, average data of the experiments from C, showing that ΔOD increases significantly in FRD with respect to CD mouse mitochondria, at the different concentrations of Ca2+ used. E, representative traces of OD in isolated mitochondria from S2814A mice, showing no significant difference in light scattering between CD and FRD mitochondria after a Ca2+ pulse. F, average data of the experiments from E, showing no difference in the ΔOD between CD and FRD in S2814A mice at the different concentrations of Ca2+ used. * P < 0.05 vs CD, n = 4–8 mice per group. G, representative photographs of isolated myocytes from CD and FRD WT and S2814A mice labelled with JC‐1. Green indicates mitochondria depolarization, and red mitochondria polarization at −180 mV. The decrease in the ratio 590/530 nm indicates mitochondria depolarization. The average data is shown at the right side of the photographs. * P < 0.05 vs CD, n = 6–12 cells from 3–5 mice per group.
FRD induces CaMKII‐dependent mitochondria–SR remodelling
The results described above prompted us to further investigate mitochondrial morphology and, in particular, SR–mitochondrial interaction and the possible involvement of CaMKII. We analysed transmission electron micrographs of WT mice and mice transgenically expressing the CaMKII inhibitory peptide (AC3I) (Zhang et al. 2005), on CD or FRD. We found that mitochondrial area and mean Feret diameter were significantly decreased in WT mice on FRD compared to CD. In contrast, none of these parameters were altered in AC3I mice on FRD when compared to CD‐AC3I or CD‐WT mice (Fig. 6). Possibly more important to the main goal of the present work, FRD hearts also showed a significant decrease in the main distance between the SR and the outer mitochondrial membrane versus CD hearts (Fig. 7). This decrease was prevented in AC3I mice on FRD vs. CD‐WT and CD‐AC3I mice, which indicates that CaMKII is involved in this remodelling. This enhanced proximity between both organelles would facilitate the Ca2+ transit from the SR to the mitochondria.
Figure 6. Fructose‐rich diet induces a CaMKII‐dependent ultrastructure remodelling.
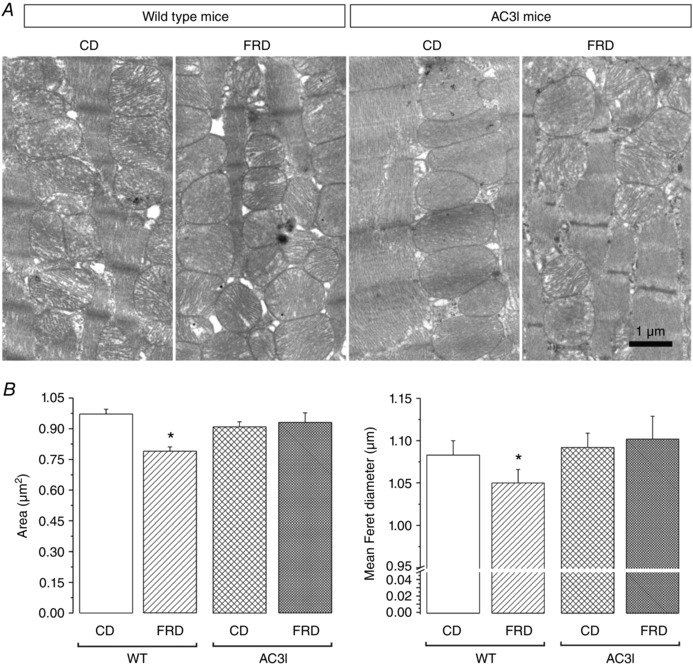
A, transmission electron micrographs of left ventricle from WT and AC3I mice expressing the CaMKII inhibitor peptide AC3 at the whole heart level. FRD induced a clear remodelling of the WT mouse mitochondria, which is prevented in the AC3I mice. B, average data of the mitochondria area and mean Feret diameter. The mitochondria from the WT FRD mice are smaller than the WT CD mitochondria. Inhibition of CaMKII prevents this remodelling. * P < 0.05 vs all other groups, n = 3 mice per group.
Figure 7. Fructose‐rich diet brings the sarcoplasmic reticulum closer to the mitochondria through a CaMKII‐mediated pathway.
