

Abstract
Key points
Heart function is critically dependent upon the balance of energy production and utilization. Sarcolemmal ATP‐sensitive potassium channels (KATP channels) in cardiac myocytes adjust contractile function to compensate for the level of available energy.
Understanding the activation of KATP channels in working myocardium during high‐stress situations is crucial to the treatment of cardiovascular disease, especially ischaemic heart disease.
Using a new optical mapping approach, we measured action potentials from the surface of excised contracting rabbit hearts to assess when sarcolemmal KATP channels were activated during physiologically relevant workloads and during gradual reductions in myocardial oxygenation.
We demonstrate that left ventricular pressure is closely linked to KATP channel activation and that KATP channel inhibition with a low concentration of tolbutamide prevents electromechanical decline when oxygen availability is reduced. As a result, KATP channel inhibition probably exacerbates a mismatch between energy demand and energy production when myocardial oxygenation is low.
Abstract
Sarcolemmal ATP‐sensitive potassium channel (KATP channel) activation in isolated cells is generally understood, although the relationship between myocardial oxygenation and KATP activation in excised working rabbit hearts remains unknown. We optically mapped action potentials (APs) in excised rabbit hearts to test the hypothesis that hypoxic changes would be more severe in left ventricular (LV) working hearts (LWHs) than Langendorff (LANG) perfused hearts. We further hypothesized that KATP inhibition would prevent those changes. Optical APs were mapped when measuring LV developed pressure (LVDP), coronary flow rate and oxygen consumption in LANG and LWHs. Hearts were paced to increase workload and perfusate was deoxygenated to study the effects of myocardial hypoxia. A subset of hearts was perfused with 1 μm tolbutamide (TOLB) to identify the level of AP duration (APD) shortening attributed to KATP channel activation. During sinus rhythm, APD was shorter in LWHs compared to LANG hearts. APD in both LWHs and LANG hearts dropped steadily during deoxygenation. With TOLB, APDs in LWHs were longer at all workloads and APD reductions during deoxygenation were blunted in both LWHs and LANG hearts. At 50% perfusate oxygenation, APD and LVDP were significantly higher in LWHs perfused with TOLB (199 ± 16 ms; 92 ± 5.3 mmHg) than in LWHs without TOLB (109 ± 14 ms, P = 0.005; 65 ± 6.5 mmHg, P = 0.01). Our results indicate that KATP channels are activated to a greater extent in perfused hearts when the LV performs pressure–volume work. The results of the present study demonstrate the critical role of KATP channels in modulating myocardial function over a wide range of physiological conditions.
Keywords: action potential, electromechanical, energetics, KATP channel, optical mapping, oxygenation
Key points
Heart function is critically dependent upon the balance of energy production and utilization. Sarcolemmal ATP‐sensitive potassium channels (KATP channels) in cardiac myocytes adjust contractile function to compensate for the level of available energy.
Understanding the activation of KATP channels in working myocardium during high‐stress situations is crucial to the treatment of cardiovascular disease, especially ischaemic heart disease.
Using a new optical mapping approach, we measured action potentials from the surface of excised contracting rabbit hearts to assess when sarcolemmal KATP channels were activated during physiologically relevant workloads and during gradual reductions in myocardial oxygenation.
We demonstrate that left ventricular pressure is closely linked to KATP channel activation and that KATP channel inhibition with a low concentration of tolbutamide prevents electromechanical decline when oxygen availability is reduced. As a result, KATP channel inhibition probably exacerbates a mismatch between energy demand and energy production when myocardial oxygenation is low.
Abbreviations
- AP
action potential
- APD
action potential duration
- CCD
charge‐coupled device
- CFR
coronary flow rate
- CL
cycle length
- GLIB
glibenclamide
- GLM
general linear model
- KATP channel
ATP‐sensitive potassium channel
- LA
left atrium
- LANG
Langendorff heart preparation
- LV
left ventricular
- LVDP
left ventricular developed pressure
- LWH
left working heart
- MVO2
oxygen consumption rate
- NSR
normal sinus rhythm
- TOLB
tolbutamide
Introduction
Sarcolemmal ATP‐sensitive potassium channels (KATP) are assumed to be predominantly closed during normal conditions and open in response to increasing [ADP]/[ATP], linking cardiac electrophysiology to the balance of ATP production and utilization (Nichols et al. 1991; Zingman et al. 2007). KATP channel activation shortens action potential (AP) duration (APD) via the efflux of K+ from the myocyte and even a small percentage of channel opening can dramatically shorten APD (Nichols et al. 1991; Weiss et al. 1992). This sensitive link between energetic state and electrical function emphasizes the importance of KATP channels in regulating the cardiac response to low oxygenation and ischaemia. Furthermore, proper gating of KATP channels is crucial in conferring myocardial protection during high‐stress situations (Barry et al. 1985; Foster & Coetzee, 2016). During ischaemia, KATP channel activation has been shown to be cardioprotective, primarily through the reduction of intracellular Ca2± cycling by the shortened APD, thereby reducing developed force, ATP utilization by the actin‐myosin ATPase and myocardial oxygen requirements (Noma, 1983; Suzuki et al. 2001). Indeed, myocardial contractile recovery is worse and ischaemic damage is more severe when KATP channels are inhibited with sulphonylureas such as glibenclamide (GLIB) or tolbutamide (TOLB) (Cole et al. 1991; Mitani et al. 1991; Yao et al. 1993).
Studies in transgenic mice have further demonstrated that cardiac KATP channels are critical for the regulation of normal cardiac function during sympathetic activation and exercise (Zingman et al. 2002) and may also be an important mechanism of APD shortening with increased heart rate (APD restitution) (Zingman et al. 2011). Intracellular ATP concentration has been reported to be between 4 and 7 mm (Swain et al. 1982; Elliott et al. 1989; Katz et al. 1989) and, considering that KATP channels operate in a compartmentalized space (Zingman et al. 2007), even slight changes in intracellular [ADP]/[ATP] can significantly alter electrical activity (Elliott et al. 1989; Kantor et al. 1990; Nichols et al. 1991). Such changes are greatly influenced by the availability of oxygen to the mitochondria because a slight oxygen limitation could impair electron transport chain flux, slow H+ extrusion from the mitochondria and result in a proton electrochemical gradient that is insufficient to maintain a constant ATP level (Nichols et al. 1991; Decking et al. 1995). Indeed, KATP channels have been shown to be potently activated during hypoxia (Takizawa et al. 1996). Oxygenation is particularly important in excised working heart studies, where hypoxia‐induced impairment of ATP production may shorten APD to promote arrhythmias, as demonstrated during increased workload for isolated rabbit hearts perfused with and without erythrocytes (Gillis et al. 1996).
In isolated myocytes, sarcolemmal KATP channel activation and the associated shortening of APD is generally understood; whereas the complex relationship between myocardial workload, oxygenation, KATP channel activation and APD shortening in excised working hearts is incompletely understood. The objective of the present study was to gain organ‐level insight into electromechanical function during increases in workload and hypoxia when measuring changes in APD that indicate KATP channel activation. Accordingly, we used a newly developed non‐contact optical mapping approach (Zhang et al. 2016) to measure epicardial APs from fully contracting rabbit hearts perfused in either left ventricular (LV) working heart (LWH) mode or Langendorff (LANG) perfusion mode. With this approach, we studied APD differences between LWHs and LANG hearts at three workloads induced by increasing pacing rate, as well as during gradual perfusate deoxygenation. We also studied the interdependence between electrical function and LV pressure development in LWHs during gradual deoxygenation with and without KATP channel inhibition with TOLB, providing new insight into the relationship between the oxygenation of working myocardium and sarcolemmal KATP activation.
The overall hypothesis that guided our studies was that, at the same heart rate and perfusate oxygenation level, APDs would be shorter in myocardium at higher workloads (LWHs) compared to APDs of myocardium at lower workloads (LANG hearts). We further hypothesized that, to a significant degree, the shorter APDs would be caused by sarcolemmal KATP channel activation and, if so, then KATP inhibition should result in significant lengthening of APDs under the same conditions (heart rate, workload and oxygenation). Finally, we hypothesized that if reductions in LV developed pressure during gradual hypoxia were tightly regulated by sarcolemmal KATP activation then the inhibition of KATP channels should prevent reductions in LV developed pressure during gradual hypoxia, at least until hypoxic elevation of [ADP]/[ATP] reached a level to overcome TOLB KATP inhibition (Venkatesh et al. 1991). Testing these hypotheses reveals the vital role of KATP channels in the electromechanical function of excised perfused rabbit hearts during normoxia and hypoxia and also indicates that, even during normal sinus rhythm (NSR), KATP channels are highly activated in perfused LWHs.
Methods
Ethical approval
All animal procedures were completed in agreement with the institutional guidelines of The George Washington University and in compliance with suggestions from the panel of Euthanasia of the American Veterinary Medical Association and the National Institutes of Health (NIH) Guide for the Care and Use of Laboratory Animals.
