

Abstract
Viewing subcellular details over large tissue volumes is becoming an essential condition of the success of large-scale projects aimed at visualizing cell connections in whole organs or tissues. However, tissue opacity remains an obstacle to deep tissue imaging. This situation has brought renewed interest for techniques of tissue clearing; new protocols, such as CLARITY (Clear Lipid-exchanged Acrylamide-hybridized Rigid Imaging/Immunostaining/In situ hybridization-compatible Tissue-hYdrogel), have recently been developed. So far, most of the tests of these techniques have been applied to brain or other soft tissues. Here we show that CLARITY clears mouse hindlimb skeletal muscles and maintains the basic structural features of muscle and its fibers. However, tagging with fluorescent markers was not successful.
Keywords: CLARITY, deep tissue imaging, optical clearing, second harmonic generation imaging, skeletal muscle
1. INTRODUCTION
Recent advances in light and electron microscopy in the form of an ever expanding spectrum of fluorescent proteins, new super-resolution techniques and electron tomography, have given us access to increasingly refined details of subcellular organization in tissues. However, the areas imaged remain limited in depth and size. Large-scale efforts such as the Brain Initiative, as well as ongoing interest in tissue development and organization, call for techniques that integrate microscopic observations at the macroscale level. Major obstacles still need to be overcome: slow image collection rate, data accumulation outpacing data management, difficulties to get uniform labeling through thick tissues, and light diffraction obscuring details deep in tissues. Light microscopy techniques such as Single Plane Illumination Microscopy (SPIM, Verveer et al., 2007) address the first of these obstacles, providing ways to image large volumes rapidly. So far, SPIM has been mostly used to follow embryos and transparent organisms such as zebrafish. Although technologies in imaging instrumentation and data management have improved dramatically, developing novel sample preparations is equally important.
The need for increased tissue transparency has spurred a large effort towards new tissue clearing approaches (Hama et al., 2011; Chung et al., 2013). A fructose-based chemical solution (SeeDB; Ke et al., 2013) was successfully used to clear a rat olfactory bulb whole-mounted for 2-photon microscopy. The tissue was rendered sufficiently transparent to observe the axonal projections of mitral dendrite cells in both longitudinal and lateral directions beyond a depth of 6 mm. The dendrite cells had been tagged with a knock-in Thy1-YFP-G and the neurons with Alexa647-dextran. This large-scale imaging would have been significantly more laborious and time-consuming with conventional mechanical sectioning followed by 3D reconstruction. Another technique, called CLARITY (Clear Lipid-exchanged Acrylamide-hybridized Rigid Imaging/Immunostaining/In situ hybridization-compatible Tissue-hYdrogel), was introduced in the same year by Chung et al. (2013). The paper shows dramatic views of completely transparent whole adult mouse brain. CLARITY is a 3-step process. First a tissue sample is embedded in a polymeric hydrogel matrix that covalently binds proteins of interest. In the second step a detergent is applied that forms micelles with the unbonded lipids, enabling their mechanical extraction from the tissue, helped by an electric field applied across the tissue. In the third step, the sample is equilibrated with a glycerol-based solution to match refractive indices between the sample’s liquid phase and the polymeric hydrogel meshwork.
The website of the Deisseroth laboratory lists about 20 publications, so far, that rely on CLARITY (http://clarityresourcecenter.org/). Most but not all, focus on the brain. One of them extends the approach to pancreas, liver, kidney, lung, and intestine (Lee at al., 2014). One noted and remarkable study reports on the whole-body clearing of a mouse (Yang et al., 2014) and another reports on PEA-CLARITY for plants (Palmer et al., 2015).
Skeletal muscle is the largest cell type in rodent or human, and is regulated by other cell types that are essential for its function – nerves and blood vessels, for example. Furthermore, the connections between muscle and these cells seem to follow a large-scale pattern that is difficult to perceive with conventional techniques and preparations. Muscle fibers are enmeshed in arbors of nerves and blood vessels, with a single synapse between nerve and each muscle fiber, called the neuromuscular junction (NMJ). The exploration of NMJ positioning (Kim & Burden, 2008; Darabid et al., 2014) and of nerve and blood vessel trajectories through muscles have so far been restricted to tissue sections, single fibers, or the thinnest of muscles such as the mouse interscutularis. Lu et al. (2009) were able to map the entire neural connectome of the interscutularis. By visualizing the intact neural network’s relation to the muscular system via the NMJ, the authors could correlate the large-scale wiring scheme of neurons with parameters like single axon caliber, arbor length, or motor unit size. These experiments uncovered several organizational principles in this muscle; they also demonstrated variability between individual muscles, a feature that does not exist in the more easily studied invertebrates. In this particular case, tissue clarification was not essential since conventional confocal microscopy could penetrate the 30 µm muscle. Nevertheless, the authors mentioned problems due to light scattering for “imaging deeper structures”. Most muscle disease studies focus on hindlimb muscles which are responsible for locomotion, show fatigue and other disease-affected properties, and have a large mass favorable for protein or RNA extraction. Most hindlimb muscles are considerably thicker than the interscutularis and cannot be viewed in their entirety with conventional microscopy.