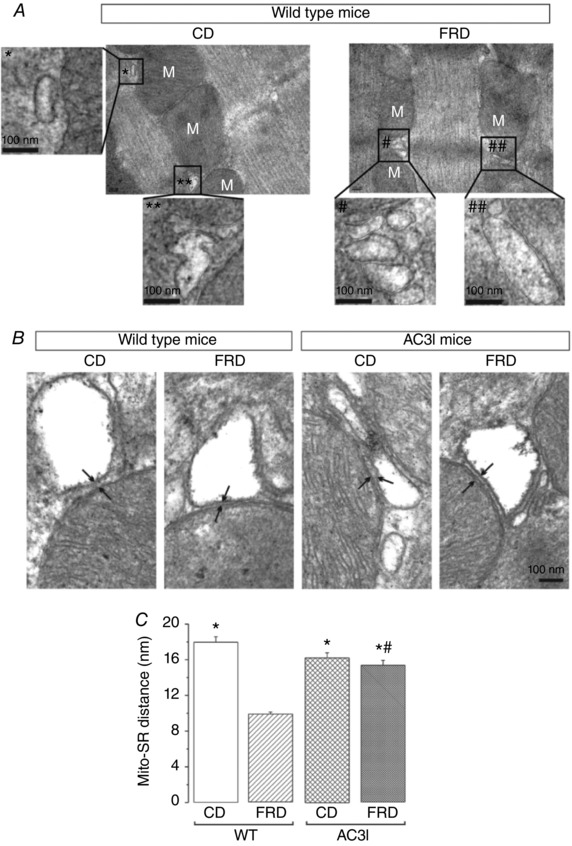
A, transmission electron micrographs of left ventricle from WT CD and FRD mice. The insets show an amplified section of the photographs at the regions marked with * or #. The amplified photographs were skeletonized to better delineate the limits of each organelle (sarcoplasmic reticulum and mitochondria, M). FRD SR/mitochondria distance is decreased with respect to the distance between CD organelles. B, transmission electron micrographs at higher magnification where the arrows point to the limits between the mitochondria and the SR in WT and AC3I mice. C, average data of the distance between the SR and the mitochondria (Mito‐SR distance). FRD significantly decreased the Mito‐SR distance in WT mice. This remodelling is greatly prevented in the AC3I mice. * P < 0.05 vs. FRD WT, # P < 0.05 vs CD WT, n = 3 mice per group.
Discussion
DCM is a disorder of the heart muscle in people with diabetes that occurs independently of hypertension or vascular disease. Impaired fasting glucose and impaired glucose tolerance are early metabolic abnormalities that precede diabetes. These alterations are thought to root irregularities at the cardiac myocyte level, ultimately contributing to structural and functional anomalies (Hajat et al. 2004). Indeed, patients with diabetes mellitus, independently of the severity of coronary artery disease, have an enhanced risk of heart failure compared to patients without diabetes mellitus (Kannel & McGee, 1979; Dandamudi et al. 2014). Although the transition from a pre‐diabetic state to overt diabetes may take years, more than 50% of pre‐diabetic individuals will eventually develop diabetes which, if untreated, will inexorably evolve to DCM. Thus, early detection and management of pre‐diabetes is mandatory to prevent the evolution of the disease. Myocardial cell death is recognized as a major event in the progression to heart failure (Kang & Izumo, 2000). Myocyte apoptosis has been demonstrated in hearts of diabetic individuals suffering from DCM (Miki et al. 2013) and in streptozotocin‐induced diabetic rats (Hostiuc et al. 2013). However, the importance of apoptosis in the early stages of the illness preceding DCM as well as the possible mechanisms involved, remain unknown.
The present experiments performed in a validated mouse model of pre‐diabetes (Alzugaray et al. 2009; Felice et al. 2014; Sommese et al. 2016) show for the first time that apoptosis is an early sign of myocardial dysfunction in the evolution of diabetic disease, preceding the increase in collagen which may lead to structural and irreversible alterations. Taking advantage of genetically modified animals, this model enabled us to delineate the signalling pathway that underlies this apoptotic cascade. The results revealed a cascade of events initiated by a CaMKII‐induced increase in SR Ca2+ leak to produce mitochondrial membrane depolarization and cardiac damage. A particularly striking finding was the CaMKII‐induced remodelling of mitochondria and SR–mitochondria interaction. The latter would strongly support SR–mitochondria dialogue, facilitating Ca2+ drain to the mitochondria and cell death, in the scenario of an increased SR Ca2+ leak.
FRD increases spontaneous CaMKII‐dependent Ca2+ release events and cardiac apoptosis
The present results clearly demonstrate that FRD promotes apoptosis, as shown by the significant increase in TUNEL‐positive nuclei and Bax/Bcl‐2 ratio, the latter suggesting the involvement of SR/mitochondria in the apoptotic process (Pinton et al. 2008). The results show that apoptosis is dependent on an increase in oxidative stress and on CaMKII activity at the SR level. Since apoptosis does not take place in mice with targeted CaMKII inhibition at the SR level (SR‐AIP mice), in which FRD also produce an increase in ROS, these findings further indicate that the increase in ROS produced in this model was not enough to evoke apoptosis by itself. In addition, the experiments revealed that ROS production is upstream of CaMKII activation in the apoptotic signalling cascade triggered by FRD. These results are in line with previous experiments which indicate that FRD increases the activity of oxidized CaMKII in rat heart to evoke arrhythmias, which like the apoptosis described in the present results, could be prevented in SR‐AIP mice (Sommese et al. 2016). These findings suggest an analogous signalling pathway for both phenomena.
