

Abstract
Objective:
To report paroxysmal episodes of cerebellar ataxia in a patient with anti–contactin-associated protein-like 2 (CASPR2) antibody-related autoimmune encephalitis and to search for similar paroxysmal ataxia in a cohort of patients with anti–CASPR2 antibody-associated autoimmune encephalitis.
Methods:
We report a patient with paroxysmal episodes of cerebellar ataxia observed during autoimmune encephalitis with anti-CASPR2 antibodies. In addition, clinical analysis was performed in a retrospective cohort of 37 patients with anti-CASPR2 antibodies to search for transient episodes of ataxia. Paroxysmal symptoms were further specified from the referral physicians, the patients, or their relatives.
Results:
A 61-year-old man with limbic encephalitis and anti-CASPR2 antibodies developed stereotyped paroxysmal episodes of cerebellar ataxia, including gait imbalance, dysarthria, and dysmetria, 1 month after the onset of the encephalitis. The ataxic episodes were specifically triggered by orthostatism and emotions. Both limbic symptoms and transient ataxic episodes resolved after treatment with steroids and IV cyclophosphamide. Among 37 other patients with anti-CASPR2 antibodies, we identified 5 additional cases with similar paroxysmal ataxic episodes that included gait imbalance (5 cases), slurred speech (3 cases), limb dysmetria (3 cases), and nystagmus (1 case). All had concomitant limbic encephalitis. Paroxysmal ataxia was not observed in patients with neuromyotonia or Morvan syndrome. Triggering factors (orthostatism or anger) were reported in 4 patients. Episodes resolved with immunomodulatory treatments in 4 patients and spontaneously in 1 case.
Conclusions:
Paroxysmal cerebellar ataxia must be added to the spectrum of the anti-CASPR2 antibody syndrome.
Episodic ataxias (EAs) are a group of hereditary channelopathies whose common feature is the occurrence of paroxysmal episodes of cerebellar ataxia.1 Ataxic episodes usually last a few minutes to a few days and can be triggered by emotions, abrupt movements, exercise, or fever. Depending on which ion channel gene is mutated, additional symptoms, such as neuromyotonia or epilepsy, can occur.2 Conversely, paroxysmal symptoms are rare in patients with antineuronal antibody-associated neurologic disorders, and EAs have not yet been reported in such cases. In this study, we report a patient with paroxysmal episodes of ataxia developed during autoimmune encephalitis with anti-CASPR2 antibodies. To assess the relevance of our case, we retrospectively searched for similar episodes of transient ataxia in a cohort of patients with anti-CASPR2 antibodies. Anti–CASPR2 antibody-related disorders encompass a wide range of neurologic autoimmune syndromes, including autoimmune encephalitis, neuromyotonia, and Morvan syndrome. A recent publication by Van Sonderen et al.3 showed that up to 77% of the patients with such antibodies had at least 3 cumulated core neurologic symptoms, including encephalic signs, cerebellar symptoms, peripheral nerve hyperexcitability, dysautonomia, neuropathic pain, insomnia, and weight loss. Our study may contribute to further delineate anti–CASPR2 antibody-related clinical presentation.
METHODS
We report a patient with autoimmune encephalitis, anti-CASPR2 antibodies, and paroxysmal cerebellar ataxia. Anti-CASPR2 antibodies were screened in serum and CSF as previously described.4 Positivity of both an immunohistofluorescent assay on rat brain slices and a cell-based binding assay with HEK-293–transfected cells were needed to confirm the presence of anti-CASPR2 antibodies. A signed patient consent-to-disclose form has been obtained from the patient for a video recording of one of the episodes.
We also studied the clinical files of 37 patients with anti-CASPR2 antibodies, detected in their CSF or sera at the Centre de Référence National pour les Syndromes Neurologiques Paranéoplasiques (Lyon, France) between March 2009 and August 2016, to search for similar transient cerebellar symptoms. Thirty-three of those patients have been previously reported.4 Written informed consent was obtained from all patients with approval of the Institutional Review Board of the Hospices Civils de Lyon. We selected all patients who had been reported by their referral physicians to have symptoms that were both transient and suggestive of cerebellar impairment, i.e., gait imbalance, slurred speech, or limb dysmetria. Further information was collected by telephone from the referral physicians, and, when possible, from the patients themselves or their relatives.
RESULTS
Index case.