We are unaware of any publication, so far, focused on skeletal muscle clearing by CLARITY, but muscle has long been the subject of clearing work (Szent-Gyorgyi, 1949; Plotnikov et al., 2006; and references therein).
In the present study we establish that CLARITY is applicable to skeletal muscle—a dense and fibrous tissue that is considerably different from the clarification method’s originally intended target of brain tissue, which comprises a higher fat content. Of the several recent tissue clarification methods already cited, we have chosen the CLARITY method over other options due to its active method of tissue clarification. We tested CLARITY on mouse hindlimb skeletal muscles, focusing specifically on the soleus and extensor digitorum longus (EDL), slow and fast muscles respectively.
2. EXPERIMENTAL
Animals and Muscle clearing
This research was done in full compliance with all the rules and regulations of the PHS Policy on Humane Care and use of Laboratory Animals, under a protocol approved by the NIAMS Institutional Animal Care and Use Committee. Two-months-old C57 BL/6 mice were euthanized with CO2 according to NIAMS guidelines. We collected the following hindlimb muscles: extensor digitorum longus (EDL), flexor digitorum brevis (FDB), and soleus. Each muscle was then incubated in a hydrogel monomer/ fixative solution of 4% (wt) PFA, 4% (wt/vol) acrylamide, 0.05% (wt/vol) bis-acrylamide, 0.25% (wt/vol) VA-044 in PBS for 3 days at 4°C (Chung et al., 2013). One muscle of each pair was then transferred to PBS and kept as control. The other hydrogel/muscle complex was submerged in a chamber built as described in Chung et al. (2013) and perfused with 4% sodium dodecyl sulfate (SDS) in PBS. Both chamber and pumping systems were built in-house, keeping the cost of the experiments down. We then applied an electric field of 23V to the hydrogel/muscle complex for 8h. The voltage selected was the highest value that did not cause sample heating above 50°C. Excess hydrogel around the muscles was removed with a scalpel, muscles were stained with Hoechst 33342 to label DNA, and then kept in PBS at 4°C. A total of 12 muscles were subjected to the protocol.
Microscopy
For immediate assessment of the treatment success, muscles were pinned to a grid-marked Petri dish coated with Sylgard and photographed in transmitted light with a Leica DFC500 digital camera mounted on a dissection microscope (Leica DMZ-6). For further analysis, the whole muscles were mounted in Vectashield (Vector, Inc.) in a chamber made with two superimposed 0.1 mm spacers glued to a slide, and sealed with a no.1.5 glass coverslip. Muscles for second harmonic generation (SHG) imaging were mounted in glycerol in the same way. Microscopy was carried out on a Leica SP5 NLO confocal microscope equipped with a Mai Tai infrared laser (Spectra-Physics, Newport Inc.) for multi-photon work. All imaging was done with a IRAPO 25× 0.95 N.A. water immersion lens. For nuclear Hoechst imaging, excitation wavelength was 770 nm and images were collected with an internal hybrid detector. For dual imaging of excited autofluorescence and nuclei, excitation wavelength was 870 nm and images were collected in a two-channel red-green non-descanned detector. For SHG, excitation wavelength was 800 nm. Emitted light was filtered through a bandpass filter centered around 405 ± 20 nm. All confocal images were collected in the Leica LASAF software, exported as .tif files, and cropped and mounted in Photoshop 5 (Adobe Inc.) for figure preparation. Images to be compared for brightness were first mounted together and then all handled simultaneously with Images/Adjust/Levels, without modification of the image gamma. For quantitative analysis, original images were analyzed in Image J (developed at NIH by Wayne Rasband, and freely downloadable from imagej.nih.gov/ij/). Results were then imported into Microsoft Excel for plotting.