Heart excision and LANG perfusion
New Zealand White rabbits (n = 25), weighing 3.3 ± 0.06 kg, were anaesthetized with an i.m. injection of ketamine (44 mg kg−1) and xylazine (10 mg kg−1). Heparin (2000 units) was administered via an i.v. ear vein injection, allowed to circulate for 15 min, and rabbits were then subjected to 5–10% isoflurane inhalation. Following cessation of pain reflex, hearts were quickly excised and LANG perfused at constant pressure (60 mmHg) using a working heart system (Hugo Sachs Elektronik, March, Germany). The perfusate was maintained at 37°C, gassed with 95%O2/5%CO2, and comprised (in mm) 115 NaCl, 3.3 KCl, 2.0 CaCl2, 1.4 MgSO4, 25.0 NaHCO3, 1.0 KH2PO4, 5.0 glucose and 1.0 lactate. The pulmonary artery was cannulated to measure coronary flow rate (CFR) and the oxygen content of the coronary venous effluent. Flow‐through optical sensors (Polestar, Needham Heights, MA, USA) measured the oxygen content (%O2) of perfusate entering the heart and leaving the pulmonary artery. A clamp‐on flow sensor (Transonic Systems, Ithaca, NY, USA) measured CFR. Aortic pressure was measured using a pressure transducer and bridge amplifier (World Precision Instruments, Sarasota, FL, USA). Nitroprusside (1 μm) was added to the perfusate of any heart with CFRs less than 30 mL min−1 to ensure adequate baseline oxygenation and myocardial oxygen consumption rate (MVO2). The experimental protocol commenced once heart function stabilized (LANG hearts). In a second set of studies, hearts were further prepared as LWH preparations.
LWH preparation
After function stabilized during LANG perfusion, hearts were prepared and transitioned to LWH mode as described by Neely et al. (1967), as well as in our previous studies (Asfour et al. 2012; Wengrowski et al. 2014). To prepare LWHs, the pulmonary veins and vena cavae were ligated and the left atrium (LA) was cannulated. Diastolic aortic pressure was consistently maintained at 60 mmHg. Aortic pressure and left atrial pressure were measured using pressure transducers and bridge amplifiers (World Precision Instruments). A catheter (SPR‐524; Millar Inc., Houston, TX, USA) was inserted into the LV via the aorta to measure LV pressure. The experimental protocol commenced once function stabilized in LWH mode.
Ratiometric optical mapping of epicardial APs
Our approach to optically map contracting rabbit hearts is similar to that recently developed by Zhang et al. (2016) for optically mapping contracting swine hearts. This approach provides high‐resolution optical mapping of APs from an area of the epicardium without the need to mechanically constrain the heart or use electromechanical uncoupling agents. Zhang et al. (2016) implemented dual wavelength excitation of di‐4‐ANEPPS by alternating epicardial illumination between blue (450 nm) and cyan (505 nm) LEDs. The fluorescence of di‐4‐ANEPPS was acquired within an emission band of 575–620 nm using a high‐speed charge‐coupled device (CCD) camera. Blue‐excited and cyan‐excited fluorescence signals were reconstructed using data from every other camera frame. A two‐step process eliminated motion artefact to enable the acquisition of optical APs from unrestrained contracting hearts. First, fiducial markers on the epicardium were tracked to remove motion artefact from the blue‐excited and cyan‐excited fluorescence signals. Then the ratio of the motion‐corrected signals removed any remaining motion artefact common to both signals to provide a final signal containing optical APs from which APDs could be measured. We implemented a version of this system appropriate for measuring optical APs from the epicardial surface of rabbit hearts (Fig. 1). With this approach, non‐contact assessments of electrophysiology can be made from hearts performing a range of physiological working conditions to more closely mimic in vivo energy supply and demand.
Figure 1. Optical mapping of transmembrane potential from the epicardial surface of unconstrained contracting rabbit hearts using motion tracking of fiducial markers and di‐4‐ANEPPS excitation ratiometry.
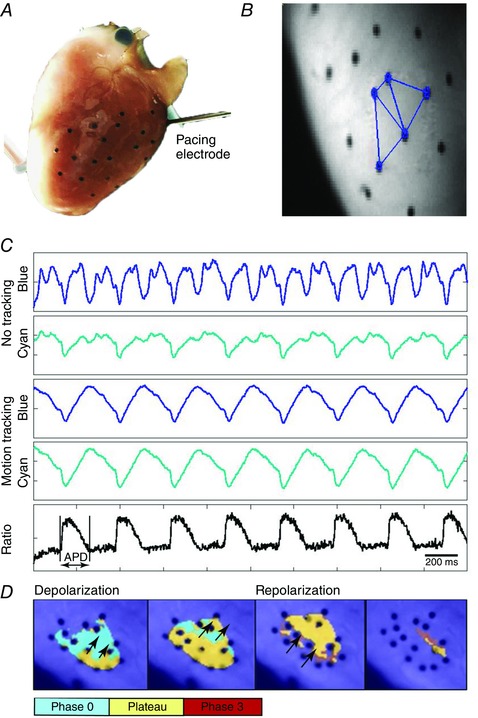
A, fiducial markers (black dots) were placed on the LV epicardium to enable tracking of local contractile motion. The LV pacing electrode is shown. B, markers were triangulated and local motion trajectories were computed for each triangle to remove motion artefact from fluorescence signals. Three such triangles are shown on a raw fluorescence image. C, di‐4‐ANNEPS fluorescence from one pixel is shown for blue and cyan excitation light before (first two signals) and after motion removal (signals three and four). The ratio of the motion corrected signals for blue and cyan excitation reveals the optical APs (last signal). D, pseudocoloured images of the upstroke (phase 0), plateau and repolarization (phase 3) of optical APs illustrate the propagation of paced waves across the epicardium on which markers were placed.
In our studies, circular fiducial markers (diameter 0.8 mm) were glued (Vetbond; 3m, St Paul, MN, USA) to the LV epicardium of rabbit hearts to track local epicardial deformation (Fig. 1 A). To stain the hearts for optical mapping, di‐4‐ANEPPS was dissolved in DMSO and diluted in 3 mL of perfusate, providing a 20 μm solution that was injected into the aorta during constant pressure LANG perfusion. Excitation light from 4 LEDs (Luxeon; Philips, Amsterdam, The Netherlands), two royal blue (450 nm) and two cyan (505 nm), was directed into a randomized fiber optic quad bundle (Schott, Auburn, NY, USA) and the output light was directed onto the region of the epicardial surface containing the fiducial markers. A 460 ± 135 nm excitation filter was attached to the output of the light guide, as was a light diffuser, which ensured uniform illumination of each excitation wavelength (Zhang et al. 2016). A Pulsemaster A3400 (World Precision Instruments) rapidly cycled the two sets of excitation LEDs to synchronize epicardial illumination with the frame acquisition trigger of a CCD camera (iXon DV860; Andor Technology, Belfast, UK). The camera was fitted with a lens (Navitar, Inc., Rochester, NY, USA) and a 635 ± 25 nm emission filter and fluorescence images (Fig. 1 B) were acquired at 750 frames s–1 (375 frames s–1 for each excitation wavelength). At the end of each study custom software tracked the fiducial markers, removed motion artefact from the signals, and computed the fluorescence ratio to provide optical APs from which APDs were measured (Fig. 1 C).
Experimental protocol
The protocol for LWH and LANG heart studies was a sequence of: (i) 60 s episodes of increased work induced by LV pacing; (ii) a stabilization period of NSR between each episode; and (iii) gradual perfusate deoxygenation, after which the study was terminated. Hearts were optically mapped at regular intervals during the pacing episodes and during perfusate deoxygenation. During the pacing episodes, the LV was paced at cycle lengths (CLs) of either 330, 220 or 170 ms to provide three discrete working conditions, as in our previous studies (Wengrowski et al. 2014). Optical APs were analysed after 60 s of continuous pacing at each CL to ensure that APD changes as a result of restitution had reached steady‐state (Dilly & Lab, 1988; Jin et al. 2008). After each pacing rate, the heart recovered for at least 3 min at NSR. To study the effects of hypoxia, the perfusate was switched from bubbling with 95%O2/5%CO2 gas to bubbling with N2 gas while also continuously pacing the heart at 330 ms CL. The oxygenation of perfusate (%O2) entering and leaving the heart was continuously measured via the oxygen sensors and the measurements were synchronized with the optically mapped APs.
Sarcolemmal KATP channel inhibition
TOLB and GLIB are hypoglycaemic sulphonylureas, a class of KATP channel inhibitors that are known to inhibit all types of KATP channels. Sulphonylureas have a wide range of efficacy that depends upon channel subunit composition and the relative expression of sulphonylurea subunits is tissue‐specific (Foster & Coetzee, 2016). For example, the SUR2B subunit is highly expressed in vascular smooth muscle and is inhibited by GLIB (EC50 = 0.3 μm) at concentrations much lower than TOLB (EC50 = 25 μm) (Dörschner et al. 1999). The SUR1 and SUR2A subunits are expressed in cardiac muscle, with SUR1 predominantly expressed in the atria (Glukhov et al. 2010), and both subunits are also inhibited by GLIB at concentrations much lower than those of TOLB (Gribble & Reimann, 2003). Studies have shown that 10 μm GLIB inhibits KATP channel activation in mouse ventricles (Glukhov et al. 2010) and lower GLIB concentrations (1 μm) cause vasoconstriction and ischaemia in isolated contracting rabbit hearts (Samaha et al. 1992). In contrast, isolated rat heart studies demonstrated that higher concentrations of TOLB (0.1–1.2 mm) had little effect on coronary flow and TOLB concentrations higher than 0.1 mm stimulated glycolysis (Kramer et al. 1983; Lampson et al. 1985).