A 61-year-old man was hospitalized for evaluation after a tonic-clonic generalized seizure. He was still active as a corporate executive, and his medical history included high blood pressure, diabetes, myocardial infarction, and a smoking habit. No prodromal or postictal symptom was reported, but the patient reported slight memory impairment, difficulties to concentrate at work, unusual emotiveness, and anxiety over a few days before the seizure. The patient was treated with levetiracetam and clobazam. However, the cognitive symptoms persisted and several partial temporal lobe seizures occurred. One month after the first seizure, the patient began to experience repeated episodes of slurred speech, gait ataxia, and slight dysmetria of the limbs (see video at Neurology.org/nn). These events occurred 3–4 times a day, were often triggered by emotions and orthostatism, and lasted less than 1 minute. No cerebellar symptom was observed between the episodes. Levetiracetam was switched to lacosamide without any improvement of the symptoms.
Brain MRI was normal. EEG found an asymmetric temporal lobe slowing. CSF analysis found 16 white blood cells/mm3, a protein level of 65 mg/dL, and no oligoclonal band. Anti-CASPR2 antibodies were detected (end-point dilution using cell-binding–based assay, 1/40,960, in serum and 1/5,120 in CSF). Brain single-photon emission CT found bilateral frontotemporal hypoperfusion with normal perfusion of basal ganglia and cerebellum. Full-body CT was normal. Sequencing of the genes involved in EA types 1 and 2, KCNA1 and CACNA1A, found that the patient carried a rare polymorphism in intron 39 of CACNA1A (c5843-14G>A).5,6 KCNA1 was normal. The patient was treated with 3 daily injections of 1 g methylprednisolone followed by 6 monthly pulses of 1 g cyclophosphamide. The frequency of the ataxia episodes decreased immediately; they had totally stopped 5 days after the end of the methylprednisolone pulses. The patient recovered all his cognitive abilities 2 weeks after the initiation of the treatment and had his last seizure 3 months after the first visit. At month 7 of follow-up, the patient remained asymptomatic and seizure free. At the peak of his disease, the patient had presented with only 2 core symptoms as defined by Van Sonderen et al.3 (encephalic signs and cerebellar ataxia).
Cohort study.
Among the cohort of 37 other patients with anti-CASPR2 antibodies (20 with encephalitis and 17 with neuromyotonia or Morvan syndrome), we retrospectively identified 5 patients with transient symptoms suggestive of cerebellar impairment. All the patients were men, with a median age of 60 years (range 57–69). Median follow-up (range) was 45.6 months (19.3–113.6). One patient had a history of surgically treated thyroid carcinoma. All these 5 patients had anti-CASPR2 antibodies in both serum and CSF and had a presentation of autoimmune encephalitis with prominent seizures and amnesia, as we previously reported.4 Three patients also developed mild permanent cerebellar symptoms, concomitantly to (2 cases) or after (1 case) the onset of paroxysmal ataxia.
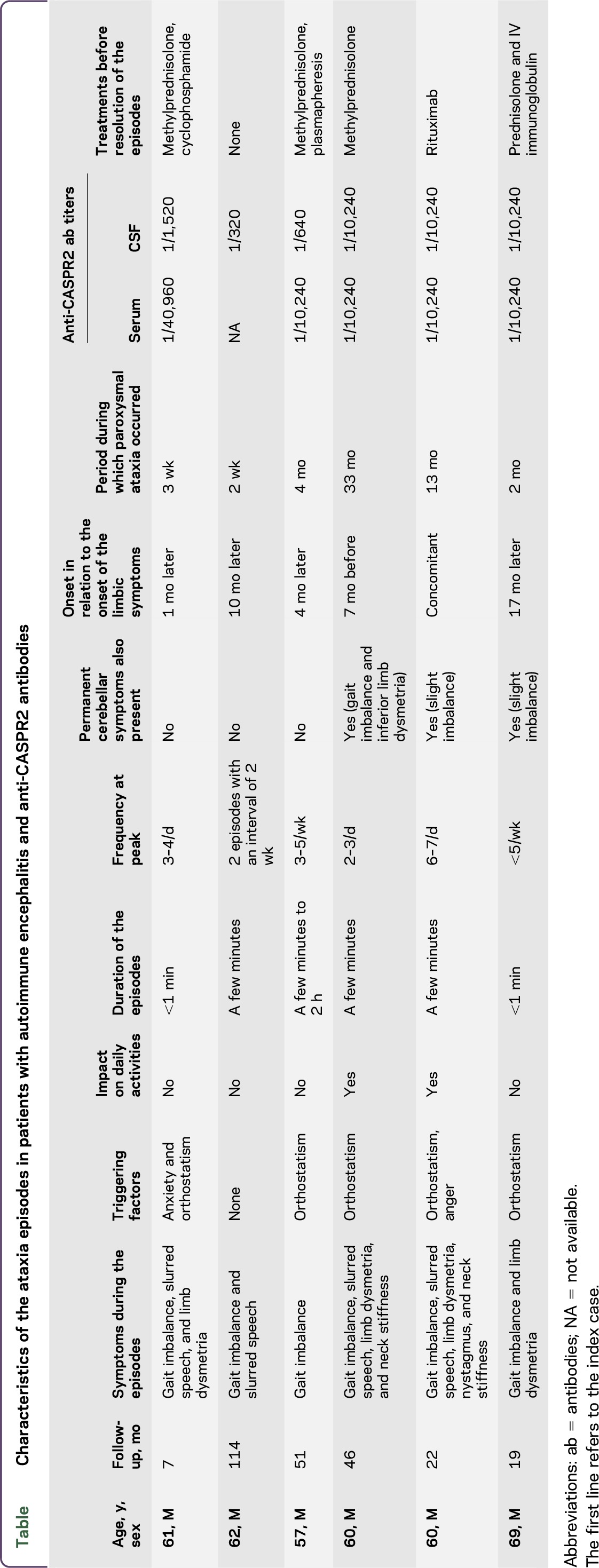
None of them exhibited neuromyotonia or Morvan syndrome. Two of the 5 patients had ≥3 core symptoms as defined in the study by Van Sonderen et al.3 The transient episodes are described in the table. They were stereotyped and included, according to the cases, gait imbalance (5/5, 100%), slurred speech (3/5, 60%), limb dysmetria (3/5, 60%), and nystagmus (1/5, 20%). Two patients (40%) reported concomitant sensations of neck stiffness. Myoclonic jerks were not observed in any of the patients. Of interest, the ataxia episodes preceded by 7 months the full development of the encephalitis in 