3. RESULTS AND DISCUSSION
At the end of the clearing procedure, a grid pattern is visible through a CLARITY-treated mouse soleus muscle, but not through the control muscle (Figure 1, arrows), indicating that clearing took place. Non-lipid based structures like tendons (arrowheads) remain opaque, however. To examine light penetration and scattering, muscles stained with Hoechst 33342 to label nuclei were examined at higher magnification (Figure 2). The staining pattern looked comparable to what we see in similarly stained single muscle fibers, indicating that the dye has access to chromatin. Stacks of images were recorded every 2 µm from the surface of the muscle to a depth of 97 µm (Figure 2A). The images are shown with the gray scale inverted. A Z-X projection of the stacks is shown in Figure 2B. Average brightness of the nuclei was quantitated and plotted (Figure 2C). Each of the three panels indicates that the fluorescence signal from the nuclei traverses the CLARITY-treated muscle much better than the control muscle. Bumps in the curves (Figure 2C) are likely due to the presence of muscle blood vessels (which have their own scattering effect) and endothelial cell nuclei, which, after staining with Hoechst, are brighter than myonuclei.
Figure 1. CLARITY treatment makes mouse muscle translucid.
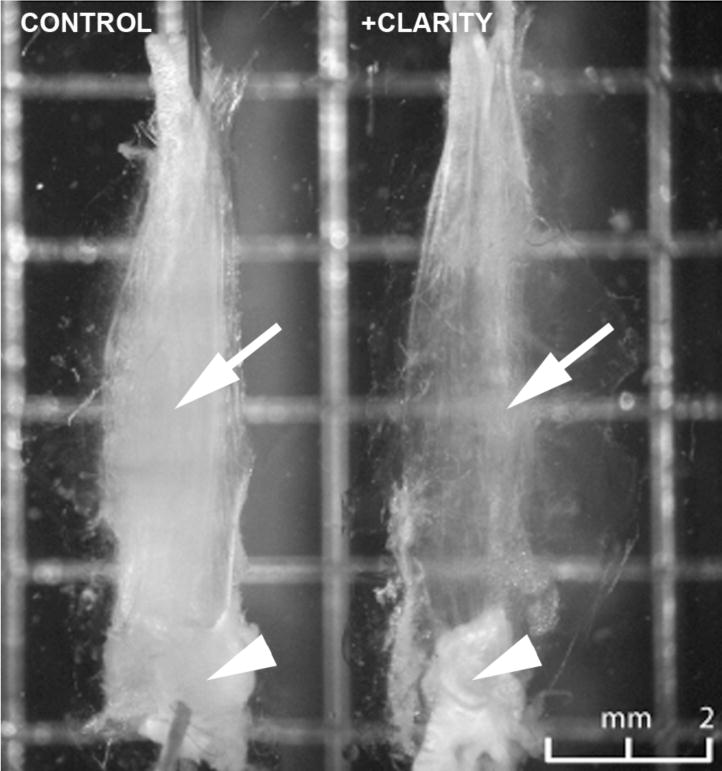
A representative pair of control and CLARITY-treated mouse soleus muscles were pinned to a Sylgard-coated gridded dish. The grid is visible through the cleared but not through the control muscle (arrows). Tendons however are not cleared (arrowheads). Bar: 2mm.
Figure 2. Fluorescence imaging through muscle is improved by CLARITY.