In light of this previous work and in consideration that the relative expression of SUR1 and SUR2A subunits in rabbit ventricular tissue remains unknown, we completed a small set of experiments to determine the effects of GLIB and TOLB on excised perfused rabbit hearts. Either GLIB (10 μm; G0639; Sigma‐Aldrich, St Louis, MO, USA) or TOLB (1 μm; T0891; Sigma‐Aldrich) was administered to LWH and LANG hearts, allowed to circulate for at least 5 min, and then heart function was monitored for at least 20 min. The conclusion of those studies was that 1 μm TOLB did not significantly alter developed pressure or coronary flow during NSR, whereas APD in LWHs with TOLB was longer, indicating a significant level of sarcolemmal KATP channel inhibition by TOLB. Therefore, 1 μm TOLB was used in all further studies.
In the second set of LWH and LANG heart studies, TOLB dissolved in ethanol was added to the perfusate to provide a circulating concentration of 1 μm. After an incubation time of 15 min, the entire experimental protocol was repeated to measure the effect of KATP channel inhibition via TOLB on electromechanical function during pacing‐induced episodes of increased workload and gradual perfusate deoxygenation.
Data analysis
The optical APD was measured as the time interval between the maximum first derivative and the secondary peak of the second derivative (Efimov et al. 2004) for pixels within optically mapped regions of the LV epicardium (Fig. 1 C). For each study, at least 20 APDs were measured for a single site and the temporal mean and SD were calculated. APDs were measured at each pacing rate and at every 5% increment of deoxygenation. In the uncommon circumstance when optical mapping data were not acquired for a specific deoxygenation level, APDs were linearly interpolated (LWH, n = 2) between measurements to provide values at that percent deoxygenation. Left ventricular developed pressure (LVDP) was calculated as the difference between systolic and diastolic LV pressures, as measured by the Millar catheter in LWHs. Contractility and relaxation were measured as the maximum and minimum values of the first derivative of the LV pressure waveform. Oxygen consumption was calculated using the formula (Schenkman et al. 2003):
where CFR is the coronary flow rate (mL g−1·min−1), CT is oxygen solubility in Krebs–Henseleit solution at 37°C (1.30 × 10−6 m mmHg−1), PA is arterial oxygen content () and PV is venous oxygen content (). The temporal mean of a 2 s interval of the CFR, MVO2 and LVDP signals was computed for comparison between groups in each experimental condition.
Statistical analysis
Specific null hypotheses were that LWHs were no different from LANG hearts, LANG hearts perfused with TOLB were no different from LANG hearts without TOLB, and LWHs perfused with TOLB were no different from LWHs without TOLB. Measurements in each group were compared using an unpaired t test, ANOVA, or a general linear model (GLM), as appropriate (Minitab, version 17; Minitab Inc., State College, PA, USA). Tukey post hoc comparisons identified significant differences between specific groups. The perfusate deoxygenation data were analysed using a GLM to evaluate the group effect and the oxygenation effect. Data comparing three groups (control, GLIB and TOLB) were analysed using an ANOVA with Tukey post hoc comparisons to identify pairwise differences between groups. All measurements were assumed to be independent and the residuals were normally distributed. P < 0.05 was considered statistically significant.
Results
Average data during NSR for each experimental condition are shown in Table 1. Average heart weight across all studies was 11.8 ± 0.35 g. Overall, sinus rate was no different between LWH and LANG hearts, although MVO2 trended higher for LWH, as expected. The general effect of pacing‐induced increases in heart rate was that MVO2 and CFR trended higher, yet LVDP trended lower. With TOLB, sinus rate was not altered significantly, although MVO2, CFR, and LVDP were greater during NSR; LVDP significantly so. With TOLB, during pacing‐induced increases in heart rate, MVO2 and CFR trended higher than values without TOLB. LVDP decreased with shorter pacing CL but was significantly greater with TOLB at each pacing CL. Specific data and comparisons for each experimental condition are reported below.
Table 1.
Average function data for LANG and LWHs during NSR and at each pacing rate when perfusing with control solution (Control) and after administering TOLB to the perfusate (1 μm)
| LANG | LWH | ||||
|---|---|---|---|---|---|
| Control | TOLB | Control | TOLB | ||
| NSR | Heart rate (beats min–1) | 122 ± 4 | 105 ± 13 | 139 ± 11 | 150 ± 17 |
| MVO2 (μmol O2 g·min–1) | 1.4 ± 0.3 | 2.1 ± 0.1 | 2.1 ± 0.2 | 2.5 ± 0.7 | |
| CFR (mL min−1) | 26 ± 4.6 | 30 ± 8.0 | 26 ± 3.0 | 37 ± 4.1 | |
| LVDP (mmHg) | – | – | 84 ± 2.9 | 93 ± 6.4 | |
| Pacing CL (ms) | |||||
| 330 | MVO2 (μmol O2 g·min–1) | 1.8 ± 0.3 | 2.0 ± 0.1 | 2.4 ± 0.1 | 2.6 ± 0.1 |
| CFR (mL min−1) | 26 ± 4.7 | 39 ± 5.3 | 27 ± 2.0 | 36 ± 1.3 | |
| LVDP (mmHg) | – | – | 71 ± 2.4 | 86 ± 4.3* | |
| 220 | MVO2 (μmol O2 g·min–1) | 2.0 ± 0.5 | 3.0 ± 0.3 | 2.8 ± 0.2 | 3.2 ± 0.2 |
| CFR (mL min−1) | 29 ± 5.9 | 56 ± 3.1* | 28 ± 1.8 | 44 ± 3.3 | |
| LVDP (mmHg) | – | – | 57 ± 4.3 | 78 ± 6.7* | |
| 170 | MVO2 (μmol O2 g·min–1) | 2.1 ± 0.4 | 3.5 ± 0.3 | 2.8 ± 0.4 | 3.0 ± 0.3 |
| CFR (mL min−1) | 30 ± 6.0 | 64 ± 6.7 | 26 ± 2.3 | 40 ± 5.3 | |
| LVDP (mmHg) | – | – | 31 ± 5.9 | 58 ± 12* | |
*Significantly different from the corresponding control.
Values are the mean ± SE. LANG control: n = 7; LANG + TOLB: n = 4. LWH control: n = 8; LWH + TOLB: n = 5. Significance differences between measurements were determined using unpaired t tests.
Function during sinus rhythm and increased workload
Average sinus rate was similar for LANG hearts (122 ± 4 beats min–1) and LWHs (139 ± 11 beats min–1, P = 0.18), as was coronary flow during sinus rhythm (LANG: 25.7 ± 4.6 mL min−1, LWH: 25.8 ± 3.0 mL min−1; P = 0.98) (Table 1). However, APD during sinus rhythm (Fig. 2 A) was shorter for LWHs (153 ± 10 ms) than for LANG hearts (246 ± 17 ms, P = 0.005). At the longest pacing CL (330 ms), APD shortened to 195 ± 16 ms in LANG hearts and 136 ± 11 ms in LWHs (Fig. 2 A) and the difference was significant (P = 0.049). APD was also shorter in LWHs at pacing CLs of 220 ms (LANG 135 ± 17 ms, LWH: 108 ± 12 ms; P = 0.517) and 170 ms (LANG: 103 ± 13 ms, LWH: 72.7 ± 2; P = 0.145), although the differences were not significant. MVO2 was significantly higher (P ≤ 0.05) in LWHs than LANG hearts (Fig. 2 B) and appeared to increase in both preparations with shorter pacing CLs, although a MANOVA did not indicate adequate significance for the effect of heart rate on MVO2 (P = 0.09).
Figure 2. APD disparity between LANG and LWHs and oxygen consumption as a function of pacing CL.
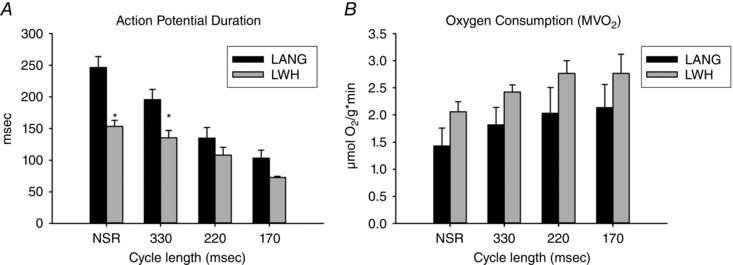
A, APD for LWHs was significantly lower than for LANG hearts at NSR and at a pacing CL of 330 ms but APDs were not significantly different at shorter pacing CLs. Values are the mean ± SE; LANG n = 5; LWH n = 6. B, oxygen consumption (MVO2) was significantly higher in LWHs than LANG hearts (MANOVA, P < 0.01) and trended higher in both preparations with shorter pacing CLs (MANOVA, P = 0.09). Values are the mean ± SE; LANG n = 6; LWH n = 7. *Significant difference (P < 0.05) between LANG and LWHs.