1 patient. The apparition of the ataxia was not subsequent to a change of antiepileptic medication in any of the patients. The attacks were reported to last from a few minutes to 2 hours, according to the cases, with triggering factors identified in 4 patients (80%; orthostatism in 4 patients and anger in 1 patient). Ataxia episodes occurred during a mean period of 4 months (range, 0.5–32.5) and stopped after the initiation of an immunomodulatory treatment in 4 patients (steroids alone, steroids and plasmapheresis, steroids and IV immunoglobulin, or rituximab) and spontaneously in 1 case. Interictal, milder cerebellar symptoms were observed in 3/5 cases (60%). Interictal brain MRI was unremarkable in all patients; magnetic resonance spectroscopy was not performed during the episodes. We did not perform genetic studies in these 5 patients.
Table.
Characteristics of the ataxia episodes in patients with autoimmune encephalitis and anti-CASPR2 antibodies
DISCUSSION
In this study, we report stereotyped episodes of paroxysmal cerebellar ataxia in a patient with anti–CASPR2 antibody-related autoimmune encephalitis. We also retrospectively identified similar paroxysmal symptoms in 5 of 20 other patients with autoimmune encephalitis and anti-CASPR2 antibodies, suggesting that episodic cerebellar ataxia could be observed in up to 25% of such patients.
As they developed during the course of encephalitis and resolved after immunomodulating therapy in most of the patients, we can speculate an immune origin of these paroxysmal ataxias. Moreover, the association of gait imbalance, slurred speech, and limb dysmetria during these episodes was highly suggestive of cerebellar impairment; although because of the retrospective collection of the data, we cannot completely exclude that the alleged cerebellar symptoms were actually of another nature (e.g., orthostatic hypotension, seizures, or peri-ictal autonomic manifestations).
Paroxysmal neurologic symptoms have already been reported in autoimmune encephalitis. For instance, faciobrachial dystonic seizures are caused by the activation of the motor cortex in patients with anti–leucine-rich glioma1 (Lgi1) antibody-associated encephalitis.7 Of interest, Lgi1 is a secreted protein that complexes with CASPR2 and the potassium channel KV1.1.5,8 More recently, orthostatic myoclonus has been observed in a patient with anti-CASPR2 antibodies.9 Although permanent ataxia without remissions is a well-described feature of anti–CASPR2 antibody-associated encephalitis, to our knowledge, EA has never been reported before in patients with autoimmune encephalitis, including for instance patients with anti-Lgi1 encephalitis.4,3,10,11
Anti-CASPR2 antibodies associate with various neurologic disorders, including limbic encephalitis, neuromyotonia, and Morvan syndrome.3,4,12–14 In a recent published cohort of 38 anti–CASPR2 antibody-positive patients, 77% of the cases had ≥3 core symptoms (encephalic signs, cerebellar symptoms, peripheral nerve hyperexcitability, dysautonomia, neuropathic pain, insomnia, and weight loss).3 In our previously published cohort of 33 patients with anti-CASPR2 antibodies, only 16/33 (48%) of the patients presented with ≥3 of those core symptoms.4 However, patients with limbic encephalitis were overrepresented and we found a greater number of patients with ≥3 core symptoms in the group of patients diagnosed with neuromyotonia or Morvan syndrome (13/15, 87%) than in the group of patients diagnosed with limbic encephalitis (3/18, 17%). Only 2/6 (33%) of our patients with paroxysmal ataxia had ≥3 core symptoms, which reflects that this paroxysmal syndrome is mostly associated with limbic encephalitis rather than neuromyotonia or Morvan syndrome.