A number of studies have investigated cell death in diabetic hearts. These studies indicate that apoptosis is enhanced and associated with left ventricular enlargement and increased fibrosis following myocardium infarction in diabetic cardiomyocytes (Cai et al. 2002; Backlund et al. 2004). Moreover, several lines of evidence indicate that CaMKII constitutes a common intermediate by which various death‐inducing stimuli trigger cardiomyocyte apoptosis (Zhu et al. 2007). Interestingly, Luo et al. (2013) described a pathway of CaMKII‐induced apoptosis in pacemaker cells of streptozotocin‐induced diabetic mice. Furthermore, different results suggest a strong link between abnormal myocyte Ca2+ handling, mitochondrial dysfunction and apoptosis (Chen et al. 2005; Vila‐Petroff et al. 2007; Salas et al. 2010). Indeed, numerous data support the hypothesis that Ca2+ movement from the endoplasmic reticulum/SR to mitochondria is a key process in some apoptotic routes (Pinton et al. 2008). In this context, experiments from the Houser laboratory revealed that mitochondria‐triggered myocyte death is increased by activation of CaMKII through an increase in L‐type Ca2+ current and SR Ca2+ release (Chen et al. 2005). A critical role for excessive cytosolic Ca2+ and CaMKII activation has also been shown under different pathological conditions (Vila‐Petroff et al. 2007; Sapia et al. 2010; Joiner et al. 2012; Di Carlo et al. 2014). Excessive [Ca2+] within mitochondria can induce apoptosis by opening the mPTP (Crow et al. 2004). This occurs at higher mitochondrial Ca2+ levels than those that match myocyte energy supply and demand in normal cardiomyocytes (Balaban et al. 2004). Finally, it is well known that over‐activation of CaMKII occurs in heart failure and that this can contribute to major dysfunctions of the failing heart (Anderson et al. 2011). However, the role of CaMKII has mainly been studied in heart failure independent of diabetes (Couchonnal & Anderson, 2008; Anderson et al. 2011; Swaminathan et al. 2012).
Our study is the first to show that in FRD mice with impaired glucose tolerance – a stage that largely precedes overt diabetes and DCM – there is a significant increase in mitochondrial damage and apoptotic death produced by an enhancement of SR Ca2+ leak due to CaMKII‐dependent phosphorylation of RyR2. Notably, a CaMKII‐dependent increase in SR Ca2+ leak inducing arrhythmias have been previously described in FRD mouse myocytes (Sommese et al. 2016), underlining the importance of CaMKII activation in the early stages of DCM evolution. The essential role of spontaneous SR Ca2+ release in CaMKII‐induced apoptosis is supported by the fact that prevention of S2814 phosphorylation of RyR2 (the CaMKII site) prevents mitochondrial damage and apoptosis.
Interestingly, recent experiments showed that acute hyperglycaemia causes covalent modification of CaMKII by O‐linked N‐acetylglucosamine, which significantly enhances CaMKII‐dependent activation of spontaneous SR Ca2+ release events and arrhythmias in the perfused intact heart (Erickson et al. 2013). In line with these seminal findings, our results in HEK293 cells subjected to hyperglycaemia showed an increase in RyR2 activity dependent on CaMKII phosphorylation. Whether the novel mechanism of CaMKII activation also takes place in our cellular model was not assessed in the present study and will be subject of future investigation. Our results in Tempol‐treated mice do clearly indicate that ROS play a significant role in the apoptotic cell death observed in the pre‐diabetic stage of the FRD mice. Moreover, the finding that apoptosis was prevented in SR‐AIP mice in spite of the fact that ROS were increased, further supports that ROS production is upstream of CaMKII activation, suggesting a role of oxidized CaMKII, according to our previous report (Sommese et al. 2016). This ROS‐induced CaMKII activity might fade other mechanisms of CaMKII activation, or act synergistically to them, as suggested by Erickson et al. (2013).
FRD evokes a CaMKII‐mediated SR–mitochondria remodelling
Mitochondrial Ca2+ homeostasis is crucial for balancing cell survival and death. On the one hand, mitochondria are essential to cardiac SR Ca2+ cycling as the source of ATP that powers several ion pumps and myosin ATPase. Moreover, Ca2+ plays a key role in mitochondria through the coordination of energy supply and demand by stimulating intra‐mitochondrial effectors, such as the Ca2+‐dependent dehydrogenases of the Krebs cycle. On the other hand, mitochondrial Ca2+ overload results in opening of a non‐specific mPTP, membrane depolarization and cell death.