Working condition determines electrical decline during deoxygenation
APD progressively shortened in LWHs and LANG hearts as perfusate was gradually deoxygenated when pacing the hearts at a CL of 330 ms. Typical optical APs recorded from the LV epicardium at 100%, 50% and 25% perfusate oxygenation are shown in Fig. 3 A. The mean of the LWH deoxygenation curve for APD was significantly lower than the LANG curve (GLM, Tukey post hoc, P < 0.05) (Fig. 3 B), probably as a result of the significantly shorter APD for LWHs at 100% oxygenation and a CL of 330 ms. However, the rate of change of deoxygenation curves was not different between LWHs and LANG hearts. APD was shorter in LWH than LANG at 100% oxygenation (154 ± 10 ms; 204 ± 6.9 ms, P = 0.002) and also at 50% oxygenation (109 ± 14 ms; 165 ± 17 ms, P = 0.034) (Fig. 3 C). APD was not significantly different between LWHs and LANG hearts at 25% oxygenation (LWH: 89 ± 7.4; LANG: 118 ± 16, P = 0.071). The mean of the LWH deoxygenation curve for coronary flow was also significantly lower than the LANG curve (GLM, Tukey post hoc, P < 0.05) (Fig. 3 D).
Figure 3. Changes in APD and coronary flow in LANG and LWHs during gradual perfusate deoxygenation when pacing at 330 ms CL.
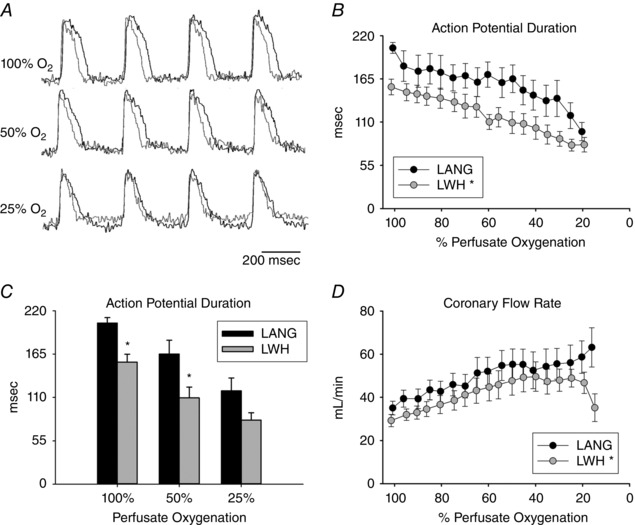
A, representative optical APs for a LANG heart (black) and a LWH (grey) at 100%, 50% and 25% perfusate oxygenation. B, APD decreases with perfusate deoxygenation in both LWHs and LANG hearts. APDs during perfusate deoxygenation were lower in LWHs than LANG hearts. C, APD disparity between LWHs and LANG hearts at 100%, 50% and 25% perfusate oxygenation. APDs for LWHs were significantly less than LANG hearts at 100% and 50% perfusate oxygenation. D, CFR increases with perfusate deoxygenation in LWHs and LANG hearts. CFR during perfusate deoxygenation was lower in LWHs than LANG hearts. Values are the mean ± SE; LANG n = 7; LWH n = 7. *Significant difference between LANG and LWHs.
Effects of GLIB
The experiments with GLIB confirmed the collective results of previous studies in canines (Zhang et al. 2003), guinea‐pigs (Daut et al. 1990) and rabbits (Samaha et al. 1992; Dhein et al. 2000) demonstrating that low concentrations of GLIB (typically 1–10 μm) alter vasomotor tone, often causing coronary vasoconstriction and downstream reductions in myocardial MVO2. The data comparing the function of LWHs and LANG hearts before and after the administration of either GLIB (10 μm) or TOLB (1 μm) are summarized in Table 2. Overall, we observed that coronary flow, MVO2 and LVDP trended lower with GLIB, whereas APD did not appear to be lengthened by GLIB (at 10 μm). In contrast, APD was longer with TOLB (significantly so for LWHs) and coronary flow, MVO2 and LVDP trended higher with TOLB.
Table 2.
Average LANG and LWH function during sinus rhythm before (Control) and after the administration of either GLIB (10 μm) or TOLB (1 μm)
| LANG | LWH | |||||
|---|---|---|---|---|---|---|
| Control | GLIB | TOLB | Control | GLIB | TOLB | |
| APD | 234 ± 17 | 237 ± 15 | 263 ± 5 | 179 ± 16 | 185 ± 26 | 200 ± 20* |
| MVO2 (μmol O2 g·min–1) | 1.4 ± 0.3 | 1.2 ± 0.3 | 2.1 ± 0.1 | 2.1 ± 0.2 | 1.2 ± 0.3*, † | 2.5 ± 0.7 |
| CFR (mL min−1) | 26 ± 4.0 | 11 ± 2.4*, † | 30 ± 8.0 | 26 ± 3.0 | 21 ± 6.2 | 37 ± 4.1 |
| LVDP (mmHg) | – | – | – | 81 ± 2.3 | 72 ± 12 | 93 ± 6.4* |
*Significantly different from the corresponding control.
†Significantly different from the corresponding TOLB measurement.
Values are the mean ± SE. LANG control: n = 7; LANG + GLIB: n = 4; LANG + TOLB: n = 4. LWH control: n = 6; LWH + GLIB: n = 3; LWH + TOLB: n = 5. Average MVO2, CFR and LVDP for Control and TOLB are the same data as shown in Table 1. Significance was tested using an ANOVA with Tukey post hoc comparisons.
KATP inhibition during NSR and increased workload
We found no difference in sinus rate before (99 ± 13 beats min–1) and after administering TOLB to LANG hearts (105 ± 13 beats min–1, P = 0.16). During sinus rhythm, APD for LANG and LANG + TOLB was not significantly different (246 ± 17 ms; 264 ± 6.3 ms, P = 0.373) (Fig. 4 A). At a pacing CL of 330 ms, APD in LANG was not different from LANG + TOLB (195 ± 16 ms; 217 ± 1.2, P = 0.239). However, at a pacing CL of 220 ms, APD in LANG shortened to 135 ± 17 ms whereas APD in LANG + TOLB only shortened to 187 ± 5.8 ms (P = 0.025) (Fig. 4 A). At 170 ms CL, APD trended higher for LANG + TOLB (155 ± 20 ms) than for LANG (103 ± 13 ms), although the difference was not significant (P = 0.115) (Fig. 4 A).
Figure 4. Effect of KATP channel inhibition (1 μm TOLB) on APD and LVDP as a function of pacing CL.
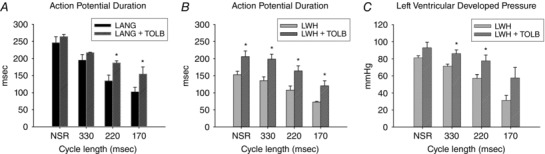
A, TOLB blunts workload‐induced APD shortening in LANG hearts when pacing at CLs of 220 ms and 170 ms (P = 0.025 and P = 0.006). B, APDs are longer in LWHs with TOLB during sinus rhythm and at all pacing CLs. C, LVDP is higher in LWHs with TOLB at pacing CLs of 330 and 220 ms. Values are the mean ± SE; LANG, n = 6; LWH, n = 7; LANG + TOLB, n = 3; LWH + TOLB, n = 4. *Significant difference with and without TOLB.
During NSR in LWHs, APD was longer with TOLB than without (206 ± 17 ms, 153 ± 10 ms, P = 0.042) (Fig. 4 B). APD was also longer at all pacing CLs in LWHs with TOLB than without (Fig. 4 B) (330 ms CL, 199 ± 14 ms, 135 ± 11 ms, P = 0.014; 220 ms CL, 164 ± 15 ms, 108 ± 12 ms, P = 0.022; and 170 ms CL, 121 ± 15 ms, 72.3 ± 1.9 ms, P = 0.047). At NSR, LVDP was not significantly higher in LWHs with TOLB than without (96.2 ± 5.9 mmHg, 81.2 ± 2.3 mmHg, P = 0.065) (Fig. 4 C). However, TOLB blunted reductions in LVDP when pacing at 330 ms CL (86.0 ± 4.3 mmHg, 71.2 ± 2.4 mmHg, P = 0.024) and 220 ms CL (77.6 ± 6.7 mmHg, 57.3 ± 4.3 mmHg, P = 0.038). There was no significant difference in LVDP with and without TOLB when pacing at 170 ms CL (57.7 ± 12 mmHg, 31.3 ± 5.9 mmHg, P = 0.121) (Fig. 4 C).
KATP inhibition during deoxygenation
At 100% oxygenation in LANG hearts, with a pacing CL of 330 ms, APD was longer with TOLB than without (230 ± 5.1 ms, 204 ± 6.9 ms, P = 0.017) (Fig. 5 A and B). During gradual deoxygenation (Fig. 5 A), APD was always longer with TOLB than without (GLM, Tukey post hoc, P < 0.05). At mid‐high levels of oxygenation (40–100%), APD shortened less with TOLB than without (Fig. 5 A). At 50% perfusate oxygenation, APD was significantly longer in LANG hearts with TOLB than without (203 ± 6.1 ms, 154 ± 18 ms, P = 0.039) (Fig. 5 B). At 25% oxygenation, APD was no different with and without TOLB (150 ± 18 ms, 118 ± 16 ms, P = 0.236). At 100% perfusate oxygenation, CFR was not significantly different with and without TOLB (40 ± 4 mL min−1, 30 ± 4 mL min−1, P = 0.856). However, during gradual deoxygenation, CFR for LANG hearts with TOLB increased much more than LANG hearts without TOLB, with flow rate peaking at 25% oxygenation and then dramatically falling with further deoxygenation (Fig. 5 C).