The resemblance of the patients' paroxysmal ataxia with manifestations observed in hereditary channelopathies such as EA is striking. Of interest, neuromyotonia can be observed in patients with EA type 1 and anti-CASPR2 antibodies, suggesting similar ion channel dysfunctions in both diseases. EA type 1 is due to mutations of KCNA1, a gene coding for the voltage-gated potassium channel KV1.1.5,13 The clustering of KV1.1 at the nodes of Ranvier depends on CASPR2, and electrophysiologic experiments have suggested an impairment of voltage-gated potassium channels in autoimmune neuromyotonia, implying that anti-CASPR2 antibodies might indirectly alter the functions of KV1.1 at the nodes of Ranvier.15,16 However, previous studies have failed to demonstrate a role of CASPR2 in KV1.1 clustering at the synaptic level, and the exact role of anti-CASPR2 antibodies in the CNS is unknown.16,17 Nevertheless, our observation supports the hypothesis of immune-mediated ion channel dysfunction in anti-CASPR2 antibody syndromes. The polymorphism of CACNA1A in the index patient is interesting because this gene codes for the CaV2.1 subunit of the P/Q type voltage-gated calcium channel that is mutated in patients with EA type 2.6 We have no data about the significance of this polymorphism, whose overall frequency in the general population is estimated at less than 1/1,000. However, we can hypothesize that it may provoke a partial impairment of CaV2.1.6 We can thus speculate that in the case, this polymorphism of CACNA1A may have led to the failure of compensatory mechanisms dependent on CaV2.1 and necessary to counterbalance the effects of anti-CASPR2 antibodies. Therefore, this polymorphism may have favored the development of episodic cerebellar symptoms in the patient.
Overall, our findings suggest that transient cerebellar ataxia should be added to the spectrum of anti–CASPR2 antibody-related symptoms. Such paroxysmal symptoms are similar to the symptoms of hereditary channelopathies, suggesting that ion channel dysfunction is involved in the pathogenesis of anti-CASPR2 antibody syndromes.
ACKNOWLEDGMENT
The authors thank Dr. Cécile Marchal, Dr. Philippe Kassiotis, Dr. Jacques Testaud, Dr. Lejla Koric, and Dr. Isabelle Lambert, who provided CSF, serum samples, and clinical data for the study.
GLOSSARY
- EA
episodic ataxia
Footnotes
Supplemental data at Neurology.org/nn
AUTHOR CONTRIBUTIONS
Dr. Joubert: study concept and design; acquisition of data; and analysis and interpretation. Dr. Gobert: acquisition of data and critical revision of the manuscript for important intellectual content. Dr. Thomas and Ms. Saint-Martin: acquisition of data. Dr. Desestret: critical revision of the manuscript for important intellectual content. Dr. Convers, Dr. Rogemond, and Ms. Picard: acquisition of data. Prof. Ducray, Dr. Psimaras, Prof. Antoine, and Prof. Delattre: critical revision of the manuscript for important intellectual content. Dr. Honnorat: study supervision; acquisition of data; study concept and design; and critical revision of the manuscript for important intellectual content.
STUDY FUNDING
This study is supported by institutional grants from Agence Nationale de la Recherche (ANR-14-CE15-0001-MECANO), Fondation pour la Recherche Médicale (Neurodon2014), and CSL Behring France.
DISCLOSURE
B. Joubert, F. Gobert, L. Thomas, M. Saint-Martin, V. Desestret, P. Convers, V. Rogemond, G. Picard, F. Ducray, and D. Psimaras report no disclosures. J.-C. Antoine served on the scientific advisory board for Pfizer; received travel funding and/or speaker honoraria from Pfizer and Laboratoire Français des Biotechnologies et du Fractionnement; served as an associate editor for Revue Neurologique; and holds a patent for diagnostic test for anti-CV2/CRMP5 andibody detection. J.-Y. Delattre served on the editorial board for The Oncologist; received research support from Institut National du Cancer; reference center for anaplastic oligodendrogliomas. J. Honnorat reports no disclosures. Go to Neurology.org/nn for full disclosure forms.