Spatial co‐localization and close contact of mitochondria with SR permit transorganelle trafficking of different compounds such as ATP, phospholipids or Ca2+. The close apposition between SR and mitochondria allows the generation of microdomains with high Ca2+ concentration, which induces accumulation of Ca2+ into the mitochondria (Giacomello et al. 2007). Decreasing the SR–mitochondria distance would increase Ca2+ concentrations in these microdomains, favouring Ca2+ influx into the mitochondrial matrix. Previous experiments have shown that the physical contact between SR and mitochondria is supported by tethering proteins. Among these, mitofusin 2 (Mfn2) has been shown to be crucial for interorganelle signalling in the myocardium (Chen et al. 2012). These authors showed that in Mfn‐knockout hearts there is a trend towards an increase in the mean distance between junctional SR and the outer mitochondrial membrane associated with an increase in mitochondria area, compared with their respective controls. This structural uncoupling correlated with decreased SR to mitochondrial Ca2+ transfer in isolated cardiomyocyte experiments. Although the role of Mfn2 as a positive regulator of SR–mitochondria interaction has been challenged (Cosson et al. 2012; Filadi et al. 2015), it is clear that the distance between the SR/endoplasmic reticulum and the outer mitochondria membrane is critical for the efficient transfer of Ca2+ (Marchi et al. 2014).
The present experiments reveal that FRD produces an early cardiac remodelling at the level of mitochondrial morphology and the SR–mitochondria interaction, which is dependent on CaMKII. Experimental evidence indicates that early during induction of apoptosis, mitochondria become largely fragmented, resulting in small and numerous organelles (Pinton et al. 2008). In our experiments the mitochondrial area of FRD myocytes was significantly decreased with respect to CD myocytes or FRD myocytes from AC3I mice, a sign that suggests mitochondrial fragmentation. Activation of the mitochondrial fission machinery and further mitochondrial fragmentation has been associated with apoptosis (Parra et al. 2008). However, the relationship between mitochondrial fusion and fission and apoptosis is complex and this point remains contentious (Martinou & Youle, 2006; Arnoult, 2007; Pinton et al. 2008). Possibly more important in the remodelling described in the present results is that the contact area between SR and mitochondria is reduced by approximately 50% in FRD mouse myocytes compared with CD myocytes, and that this enhanced proximity is greatly prevented in mouse myocytes with CaMKII inhibition at the heart level. This increased structural coupling correlated with an increase in mitochondria damage and apoptosis, suggesting that this alteration may facilitate Ca2+ transit from the SR to the mitochondria. Further studies are required to explore the intrinsic nature of this increased interaction.
In conclusion, this study is the first to investigate apoptosis and its underlying mechanisms in a pre‐diabetes model. The elucidation of these early events preceding DCM is necessary and may lead to the design of novel targets to prevent the evolution to more critical stages of the illness.
Additional information
Conflict of interest
XHTW is a co‐founder and co‐owner of Elex Biotech, a small biotech company dedicated to the development of drug molecules for the treatment of heart disease.
Author contributions
JP conceived and designed the experiments. JP, AM, FA and ELP interpreted the data. MF, ELP, CNZ, FA, PGB and LS performed the experiments and were involved in collection analysis and interpretation of the data. ELP was involved in image analysis and revision of the manuscript; JD and MK provided the SR‐AIP mice; XHTW provided the S2814A mice and was involved in critically revising the manuscript. JP and AM wrote the manuscript. All authors approved the final version of the manuscript. All persons designated as authors qualify for authorship and all those who qualify for authorship are listed.
Funding
This work was supported by PICT 2015‐3009 (FONCyT, Argentina) to JP, 2014‐2524 (FONCyT, Argentina) and PIP 0890 (CONICET, Argentina) to AM; PICT 2012‐0574 (FONCyT, Argentina) to ELP and supported by NIH/NHLBI grants HL089598, HL091947, HL117641, HL129570 (XHTW), and American Heart Association grant 13EIA14560061 (XHTW).
Acknowledgements
We are very grateful to Dr Mark Anderson for the AC3I mice and Dr Héctor Valdivia for his selfless collaboration. We also acknowledge Mrs Mónica Rando, Mr Omar Castillo, Mr Leandro Di Ciani, Mrs Alfonsina Morales and veterinary Juan Lofeudo for their technical assistance.