Figure 5. Effect of KATP channel inhibition (1 μm TOLB) on APD and CFR during gradual perfusate deoxygenation in LANG hearts when pacing at 330 ms CL.
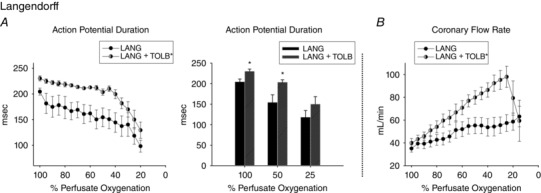
A, TOLB blunts APD shortening during perfusate deoxygenation in LANG hearts. With TOLB, APD remained relatively constant until 40% oxygenation, after which APD dramatically shortened. Without TOLB, APD gradually shortened with deoxygenation until 30% oxygenation, after which APD dramatically shortened. B, with TOLB, APD was significantly longer at 100% and 50% perfusate oxygenation. C, coronary flow of LANG hearts with TOLB has higher during perfusate deoxygenation and increased to a greater extent than the coronary flow of LANG hearts without TOLB. Values are the mean ± SE; LANG, n = 7; LANG + TOLB, n = 4. *Significant difference in APD between LANG hearts with and without TOLB.
The response of LWHs to TOLB (Fig. 6) was generally the same as that of LANG hearts (Fig. 5) but with additional insight provided by measurements of LVDP. At 100% oxygenation in LWHs hearts, APD was longer with TOLB than without (229 ± 22 ms, 154 ± 10 ms, P = 0.03) (Fig. 6 A). During gradual deoxygenation, APD was always longer with TOLB than without (GLM, Tukey post hoc, P < 0.05). At mid‐high levels of oxygenation (40–100%), as observed for LANG hearts, APD in LWHs shortened less with TOLB than without (Fig. 6 A). At 50% perfusate oxygenation, APD was significantly longer with TOLB than without (200 ± 16 ms, 109 ± 14 ms, P = 0.003). At 25% oxygenation, APD was not significantly different with and without TOLB (118 ± 17 ms, 81.6 ± 7.2 ms, P = 0.165) (Fig. 6 A).
Figure 6. Effect of KATP channel inhibition (1 μm TOLB) on APD, CFR, LVDP, contractility, and relaxation while pacing LWHs at a CL of 330 ms.
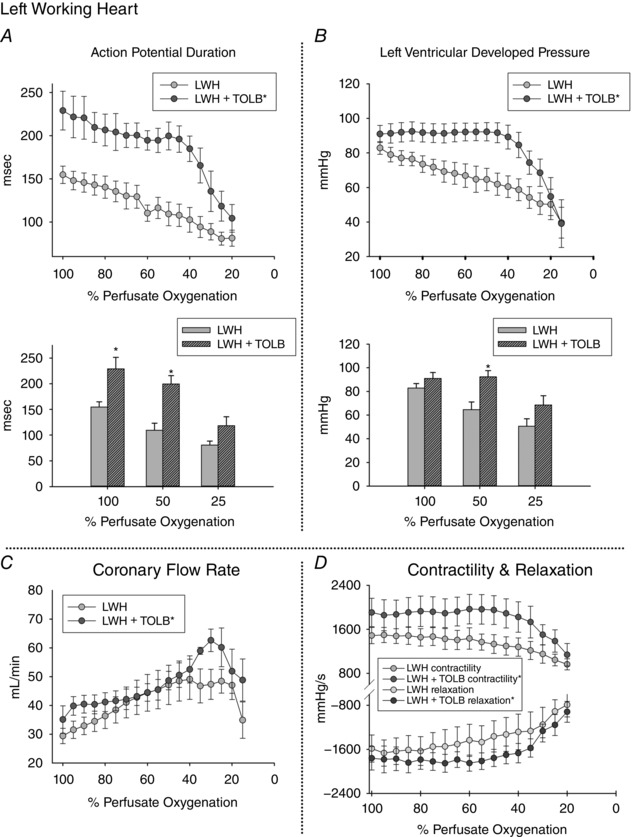
A, APDs were longer in LWHs with TOLB than for APDs measured without TOLB at all levels of perfusate oxygenation above 25%. With TOLB, APD shortened dramatically after 40% perfusate oxygenation. Without TOLB, APD shortened in a linear fashion throughout deoxygenation. At 25% oxygenation, APDs are the same for LWHs with and without TOLB. B, LVDP was higher in LWHs with TOLB than for LVDP measured without TOLB at all mid‐levels of perfusate oxygenation. With TOLB, LVDP for the high and mid‐levels of deoxygenation was constant, yet dropped dramatically after 40% perfusate oxygenation. Without TOLB, LVDP dropped in a linear fashion throughout deoxygenation. At 25% oxygenation, LVDP was the same for LWHs with and without TOLB. C, throughout deoxygenation, average coronary flow was significantly higher for LWHs with TOLB. With TOLB, coronary flow increased to a greater extent within the lower levels of oxygenation and dropped dramatically after 30% oxygenation. Without TOLB, coronary flow increased until 40% oxygenation, stopped increasing at oxygenation levels between 20% and 40% and dropped dramatically after 20% oxygenation. D, with TOLB, left ventricular contractility and relaxation curves were more positive (contractility) and more negative (relaxation) than for curves for LWHs without TOLB. Values are the mean ± SE; LWH, n = 6; LWH + TOLB, n = 5. *Significant difference between values measured with and without TOLB.
At oxygenation levels greater than 40%, developed pressure was higher in LWHs with TOLB than without (GLM, Tukey post hoc, P < 0.05) and dropped very little until oxygenation reached a level of ∼40%, at which point LVDP dropped dramatically (Fig. 6 B). Without TOLB, LVDP decreased in a linear fashion throughout deoxygenation. At 50% oxygenation, LVDP was higher for LWHs with TOLB than without (92.3 ± 5.3 mmHg, 64.6 ± 6.5 mmHg, P = 0.011). At 25% oxygenation, LVDP was the same with and without TOLB (68.5 ± 7.9 mmHg, 50.6 ± 8.8 mmHg, P = 0.112). Coronary flow increased, almost doubling, during deoxygenation with and without TOLB, and average flow was consistently higher with TOLB (GLM, Tukey post hoc, P < 0.05) (Fig. 6 C). With TOLB, CFR began to drop at 30% oxygenation; without TOLB, flow began to drop at 20% oxygenation. Such changes in coronary flow indicate a substantial change in vasomotor tone at low levels of oxygenation. On average, LV contractility and relaxation during deoxygenation were higher with TOLB than without (GLM, Tukey post hoc, P < 0.05) (Fig. 6 D), with both sets of curves exhibiting a similar trend during deoxygenation.
Discussion
Using a new non‐contact optical mapping approach, these studies are the first to compare APs mapped from contracting LANG perfused hearts with APs mapped from LWHs to identify the effect of sarcolemmal KATP channel activation over a wide range of physiological conditions. We have demonstrated that LANG hearts and LWHs have different time courses of repolarization and that those differences are brought about by sarcolemmal KATP channel activation. The administration of a relatively low concentration of the sulphonylurea TOLB to LWHs blunted reductions in LVDP and APD when pacing CL was shortened, indicating that such reductions are driven by [ADP]/[ATP] elevations, probably caused by demand‐induced oxygen deprivation in crystalloid‐perfused myocardium when workload is elevated. We additionally analysed functional differences between LWHs and LANG hearts during progressive perfusate deoxygenation and found that reductions in APD (LANG and LWHs) and LVDP (LWHs) were blunted when KATP channels were inhibited with TOLB. In LWHs, we found that TOLB substantially preserved APD, LVDP, contractility and relaxation until perfusate oxygenation levels dropped to 40%. Our results highlight the major influence of KATP channel activation in modulating myocardial energy utilization and may indicate that oxygenation of perfused myocardium should be carefully considered in almost all studies using excised contracting hearts.
Advantages of optical mapping of unconstrained contracting hearts
Recent advances in optical mapping of contracting hearts (Zhang et al. 2016) enable electrophysiological studies that encompass a wide range of metabolic perturbations, such as those reported in the present study. In conventional optical mapping studies, LANG perfused hearts are electromechanically uncoupled to prevent motion artefact from distorting optical APs. As expected, electromechanical uncoupling dramatically reduces myocardial energetics (Wengrowski et al. 2014) and the rate of oxygen consumption drops by at least 75% (Yaku et al. 1993; Schramm et al. 1994; Kuzmiak‐Glancy et al. 2015). As a result of the inarguable link between mechanical contraction and electrophysiological function, the translational applicability of results from non‐contracting hearts is bounded by the limitations of the experimental preparation. Indeed, in our previous work, we have demonstrated that biventricular working rabbit hearts have greater oxygen demand and are more sensitive to increases in workload than either unloaded or electromechanically uncoupled hearts (Wengrowski et al. 2014). Non‐contact optical mapping of contracting hearts removes the requirement of electromechanical uncoupling during optical mapping so that interaction between metabolism, contraction and electrophysiology can be studied with all the advantages of high spatiotemporal resolution imaging. Furthermore, in contrast to previous studies that measured monophasic APs in excised contracting hearts (Gillis et al. 1996), our non‐contact optical mapping approach does not introduce any constraint of contractile motion or potentially cause local ischaemia by pressing a monophasic AP electrode on to the surface of the myocardium.