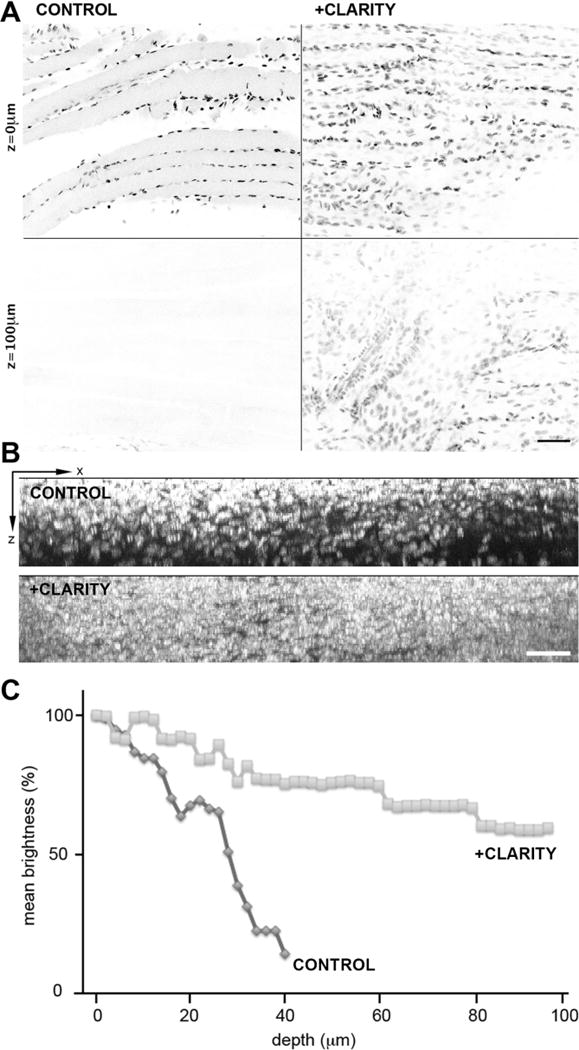
Hoechst-stained nuclei of representative soleus muscles were imaged in 2-photon fluorescence on a Leica SP5 NLO confocal setup; optical sections were recorded every 2 µm from the surface to a depth of 97 µm. A: X-Y single optical sections recorded with identical settings (laser power, image acquisition gain) show brighter nuclei at the surface of the control compared to the treated muscle, but the control shows a rapid decrease in fluorescence through the sample. Panel B shows an X-Z projection of the images over the same 97 µm and panel C shows quantitation of the fluorescence expressed in % of the fluorescence at the surface. Bars: A: 75 µm; B: 50 µm.
To assess conservation of the structural features of muscle fibers, we also imaged muscles in 2P-excited autofluorescence (Figure 3). Individual cleared fibers show the characteristic autofluorescence striations that originate in mitochondria (Ralston et al., 2008); the Hoechst-stained myonuclei (shown in red) are positioned normally, i.e. along the plasma membrane of the muscle fibers and along blood vessels (Figure 3, A–B, arrows; also see Ralston et al., 2006). We conclude from these tests that the basic organization of muscle fibers is well preserved.
Fig. 3. Characteristic structural features of muscle fibers are conserved after CLARITY treatment of EDL muscles while some modifications of fat cells and tendons are observed.
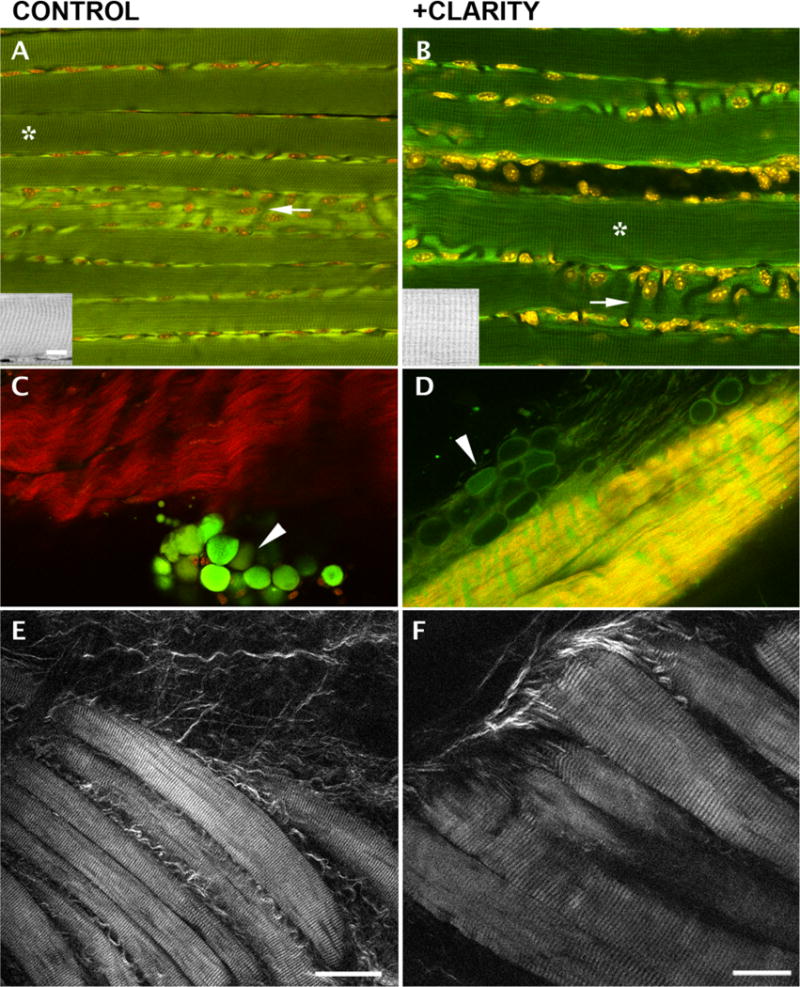
A–B: 2P fluorescence shows characteristic striated autofluorescence in both control and treated fibers. The insets reproduce the areas marked with an asterisk. Nuclei are positioned, as expected, along plasma membrane and along blood vessels that appear as dark channels (arrows); (C–D): SHG imaging (red) and 2-photon autofluorescence (green) of tendon and fat cells (arrowheads) show that the former keep their basic organization while fat cells are depleted and flattened by the treatment; E–F: SHG imaging of unstained EDL fiber bundles shows conservation of the striated pattern. Bars: A–E: 50 µm (see bar in E); F: 25 µm; insets 10 µm (bar in A).