References
- Alzugaray ME, Garcia ME, Del Zotto HH, Raschia MA, Palomeque J, Rossi JP, Gagliardino JJ & Flores LE (2009). Changes in islet plasma membrane calcium‐ATPase activity and isoform expression induced by insulin resistance. Arch Biochem Biophys 490, 17–23. [DOI] [PubMed] [Google Scholar]
- Anderson ME, Brown JH & Bers DM (2011). CaMKII in myocardial hypertrophy and heart failure. J Mol Cell Cardiol 51, 468–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ares‐Carrasco S, Picatoste B, Benito‐Martin A, Zubiri I, Sanz AB, Sanchez‐Nino MD, Ortiz A, Egido J, Tunon J & Lorenzo O (2009). Myocardial fibrosis and apoptosis, but not inflammation, are present in long‐term experimental diabetes. Am J Physiol 297, H2109–2119. [DOI] [PubMed] [Google Scholar]
- Arnoult D (2007). Mitochondrial fragmentation in apoptosis. Trends Cell Biol 17, 6–12. [DOI] [PubMed] [Google Scholar]
- Backlund T, Palojoki E, Saraste A, Eriksson A, Finckenberg P, Kyto V, Lakkisto P, Mervaala E, Voipio‐Pulkki LM, Laine M & Tikkanen I (2004). Sustained cardiomyocyte apoptosis and left ventricular remodelling after myocardial infarction in experimental diabetes. Diabetologia 47, 325–330. [DOI] [PubMed] [Google Scholar]
- Balaban P, Chistiakova M, Malyshev A & Volgushev M (2004). Dependence of calcium influx in neocortical cells on temporal structure of depolarization, number of spikes, and blockade of NMDA receptors. J Neurosci Res 76, 481–487. [DOI] [PubMed] [Google Scholar]
- Biesmans L, Macquaide N, Heinzel FR, Bito V, Smith GL & Sipido KR (2011). Subcellular heterogeneity of ryanodine receptor properties in ventricular myocytes with low T‐tubule density. PloS One 6, e25100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai L, Li W, Wang G, Guo L, Jiang Y & Kang YJ (2002). Hyperglycemia‐induced apoptosis in mouse myocardium: mitochondrial cytochrome C‐mediated caspase‐3 activation pathway. Diabetes 51, 1938–1948. [DOI] [PubMed] [Google Scholar]
- Chelu MG, Sarma S, Sood S, Wang S, van Oort RJ, Skapura DG, Li N, Santonastasi M, Muller FU, Schmitz W, Schotten U, Anderson ME, Valderrabano M, Dobrev D & Wehrens XH (2009). Calmodulin kinase II‐mediated sarcoplasmic reticulum Ca2+ leak promotes atrial fibrillation in mice. J Clin Invest 119, 1940–1951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Zhang X, Kubo H, Harris DM, Mills GD, Moyer J, Berretta R, Potts ST, Marsh JD & Houser SR (2005). Ca2+ influx‐induced sarcoplasmic reticulum Ca2+ overload causes mitochondrial‐dependent apoptosis in ventricular myocytes. Circ Res 97, 1009–1017. [DOI] [PubMed] [Google Scholar]
- Chen Y, Csordas G, Jowdy C, Schneider TG, Csordas N, Wang W, Liu Y, Kohlhaas M, Meiser M, Bergem S, Nerbonne JM, Dorn GW, 2nd & Maack C (2012). Mitofusin 2‐containing mitochondrial‐reticular microdomains direct rapid cardiomyocyte bioenergetic responses via interorganelle Ca2+ crosstalk. Circ Res 111, 863–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chowdhry MF, Vohra HA & Galinanes M (2007). Diabetes increases apoptosis and necrosis in both ischemic and nonischemic human myocardium: role of caspases and poly‐adenosine diphosphate‐ribose polymerase. J Thorac Cardiovasc Surg 134, 124–131, 131.e1–3. [DOI] [PubMed] [Google Scholar]
- Colagiuri S (2011). Epidemiology of prediabetes. Med Clin North Am 95, 299–307, vii. [DOI] [PubMed] [Google Scholar]
- Cosson P, Marchetti A, Ravazzola M & Orci L (2012). Mitofusin‐2 independent juxtaposition of endoplasmic reticulum and mitochondria: an ultrastructural study. PloS One 7, e46293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couchonnal LF & Anderson ME (2008). The role of calmodulin kinase II in myocardial physiology and disease. Physiology (Bethesda) 23, 151–159. [DOI] [PubMed] [Google Scholar]
- Crow MT, Mani K, Nam YJ & Kitsis RN (2004). The mitochondrial death pathway and cardiac myocyte apoptosis. Circ Res 95, 957–970. [DOI] [PubMed] [Google Scholar]
- Dandamudi S, Slusser J, Mahoney DW, Redfield MM, Rodeheffer RJ & Chen HH (2014). The prevalence of diabetic cardiomyopathy: a population‐based study in Olmsted County, Minnesota. J Card Fail 20, 304–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Carlo MN, Said M, Ling H, Valverde CA, De Giusti VC, Sommese L, Palomeque J, Aiello EA, Skapura DG, Rinaldi G, Respress JL, Brown JH, Wehrens XH, Salas MA & Mattiazzi A (2014). CaMKII‐dependent phosphorylation of cardiac ryanodine receptors regulates cell death in cardiac ischemia/reperfusion injury. J Mol Cell Cardiol 74, 274–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz ME, Graham HK & Trafford AW (2004). Enhanced sarcolemmal Ca2+ efflux reduces sarcoplasmic reticulum Ca2+ content and systolic Ca2+ in cardiac hypertrophy. Cardiovasc Res 62, 538–547. [DOI] [PubMed] [Google Scholar]