Repolarization differences between LWHs and LANG hearts
We observed significant differences in APD between LWHs and LANG hearts during sinus rhythm, pacing at a CL of 330 ms (Fig. 2 A), and for perfusate oxygen levels above 25% (Fig. 3). Sarcolemmal KATP channel activation was the probable mechanism for APD shortening in both LANG and LWH, as indicated by the significant lengthening of APD after administering TOLB. Interestingly, for LANG hearts paced at a CL of 220 ms, APD with TOLB was significantly longer than APD without TOLB (Fig. 4 A), indicating significant KATP channel activation during elevated heart rates in unloaded hearts. It is also interesting that APD differences between LWHs and LANG hearts were attenuated during shorter pacing CLs (170 ms) and at oxygen levels below 25%, representing situations of significant hypoxia or anoxia. Indeed, our previous studies demonstrated that a pacing CL of 170 ms caused demand‐induced hypoxia in biventricular working hearts, which we suggested was the result of the limited oxygen buffering capacity of crystalloid perfusate (Wengrowski et al. 2014). Furthermore, previous studies of ischaemia have shown that APD shortens by ∼50% in contracting LANG and LWHs during no‐flow ischaemia (Botsford & Lukas, 1998; Wolk et al. 1999, 2000; Horimoto et al. 2002; Saltman et al. 2002). We observed a similar result: at 20% perfusate oxygenation, APD in LWHs and LANG hearts shortened by ∼50% (LANG: 51%, LWH: 47%) compared to the APD at 100% oxygenation (Figs 5 and 6), indicating the convergence of full KATP activation at low levels of oxygenation in both heart preparations.
Effect of KATP channel inhibition on electrical function in LANG hearts
The mismatch of ATP supply and demand at faster pacing CLs in LANG hearts activates KATP channels, as demonstrated by our finding of prolonged APD at 220 ms CL with TOLB (Fig. 4 A). Similarly, during gradual deoxygenation in LANG hearts, APD shortened sooner without KATP channel inhibition (Fig. 5). Tissue with longer APs has been suggested to be more sensitive to sarcolemmal KATP channel activation than tissue with shorter APs (Zhang et al. 2013). The longer AP of rabbit epicardium, relative to that of the rat, may explain the relatively large APD differences that we measured with and without TOLB, particularly in consideration of the low TOLB concentration used in our experiments. Interestingly, when KATP channels were inhibited with TOLB, APD was preserved until ∼40% perfusate oxygenation, after which APD dropped rapidly. It is well documented that sulphonylurea inhibition of KATP channels is ineffective during severely compromised metabolic states, as would occur at perfusate oxygenation levels below 40%, explaining the rapid shortening of APD at those levels (Weiss et al. 1992; Foster & Coetzee, 2016).
Effect of KATP channel inhibition on electromechanical function in LWHs
We found that, at 100% perfusate oxygenation, APD was longer at all pacing CLs in LWHs perfused with TOLB (Fig. 4 B). In particular, during sinus rhythm and when pacing at a CL of 330 ms, the APD of LWHs was longer with TOLB than without. This observation suggests that sarcolemmal KATP channels are activated in normal working myocardium during standard conditions of perfusion. This emphasizes the importance of maintaining sufficient oxygenation in LWHs and the careful interpretation of working heart function during physiological workloads. In LWHs with TOLB, APD during the shortest pacing CL of 170 ms (120 ± 15 ms) was within the range of APDs measured without TOLB during sinus rhythm (141 ± 8.6 ms). TOLB also significantly preserved LVDP when pacing at CLs of 330 and 220 ms (Fig. 4 C). The preservation of both APD and LVDP at these mid‐range pacing rates indicates the importance of KATP channels in modulating electromechanical function of perfused hearts during increased workload.
The energy dependence of working myocardium necessitates a balance between ATP supply and demand and KATP channels participate in regulating that balance (Zingman et al. 2007). Our deoxygenation measurements revealed a synchrony between APD and LVDP, which was apparent by the maintenance of electromechanical function during KATP inhibition (Figs 6 A and B). As we observed in LWHs during deoxygenation, within the setting of impaired ATP production, shortening of APD is an important mechanism for reducing contractile function and thus lowering energy utilization. APD shortening reduces intracellular Ca2+ cycling and prevents diastolic Ca2+ overload, thereby preserving the ratio of systolic to diastolic calcium, thus reducing myosin ATPase activity at the same time as maintaining normal cardiac relaxation (Kane et al. 2004; Yamada et al. 2006; Zingman et al. 2007). APD shortening has also been shown to lower Ca2± entry and SR Ca2± loading per beat, reiterating the modulation of Ca2± handling through electrical means (Zhou et al. 2009). Overall, these are mechanisms explaining the observed synchrony between APD and LVDP in LWHs. Our observation of maintained electromechanical function within the setting of significant hypoxia with KATP channel inhibition indicates a probable mismatch between energy demand and energy production during this condition, with such a conjecture being supported by previous work demonstrating that KATP channels have a protective role in ischaemia/reperfusion injury (Cole et al. 1991; McPherson et al. 1993; Suzuki et al. 2002).
Vasomodulation and coronary flow during hypoxia
Arterial smooth muscle relaxation is modulated by multiple factors, including KATP channel activation (Daut et al. 1990; Nelson et al. 1990), adenosine (Katori & Berne, 1966) and the release of endothelium‐derived nitric oxide (Quyyumi et al. 1995). LANG and LWH hearts with TOLB exhibited a dramatic CFR increase during gradual perfusate deoxygenation, recapitulating the well‐known vasodilatory response of coronary arteries to hypoxia (Laurent et al. 1956; Katori & Berne, 1966; Daut et al. 1990; Heineman et al. 1992; Cannon, 1998). In studies conducted by Duncker et al. (1995) KATP inhibition with GLIB reduced CFR at rest; however, increases in CFR with exercise were still observed (Duncker et al. 1995). This suggests that during KATP channel inhibition, adenosine production may significantly increase to provide another mechanism of vasodilatation other than KATP channel activation (Duncker et al. 1995). In our experiments, we postulate that the adenosine concentration increased during deoxygenation to cause the impressive increase in CFR that we observed in hearts perfused with TOLB (Fig. 5 B and 6 C). Interestingly, following the CFR increase with hypoxia, coronary flow dramatically dropped as the oxygen content of the perfusate lowered further, particularly in hearts with TOLB in both LANG and LWH (Fig. 5 B and 6 C). We surmise that this finding is similar to the isolated coronary artery studies in which canine coronary artery rings progressively relaxed as the oxygen content of the bathing solution was dropped to 5%, yet the rings contracted with a force greater than their normoxic tension when the bath oxygen content was reduced to 0% (Rubanyi & Vanhoutte, 1985). This hypercontracture is considered to be mediated primarily by endothelial cells through anoxia‐induced withdrawal of nitric oxide release (Gräser & Vanhoutte, 1991; Vanhoutte et al. 2005). Indeed, our observations are consistent with the sudden withdrawal of nitric oxide production, and subsequent vasoconstriction, once the hearts in our studies reached states of severe hypoxia or anoxia. Interestingly, a dramatic drop in CFR was not observed in LANG hearts (Fig. 3 D), perhaps because, as a result of their lower workload, the impact of hypoxia/anoxia was less in LANG hearts compared to LWHs, which may have slowed the withdrawal of nitric oxide, and thus slowed the resulting vasoconstriction.
Mechanoelectrical feedback
Mechanoelectrical feedback reduces APD with increases in stretch (Franz et al. 1992; Van Wagoner, 1993). However, we consider that, compared to increased I KATP current, an increase in stretch‐activated membrane current was not the mechanism that dominated the APD differences observed between LANG and LWHs. In a canine model, increases of end diastolic volume caused APD shortening (Hansen, 1993); however, in our studies, LWH preloads were not sufficiently large to cause such shortening of APD. Additionally, the addition of TOLB to LWH resulted in a lengthening of APD at baseline despite similar LVDPs, indicating no change in stretch, further suggesting the observed differences were primarily a result of KATP channel activation.