REFERENCES
- 1.Tomlinson SE, Hanna MG, Kullmann DM, Tan SV, Burke D. Clinical neurophysiology of the episodic ataxias: insights into ion channel dysfunction in vivo. Clin Neurophysiol 2009;120:1768–1776. [DOI] [PubMed] [Google Scholar]
- 2.D'Adamo MC, Hasan S, Guglielmi L, et al. New insights into the pathogenesis and therapeutics of episodic ataxia type 1. Front Cell Neurosci 2015;9:317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Van Sonderen A, Ariño H, Petit-Pedrol M, et al. The clinical spectrum of Caspr2 antibody-associated disease. Neurology 2016;87:521–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Joubert B, Saint-Martin M, Noraz N, et al. Characterization of a subtype of autoimmune encephalitis with anti-contactin-associated protein-like 2 antibodies in the cerebrospinal fluid, prominent limbic symptoms, and seizures. JAMA Neurol 2016;73:1115–1124. [DOI] [PubMed] [Google Scholar]
- 5.Browne DL, Gancher ST, Nutt JG, et al. Episodic ataxia/myokymia syndrome is associated with point mutations in the human potassium channel gene, KCNA1. Nat Genet 1994;8:136–140. [DOI] [PubMed] [Google Scholar]
- 6.Terwindt GM, Ophoff RA, Haan J, et al. Variable clinical expression of mutations in the P/Q-type calcium channel gene in familial hemiplegic migraine. Dutch Migraine Genetics Research Group. Neurology 1998;50:1105–1110. [DOI] [PubMed] [Google Scholar]
- 7.Navarro V, Kas A, Apartis E, et al. Motor cortex and hippocampus are the two main cortical targets in LGI1-antibody encephalitis. Brain J Neurol 2016;139:1079–1093. [DOI] [PubMed] [Google Scholar]
- 8.Lai M, Huijbers MGM, Lancaster E, et al. Investigation of LGI1 as the antigen in limbic encephalitis previously attributed to potassium channels: a case series. Lancet Neurol 2010;9:776–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gövert F, Witt K, Erro R, et al. Orthostatic myoclonus associated with Caspr2 antibodies. Neurology 2016;86:1353–1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Becker EBE, Zuliani L, Pettingill R, et al. Contactin-associated protein-2 antibodies in non-paraneoplastic cerebellar ataxia. J Neurol Neurosurg Psychiatry 2012;83:437–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Balint B, Regula JU, Jarius S, Wildemann B. Caspr2 antibodies in limbic encephalitis with cerebellar ataxia, dyskinesias and myoclonus. J Neurol Sci 2013;327:73–74. [DOI] [PubMed] [Google Scholar]
- 12.Irani SR, Pettingill P, Kleopa KA, et al. Morvan syndrome: clinical and serological observations in 29 cases. Ann Neurol 2012;72:241–255. [DOI] [PubMed] [Google Scholar]
- 13.Lancaster E, Huijbers MG, Bar V, et al. Investigations of Caspr2, an autoantigen of encephalitis and neuromyotonia. Ann Neurol 2011;69:303–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bien CG, Mirzadjanova Z, Baumgartner C, et al. Anti-contactin-associated protein-2 encephalitis: relevance of antibody titres, presentation and outcome. Eur J Neurol 2017;24:175–186. [DOI] [PubMed] [Google Scholar]
- 15.Horresh I, Poliak S, Grant S, Bredt D, Rasband MN, Peles E. Multiple molecular interactions determine the clustering of Caspr2 and Kv1 channels in myelinated axons. J Neurosci 2008;28:14213–14222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sinha S, Newsom-Davis J, Mills K, Byrne N, Lang B, Vincent A. Autoimmune aetiology for acquired neuromyotonia (Isaacs' syndrome). Lancet 1991;338:75–77. [DOI] [PubMed] [Google Scholar]
- 17.Ogawa Y, Horresh I, Trimmer JS, Bredt DS, Peles E, Rasband MN. Postsynaptic density-93 clusters Kv1 channels at axon initial segments independently of Caspr2. J Neurosci 2008;28:5731–5739. [DOI] [PMC free article] [PubMed] [Google Scholar]