- Dobrin JS & Lebeche D (2010). Diabetic cardiomyopathy: signaling defects and therapeutic approaches. Expert Rev Cardiovasc Ther 8, 373–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erickson JR, Pereira L, Wang L, Han G, Ferguson A, Dao K, Copeland RJ, Despa F, Hart GW, Ripplinger CM & Bers DM (2013). Diabetic hyperglycaemia activates CaMKII and arrhythmias by O‐linked glycosylation. Nature 502, 372–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felice JI, Gangoiti MV, Molinuevo MS, McCarthy AD & Cortizo AM (2014). Effects of a metabolic syndrome induced by a fructose‐rich diet on bone metabolism in rats. Metabolism 63, 296–305. [DOI] [PubMed] [Google Scholar]
- Filadi R, Greotti E, Turacchio G, Luini A, Pozzan T & Pizzo P (2015). Mitofusin 2 ablation increases endoplasmic reticulum‐mitochondria coupling. Proc Natl Acad Sci U S A 112, E2174–2181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiordaliso F, Bianchi R, Staszewsky L, Cuccovillo I, Doni M, Laragione T, Salio M, Savino C, Melucci S, Santangelo F, Scanziani E, Masson S, Ghezzi P & Latini R (2004). Antioxidant treatment attenuates hyperglycemia‐induced cardiomyocyte death in rats. J Mol Cell Cardiol 37, 959–968. [DOI] [PubMed] [Google Scholar]
- Frustaci A, Kajstura J, Chimenti C, Jakoniuk I, Leri A, Maseri A, Nadal‐Ginard B & Anversa P (2000). Myocardial cell death in human diabetes. Circ Res 87, 1123–1132. [DOI] [PubMed] [Google Scholar]
- Ghosh S, Pulinilkunnil T, Yuen G, Kewalramani G, An D, Qi D, Abrahani A & Rodrigues B (2005). Cardiomyocyte apoptosis induced by short‐term diabetes requires mitochondrial GSH depletion. Am J Physiol 289, H768–776. [DOI] [PubMed] [Google Scholar]
- Ghosh S & Rodrigues B (2006). Cardiac cell death in early diabetes and its modulation by dietary fatty acids. Biochem Biophys Acta 1761, 1148–1162. [DOI] [PubMed] [Google Scholar]
- Giacomello M, Drago I, Pizzo P & Pozzan T (2007). Mitochondrial Ca2+ as a key regulator of cell life and death. Cell Death Differ 14, 1267–1274. [DOI] [PubMed] [Google Scholar]
- Gonano LA, Sepulveda M, Rico Y, Kaetzel M, Valverde CA, Dedman J, Mattiazzi A & Vila Petroff M (2011). Calcium‐calmodulin kinase II mediates digitalis‐induced arrhythmias. Circ Arrhythm Electrophysiol 4, 947–957. [DOI] [PubMed] [Google Scholar]
- Grundy D (2015). Principles and standards for reporting animal experiments in The Journal of Physiology and Experimental Physiology . J Physiol 593,2547–2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajat C, Tilling K, Stewart JA, Lemic‐Stojcevic N & Wolfe CD (2004). Ethnic differences in risk factors for ischemic stroke: a European case‐control study. Stroke 35, 1562–1567. [DOI] [PubMed] [Google Scholar]
- Helms AS, Alvarado FJ, Yob J, Tang VT, Pagani F, Russell MW, Valdivia HH & Day SM (2016). Genotype‐dependent and independent calcium signaling dysregulation in human hypertrophic cardiomyopathy. Circulation 134, 1738–1748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hostiuc S, Popescu A, Gutu ED, Rusu MC & Pop F (2013). Electrical conduction system apoptosis in type II diabetes mellitus. Rom J Morphol Embryol 54, 953–959. [PubMed] [Google Scholar]
- Ji Y, Li B, Reed TD, Lorenz JN, Kaetzel MA & Dedman JR (2003). Targeted inhibition of Ca2+/calmodulin‐dependent protein kinase II in cardiac longitudinal sarcoplasmic reticulum results in decreased phospholamban phosphorylation at threonine 17. J Biol Chem 278, 25063–25071. [DOI] [PubMed] [Google Scholar]
- Joiner ML, Koval OM, Li J, He BJ, Allamargot C, Gao Z, Luczak ED, Hall DD, Fink BD, Chen B, Yang J, Moore SA, Scholz TD, Strack S, Mohler PJ, Sivitz WI, Song LS & Anderson ME (2012). CaMKII determines mitochondrial stress responses in heart. Nature 491, 269–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang PM & Izumo S (2000). Apoptosis and heart failure: a critical review of the literature. Circ Res 86, 1107–1113. [DOI] [PubMed] [Google Scholar]
- Kannel WB & McGee DL (1979). Diabetes and cardiovascular risk factors: the Framingham study. Circulation 59, 8–13. [DOI] [PubMed] [Google Scholar]