Conclusions
Our assessments of changes in electromechanical function resulting from increased workload and gradual deoxygenation provide new insight into the dynamics of KATP channel activation within perfused myocardial tissue. We found that, at the same heart rate and perfusate oxygenation level, APDs were shorter in LWHs than in LANG perfused hearts and, furthermore, that APD differences between the two heart preparations diminished at high heart rates and at low levels of perfusate oxygenation. These results are consistent with a higher level of sarcolemmal KATP channel activation when excised perfused myocardium performs pressure–volume work, indicating elevated [ADP]/[ATP], probably as a result of inadequate delivery of oxygen to the myocardium by crystalloid perfusate (Gillis et al. 1996; Kuzmiak‐Glancy et al. 2015). The results also indicate that KATP channels can be highly activated in perfused rabbit hearts at high heart rates. Significant KATP channel activation in LWHs and LANG perfused hearts was confirmed by the finding that TOLB blunted APD shortening during increased workload in both heart preparations. During gradual deoxygenation, TOLB also blunted APD shortening in both heart preparations and almost abolished reductions in LVDP in LWHs until 60% perfusate deoxygenation. This result is consistent with a tight coupling between KATP activation and ventricular pressure development and the general notion that KATP channel inhibition prevents a loss of function when oxygen availability is reduced, although such inhibition will probably exacerbate a mismatch between myocyte energy demand and myocyte energy production.
Additional information
Competing interests
The authors declare that they have no competing interests.
Author contributions
All experiments were performed in the laboratory of M. W. Kay at the George Washington University in Washington, DC. Study design and the experimental approach were discussed among all of the authors. KG, SK‐G and MWK were responsible for data acquisition. HZ and JMR guided the optical mapping approach and assisted in analysis and interpretation of the data. All authors critically revised the work for intellectual content. All authors listed approved the final version of the manuscript submitted for publication, support the integrity and accuracy of the studies, and qualify for authorship.
Funding
This work was supported by grants from the National Institutes of Health (R01‐HL095828 and R21‐HL132618 to MWK and R01‐HL115108 JMR) and the American Heart Association (14POST20490181 to SK‐G).
Acknowledgements
The authors thank Mary Kate Dwyer for many insightful conversations about the results and for her valuable assistance during the studies.
References
- Asfour H, Wengrowski A, Jaimes R III, Swift L & Kay M (2012). NADH fluorescence imaging of isolated biventricular working rabbit hearts. J Vis Exp 65, e4115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barry WH, Hasin Y & Smith TW (1985). Sodium pump inhibition, enhanced calcium influx via sodium‐calcium exchange, and positive inotropic response in cultured heart cells. Circ Res 56, 231–241. [DOI] [PubMed] [Google Scholar]
- Botsford MW & Lukas A (1998). Ischemic Preconditioning and arrhythmogenesis in the rabbit heart: effects on epicardium versus endocardium. J Mol Cell Cardiol 30, 1723–1733. [DOI] [PubMed] [Google Scholar]
- Cannon RO (1998). Role of nitric oxide in cardiovascular disease: focus on the endothelium. Clin Chem 44, 1809–1819. [PubMed] [Google Scholar]
- Cole WC, McPherson CD & Sontag D (1991). ATP‐regulated K+ channels protect the myocardium against ischemia/reperfusion damage. Circ Res 69, 571–581. [DOI] [PubMed] [Google Scholar]
- Daut J, Maier‐Rudolph W, von Beckerath N, Mehrke G, Günther K & Goedel‐Meinen L (1990). Hypoxic dilation of coronary arteries is mediated by ATP‐sensitive potassium channels. Science 247, 1341–1344. [DOI] [PubMed] [Google Scholar]
- Decking UK, Reffelmann T, Schrader J & Kammermeier H (1995). Hypoxia‐induced activation of KATP channels limits energy depletion in the guinea pig heart. Am J Physiol Heart Circ Physiol 269, H734–H742. [DOI] [PubMed] [Google Scholar]
- Dhein S, Pejman P & Krüsemann K (2000). Effects of the IK.ATP blockers glibenclamide and HMR1883 on cardiac electrophysiology during ischemia and reperfusion. Eur J Pharmacol 398, 273–284. [DOI] [PubMed] [Google Scholar]
- Dilly SG & Lab MJ (1988). Electrophysiological alternans and restitution during acute regional ischaemia in myocardium of anaesthetized pig. J Physiol 402, 315–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dörschner H, Brekardin E, Uhde I, Schwanstecher C & Schwanstecher M (1999). Stoichiometry of sulfonylurea‐induced ATP‐sensitive potassium channel closure. Mol Pharmacol 55, 1060–1066. [DOI] [PubMed] [Google Scholar]
- Duncker DJ, van Zon NS, Pavek TJ, Herrlinger SK & Bache RJ (1995). Endogenous adenosine mediates coronary vasodilation during exercise after K(ATP)+ channel blockade. J Clin Invest 95, 285–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efimov IR, Nikolski VP & Salama G (2004). Optical imaging of the heart. Circ Res 95, 21–33. [DOI] [PubMed] [Google Scholar]
- Elliott AC, Smith GL & Allen DG (1989). Simultaneous measurements of action potential duration and intracellular ATP in isolated ferret hearts exposed to cyanide. Circ Res 64, 583–591. [DOI] [PubMed] [Google Scholar]
- Foster MN & Coetzee WA (2016). KATP channels in the cardiovascular system. Physiol Rev 96, 177–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franz MR, Cima R, Wang D, Profitt D & Kurz R (1992). Electrophysiological effects of myocardial stretch and mechanical determinants of stretch‐activated arrhythmias. Circulation 86, 968–978. [DOI] [PubMed] [Google Scholar]
- Gillis AM, Kulisz E & Mathison HJ (1996). Cardiac electrophysiological variables in blood‐perfused and buffer‐perfused, isolated, working rabbit heart. Am J Physiol Heart Circ Physiol 271, H784–H789. [DOI] [PubMed] [Google Scholar]
- Glukhov AV, Flagg TP, Fedorov VV, Efimov IR & Nichols CG (2010). Differential K(ATP) channel pharmacology in intact mouse heart. J Mol Cell Cardiol 48, 152–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gräser T & Vanhoutte PM (1991). Hypoxic contraction of canine coronary arteries: role of endothelium and cGMP. Am J Physiol Heart Circ Physiol 261, H1769–H1777. [DOI] [PubMed] [Google Scholar]
- Gribble FM & Reimann F (2003). Sulphonylurea action revisited: the post‐cloning era. Diabetologia 46, 875–891. [DOI] [PubMed] [Google Scholar]
- Hansen DE (1993). Mechanoelectrical feedback effects of altering preload, afterload, and ventricular shortening. Am J Physiol Heart Circ Physiol 264, H423–H432. [DOI] [PubMed] [Google Scholar]
- Heineman FW, Kupriyanov VV, Marshall R, Fralix TA & Balaban RS (1992). Myocardial oxygenation in the isolated working rabbit heart as a function of work. Am J Physiol Heart Circ Physiol 262, H255–H267. [DOI] [PubMed] [Google Scholar]
- Horimoto H, Nakai Y, Mieno S, Nomura Y, Nakahara K & Sasaki S (2002). Oral hypoglycemic sulfonylurea glimepiride preserves the myoprotective effects of ischemic preconditioning. J Surg Res 105, 181–188. [DOI] [PubMed] [Google Scholar]
- Jin Q, Chen X, Smith WM, Ideker RE & Huang J (2008). Effects of procainamide and sotalol on restitution properties, dispersion of refractoriness, and ventricular fibrillation activation patterns in pigs. J Cardiovasc Electrophysiol 19, 1090–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kane GC, Behfar A, Yamada S, Perez‐Terzic C, O'Cochlain F, Reyes S, Dzeja PP, Miki T, Seino S & Terzic A (2004). ATP‐sensitive K+ channel knockout compromises the metabolic benefit of exercise training, resulting in cardiac deficits. Diabetes 53 (Suppl 3), S169–S175. [DOI] [PubMed] [Google Scholar]
- Kantor PF, Coetzee WA, Carmeliet EE, Dennis SC & Opie LH (1990). Reduction of ischemic K+ loss and arrhythmias in rat hearts. Effect of glibenclamide, a sulfonylurea. Circ Res 66, 478–485. [DOI] [PubMed] [Google Scholar]
- Katori M & Berne RM (1966). Release of adenosine from anoxic hearts: relationship to coronary flow. Circ Res 19, 420–425. [DOI] [PubMed] [Google Scholar]
- Katz LA, Swain JA, Portman MA & Balaban RS (1989). Relation between phosphate metabolites and oxygen consumption of heart in vivo. Am J Physiol Heart Circ Physiol 256, H265–H274. [DOI] [PubMed] [Google Scholar]
- Kramer JH, Lampson WG & Schaffer SW (1983). Effect of tolbutamide on myocardial energy metabolism. Am J Physiol Heart Circ Physiol 245, H313–H319. [DOI] [PubMed] [Google Scholar]