- Kuethe F, Sigusch HH, Bornstein SR, Hilbig K, Kamvissi V & Figulla HR (2007). Apoptosis in patients with dilated cardiomyopathy and diabetes: a feature of diabetic cardiomyopathy? Horm Metab Res 39, 672–676. [DOI] [PubMed] [Google Scholar]
- Lang RM, Bierig M, Devereux RB, Flachskampf FA, Foster E, Pellikka PA, Picard MH, Roman MJ, Seward J, Shanewise JS, Solomon SD, Spencer KT, Sutton MS, Stewart WJ, Chamber Quantification Writing Group, American Society of Echocardiography's Guidelines and Standards Committee & European Association of Echocardiography (2005). Recommendations for chamber quantification: a report from the American Society of Echocardiography's Guidelines and Standards Committee and the Chamber Quantification Writing Group, developed in conjunction with the European Association of Echocardiography, a branch of the European Society of Cardiology. J Am Soc Echocardiogr 18, 1440–1463. [DOI] [PubMed] [Google Scholar]
- Lebeche D, Davidoff AJ & Hajjar RJ (2008). Interplay between impaired calcium regulation and insulin signaling abnormalities in diabetic cardiomyopathy. Nat Clin Pract Cardiovasc Med 5, 715–724. [DOI] [PubMed] [Google Scholar]
- Luo M, Guan X, Luczak ED, Lang D, Kutschke W, Gao Z, Yang J, Glynn P, Sossalla S, Swaminathan PD, Weiss RM, Yang B, Rokita AG, Maier LS, Efimov IR, Hund TJ & Anderson ME (2013). Diabetes increases mortality after myocardial infarction by oxidizing CaMKII. J Clin Invest 123, 1262–1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchi S, Patergnani S & Pinton P (2014). The endoplasmic reticulum–mitochondria connection: one touch, multiple functions. Biochim Biophys Acta 1837, 461–469. [DOI] [PubMed] [Google Scholar]
- Martinou JC & Youle RJ (2006). Which came first, the cytochrome c release or the mitochondrial fission? Cell Death Differ 13, 1291–1295. [DOI] [PubMed] [Google Scholar]
- Mazzocchi G, Sommese L, Palomeque J, Felice JI, Di Carlo MN, Fainstein D, Gonzalez P, Contreras P, Skapura D, McCauley MD, Lascano EC, Negroni JA, Kranias EG, Wehrens XH, Valverde CA & Mattiazzi A (2016). Phospholamban ablation rescues the enhanced propensity to arrhythmias of mice with CaMKII‐constitutive phosphorylation of RyR2 at site S2814. J Physiol 594, 3005–3030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miki T, Yuda S, Kouzu H & Miura T (2013). Diabetic cardiomyopathy: pathophysiology and clinical features. Heart Fail Rev 18, 149–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mokdad AH, Bowman BA, Ford ES, Vinicor F, Marks JS & Koplan JP (2001). The continuing epidemics of obesity and diabetes in the United States. JAMA 286, 1195–1200. [DOI] [PubMed] [Google Scholar]
- Montes GS (1996). Structural biology of the fibres of the collagenous and elastic systems. Cell Biol Int 20, 15–27. [DOI] [PubMed] [Google Scholar]
- Mundina‐Weilenmann C, Vittone L, Ortale M, de Cingolani GC & Mattiazzi A (1996). Immunodetection of phosphorylation sites gives new insights into the mechanisms underlying phospholamban phosphorylation in the intact heart. J Biol Chem 271, 33561–33567. [DOI] [PubMed] [Google Scholar]
- Nguyen QM, Srinivasan SR, Xu JH, Chen W & Berenson GS (2010). Fasting plasma glucose levels within the normoglycemic range in childhood as a predictor of prediabetes and type 2 diabetes in adulthood: the Bogalusa Heart Study. Arch Pediatr Adolesc Med 164, 124–128. [DOI] [PubMed] [Google Scholar]
- Olichon A, Baricault L, Gas N, Guillou E, Valette A, Belenguer P & Lenaers G (2003). Loss of OPA1 perturbates the mitochondrial inner membrane structure and integrity, leading to cytochrome c release and apoptosis. J Biol Chem 278, 7743–7746. [DOI] [PubMed] [Google Scholar]
- Palomeque J, Rueda OV, Sapia L, Valverde CA, Salas M, Petroff MV & Mattiazzi A (2009). Angiotensin II‐induced oxidative stress resets the Ca2+ dependence of Ca2+‐calmodulin protein kinase II and promotes a death pathway conserved across different species. Circ Res 105, 1204–1212. [DOI] [PubMed] [Google Scholar]
- Pan XR, Yang WY, Li GW & Liu J (1997). Prevalence of diabetes and its risk factors in China, 1994. National Diabetes Prevention and Control Cooperative Group. Diabetes Care 20, 1664–1669. [DOI] [PubMed] [Google Scholar]
- Pardo AC, Rinaldi GJ & Mosca SM (2015). Mitochondrial calcium handling in normotensive and spontaneously hypertensive rats: correlation with systolic blood pressure levels. Mitochondrion 20, 75–81. [DOI] [PubMed] [Google Scholar]
- Parra V, Eisner V, Chiong M, Criollo A, Moraga F, Garcia A, Hartel S, Jaimovich E, Zorzano A, Hidalgo C & Lavandero S (2008). Changes in mitochondrial dynamics during ceramide‐induced cardiomyocyte early apoptosis. Cardiovasc Res 77, 387–397. [DOI] [PubMed] [Google Scholar]
- Pinton P, Giorgi C, Siviero R, Zecchini E & Rizzuto R (2008). Calcium and apoptosis: ER‐mitochondria Ca2+ transfer in the control of apoptosis. Oncogene 27, 6407–6418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds ES (1963). The use of lead citrate at high pH as an electron‐opaque stain in electron microscopy. J Cell Biol 17, 208–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salas MA, Valverde CA, Sanchez G, Said M, Rodriguez JS, Portiansky EL, Kaetzel MA, Dedman JR, Donoso P, Kranias EG & Mattiazzi A (2010). The signalling pathway of CaMKII‐mediated apoptosis and necrosis in the ischemia/reperfusion injury. J Mol Cell Cardiol 48, 1298–1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sapia L, Palomeque J, Mattiazzi A & Petroff MV (2010). Na+/K+‐ATPase inhibition by ouabain induces CaMKII‐dependent apoptosis in adult rat cardiac myocytes. J Mol Cell Cardiol 49, 459–468. [DOI] [PubMed] [Google Scholar]
- Sommese L, Valverde CA, Blanco P, Castro MC, Rueda OV, Kaetzel M, Dedman J, Anderson ME, Mattiazzi A & Palomeque J (2016). Ryanodine receptor phosphorylation by CaMKII promotes spontaneous Ca2+ release events in a rodent model of early stage diabetes: the arrhythmogenic substrate. Int J Cardiol 202, 394–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spurr AR (1969). A low‐viscosity epoxy resin embedding medium for electron microscopy. J Ultrastruct Res 26, 31–43. [DOI] [PubMed] [Google Scholar]
- Swaminathan PD, Purohit A, Hund TJ & Anderson ME (2012). Calmodulin‐dependent protein kinase II: linking heart failure and arrhythmias. Circ Res 110, 1661–1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Oort RJ, McCauley MD, Dixit SS, Pereira L, Yang Y, Respress JL, Wang Q, De Almeida AC, Skapura DG, Anderson ME, Bers DM & Wehrens XH (2010). Ryanodine receptor phosphorylation by calcium/calmodulin‐dependent protein kinase II promotes life‐threatening ventricular arrhythmias in mice with heart failure. Circulation 122, 2669–2679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velez Rueda JO, Palomeque J & Mattiazzi A (2012). Early apoptosis in different models of cardiac hypertrophy induced by high renin‐angiotensin system activity involves CaMKII. J Appl Physiol 112, 2110–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vila‐Petroff M, Salas MA, Said M, Valverde CA, Sapia L, Portiansky E, Hajjar RJ, Kranias EG, Mundina‐Weilenmann C & Mattiazzi A (2007). CaMKII inhibition protects against necrosis and apoptosis in irreversible ischemia‐reperfusion injury. Cardiovasc Res 73, 689–698. [DOI] [PubMed] [Google Scholar]
- Wang H, Shara NM, Calhoun D, Umans JG, Lee ET & Howard BV (2010). Incidence rates and predictors of diabetes in those with prediabetes: the Strong Heart Study. Diabetes Metab Res Rev 26, 378–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whelan RS, Kaplinskiy V & Kitsis RN (2010). Cell death in the pathogenesis of heart disease: mechanisms and significance. Annu Rev Physiol 72, 19–44. [DOI] [PubMed] [Google Scholar]
- Williamson CL, Dabkowski ER, Baseler WA, Croston TL, Alway SE & Hollander JM (2010). Enhanced apoptotic propensity in diabetic cardiac mitochondria: influence of subcellular spatial location. Am J Physiol Heart Circ Physiol 298, H633–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Zhu WZ, Joiner ML, Zhang R, Oddis CV, Hou Y, Yang J, Price EE, Gleaves L, Eren M, Ni G, Vaughan DE, Xiao RP & Anderson ME (2006). Calmodulin kinase II inhibition protects against myocardial cell apoptosis in vivo. Am J Physiol Heart Circ Physiol 291, H3065–3075. [DOI] [PubMed] [Google Scholar]
- Zhang R, Khoo MS, Wu Y, Yang Y, Grueter CE, Ni G, Price EE, Jr , Thiel W, Guatimosim S, Song LS, Madu EC, Shah AN, Vishnivetskaya TA, Atkinson JB, Gurevich VV, Salama G, Lederer WJ, Colbran RJ & Anderson ME (2005). Calmodulin kinase II inhibition protects against structural heart disease. Nat Med 11, 409–417. [DOI] [PubMed] [Google Scholar]
- Zhu W, Woo AY, Yang D, Cheng H, Crow MT & Xiao RP (2007). Activation of CaMKIIδC is a common intermediate of diverse death stimuli‐induced heart muscle cell apoptosis. J Biol Chem 282, 10833–10839. [DOI] [PubMed] [Google Scholar]