- Kuzmiak‐Glancy S, Jaimes R III, Wengrowski AM & Kay MW (2015). Oxygen demand of perfused heart preparations: how electromechanical function and inadequate oxygenation affect physiology and optical measurements. Exp Physiol 100, 603–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lampson WG, Kramer JH & Schaffer SW (1985). Effect of tolbutamide on myocardial energy metabolism of the ischemic heart. Biochem Pharmacol 34, 803–809. [DOI] [PubMed] [Google Scholar]
- Laurent D, Bolene‐Williams C, Williams FL & Katz LN (1956). Effects of heart rate on coronary flow and cardiac oxygen consumption. Am J Physiol 185, 355–364. [DOI] [PubMed] [Google Scholar]
- McPherson CD, Pierce GN & Cole WC (1993). Ischemic cardioprotection by ATP‐sensitive K+ channels involves high‐energy phosphate preservation. Am J Physiol 265, H1809–H1818. [DOI] [PubMed] [Google Scholar]
- Mitani A, Kinoshita K, Fukamachi K, Sakamoto M, Kurisu K, Tsuruhara Y, Fukumura F, Nakashima A & Tokunaga K (1991). Effects of glibenclamide and nicorandil on cardiac function during ischemia and reperfusion in isolated perfused rat hearts. Am J Physiol 261, H1864–H1871. [DOI] [PubMed] [Google Scholar]
- Neely JR, Liebermeister H, Battersby EJ & Morgan HE (1967). Effect of pressure development on oxygen consumption by isolated rat heart. Am J Physiol 212, 804–814. [DOI] [PubMed] [Google Scholar]
- Nelson MT, Patlak JB, Worley JF & Standen NB (1990). Calcium channels, potassium channels, and voltage dependence of arterial smooth muscle tone. Am J Physiol Cell Physiol 259, C3–C18. [DOI] [PubMed] [Google Scholar]
- Nichols CG, Ripoll C & Lederer WJ (1991). ATP‐sensitive potassium channel modulation of the guinea pig ventricular action potential and contraction. Circ Res 68, 280–287. [DOI] [PubMed] [Google Scholar]
- Noma A (1983). ATP‐regulated K+ channels in cardiac muscle. Nature 305, 147–148. [DOI] [PubMed] [Google Scholar]
- Quyyumi AA, Dakak N, Andrews NP, Gilligan DM, Panza JA & Cannon RO (1995). Contribution of nitric oxide to metabolic coronary vasodilation in the human heart. Circulation 92, 320–326. [DOI] [PubMed] [Google Scholar]
- Rubanyi GM & Vanhoutte PM (1985). Hypoxia releases a vasoconstrictor substance from the canine vascular endothelium. J Physiol 364, 45–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saltman AE, Aksehirli TO, Valiunas V, Gaudette GR, Matsuyama N, Brink P & Krukenkamp IB (2002). Gap junction uncoupling protects the heart against ischemia. J Thorac Cardiovasc Surg 124, 371–376. [DOI] [PubMed] [Google Scholar]
- Samaha FF, Heineman FW, Ince C, Fleming J & Balaban RS (1992). ATP‐sensitive potassium channel is essential to maintain basal coronary vascular tone in vivo. Am J Physiol Cell Physiol 262, C1220–C1227. [DOI] [PubMed] [Google Scholar]
- Schenkman KA, Beard DA, Ciesielski WA, Feigl EO, Kenneth A & Wayne A (2003). Comparison of buffer and red blood cell perfusion of guinea pig heart oxygenation. Am J Physiol Heart Circ Physiol 285, H1819–H1825. [DOI] [PubMed] [Google Scholar]
- Schramm M, Klieber HG & Daut J (1994). The energy expenditure of actomyosin‐ATPase, Ca2+‐ATPase and Na+,K+‐ATPase in guinea‐pig cardiac ventricular muscle. J Physiol 481.3, 647–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki M, Li RA, Miki T, Uemura H, Sakamoto N, Ohmoto‐Sekine Y, Tamagawa M, Ogura T, Seino S, Marban E, Nakaya H, Marbán E & Nakaya H (2001). Functional roles of cardiac and vascular ATP‐sensitive potassium channels clarified by Kir6.2‐knockout mice. Circ Res 88, 570–577. [DOI] [PubMed] [Google Scholar]
- Suzuki M, Sasaki N, Miki T, Sakamoto N, Ohmoto‐Sekine Y, Tamagawa M, Seino S, Marbán E & Nakaya H (2002). Role of sarcolemmal K(ATP) channels in cardioprotection against ischemia/reperfusion injury in mice. J Clin Invest 109, 509–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swain JL, Sabina RL, McHale PA, Greenfield JC Jr. & Holmes EW (1982). Prolonged myocardial nucleotide depletion after brief ischemia in the open‐chest dog. Am J Physiol Heart Circ Physiol 242, H818–H826. [DOI] [PubMed] [Google Scholar]
- Takizawa T, Hara Y, Saito T, Masuda Y & Nakaya H (1996). Alpha 1‐Adrenoceptor stimulation partially inhibits ATP‐sensitive K+ current in guinea pig ventricular cells: attenuation of the action potential shortening induced by hypoxia and K+ channel openers. J Cardiovasc Pharmacol 28, 799–808. [DOI] [PubMed] [Google Scholar]
- Vanhoutte PM, Feletou M & Taddei S (2005). Endothelium‐dependent contractions in hypertension. Br J Pharmacol 144, 449–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatesh N, Lamp ST & Weiss JN (1991). Sulfonylureas, ATP‐sensitive K+ channels, and cellular K+ loss during hypoxia, ischemia, and metabolic inhibition in mammalian ventricle. Circ Res 69, 623–637. [DOI] [PubMed] [Google Scholar]
- Van Wagoner DRR (1993). Mechanosensitive gating of atrial ATP‐sensitive potassium channels. Circ Res 72, 973–983. [DOI] [PubMed] [Google Scholar]
- Weiss JN, Venkatesh N & Lamp ST (1992). ATP‐sensitive K+ channels and cellular K+ loss in hypoxic and ischaemic mammalian ventricle. J Physiol 447, 649–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wengrowski AM, Kuzmiak‐Glancy S, Jaimes R & Kay MW (2014). NADH changes during hypoxia, ischemia, and increased work differ between isolated heart preparations. Am J Physiol Heart Circ Physiol 306, H529–H537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolk R, Kane KA, Cobbe SM & Hicks MN (1999). Facilitation of spontaneous defibrillation by moxonidine during regional ischaemia in an isolated working rabbit heart model. Eur J Pharmacol 367, 25–32. [DOI] [PubMed] [Google Scholar]
- Wolk R, Sneddon KP, Dempster J, Kane KA, Cobbe SM & Hicks MN (2000). Regional electrophysiological effects of left ventricular hypertrophy in isolated rabbit hearts under normal and ischaemic conditions. Cardiovasc Res 48, 120–128. [DOI] [PubMed] [Google Scholar]
- Yaku H, Slinker BK, Mochizuki T, Lorell BH & LeWinter MM (1993). Use of 2,3‐butanedione monoxime to estimate nonmechanical VO2 in rabbit hearts. Am J Physiol Heart Circ Physiol 34, H834–H842. [DOI] [PubMed] [Google Scholar]
- Yamada S, Kane GC, Behfar A, Liu X‐K, Dyer RB, Faustino RS, Miki T, Seino S & Terzic A (2006). Protection conferred by myocardial ATP‐sensitive K+ channels in pressure overload‐induced congestive heart failure revealed in KCNJ11 Kir6.2‐null mutant. J Physiol 577, 1053–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao Z, Cavero I & Gross GJ (1993). Activation of cardiac KATP channels: an endogenous protective mechanism during repetitive ischemia. Am J Physiol Heart Circ Physiol 264, H495–H504. [DOI] [PubMed] [Google Scholar]
- Zhang H, Iijima K, Huang J, Walcott GP & Rogers JM (2016). Optical mapping of membrane potential and epicardial deformation in beating hearts. Biophys J 111, 438–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang HX, Silva JR, Lin Y‐W, Verbsky JW, Lee US, Kanter EM, Yamada KA, Schuessler RB & Nichols CG (2013). Heterogeneity and function of K(ATP) channels in canine hearts. Hear Rhythm 10, 1576–1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, From AHL, Ugurbil K & Bache RJ (2003). Myocardial oxygenation and high‐energy phosphate levels during KATP channel blockade. Am J Physiol Heart Circ Physiol 285, H1420–H1427. [DOI] [PubMed] [Google Scholar]
- Zhou L, Cortassa S, Wei A‐C, Aon MA, Winslow RL & O'Rourke B (2009). Modeling cardiac action potential shortening driven by oxidative stress‐induced mitochondrial oscillations in guinea pig cardiomyocytes. Biophys J 97, 1843–1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zingman LV, Alekseev AE, Hodgson‐Zingman DM & Terzic A (2007). ATP‐sensitive potassium channels: metabolic sensing and cardioprotection. J Appl Physiol 103, 1888–1893. [DOI] [PubMed] [Google Scholar]
- Zingman LV, Hodgson DM, Bast PH, Kane GC, Perez‐Terzic C, Gumina RJ, Pucar D, Bienengraeber M, Dzeja PP, Miki T, Seino S, Alekseev AE & Terzic A (2002). Kir6.2 is required for adaptation to stress. Proc Natl Acad Sci USA 99, 13278–13283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zingman LV, Zhu Z, Sierra A, Stepniak E, Burnett CM‐L, Maksymov G, Anderson ME, Coetzee WA & Hodgson‐Zingman DM (2011). Exercise‐induced expression of cardiac ATP‐sensitive potassium channels promotes action potential shortening and energy conservation. J Mol Cell Cardiol 51, 72–81. [DOI] [PMC free article] [PubMed] [Google Scholar]