We also used SHG microscopy coupled with 2P-excited fluorescence (Plotnikov et al., 2006; Ralston et al., 2008) to visualize other cell types in mouse muscles. SHG is a 2-photon technique akin to polarization microscopy. Endogenous molecules that are organized in non-centrosymmetric arrays can be visualized by SHG in unstained, unfixed tissue and collagen and myosin are the best SHG emitters. Panels C–D present SHG in red and autofluorescence in green. Fat cells (arrowheads) appear as fluorescent globules in control samples but as empty envelopes in cleared tissue. The SHG signal of tendons is similar in both samples but there is increased autofluorescence of the tendon in the CLARITY-treated sample—hence the yellow rather than red shade. The SHG striated signal recorded from both control and treated muscles (panels E–F) implies preservation of the myosin heavy chain organization. However, the signal from the cleared fibers was considerably weaker than that of the control fibers. Plotnikov et al. (2006) showed that skeletal muscle clearing by glycerol improves myosin SHG while reducing collagen SHG, but their work was carried out on unfixed muscle.
CLARITY should give access to molecules that are kept in the cleared tissue. We were not successful, however, in tagging muscle components: three separate attempts at labeling neuromuscular junctions with fluorescently labeled α-bungarotoxin were negative. Since even the most exposed fibers gave no label, we assume that the cross-linking and fixation prevented access of the toxin to the acetylcholine receptors. Similar attempts to label actin with fluorescently labeled phalloidin were unsuccessful. The publication of a new, faster CLARITY protocol after this work was finished (Poguzhelskaya et al., 2014) as well as the recent use of a hydrogel to expand cell volumes for diffraction-limited light microscopy (Chen et al., 2015), indicate that this area of research is very much alive. Furthermore, a recent publication (Calve et al., 2015) on clearing of bovine and rodent musculoskeletal tissues by the already mentioned fructose-based technique SeeDB (Ke et al., 2013) provides data that complement ours. Calve et al. (2015) focused mostly on the clearing of bovine and rodent bone and connective tissues (cartilage, ligaments, meniscus). Transmitted light photography of a grid pattern through a fragment of each of these three tissues shows clearing with cartilage only. Nevertheless their fluorescence imaging of all 3 connective tissues labeled with small molecular weight dyes indicates a 2-fold improvement of light penetration tested down to 50 µm. Their study also considered mouse leg muscles that appeared cleared by transmitted light photography. However, there was no quantitation or grid test of the improvement of light penetration and their images of a fluorescent Pax3 construct genetically expressed in the mouse muscle was not pursued at high resolution. It is not possible to declare one of the techniques superior, since the tests performed were different but we feel that our demonstration that CLARITY works on skeletal muscle further opens the field.
Cardiac muscle could benefit from clearing techniques as well. In a comparison between healthy and infarcted cardiac tissue, Freeman et al. (2014) demonstrated how the geometry and directionality of axons are adversely affected after infarction. However, only the most superficial layer of tissue was imaged. Imaging deeper could reveal more about neural remodeling after infarction. Indeed, Freeman et al. (2014) noted that technical innovations such as tissue clarification and adaptive optics might overcome some of the limitations they faced.
4. CONCLUSION
In conclusion, we show that adult mouse skeletal muscle can be cleared by the CLARITY protocol and that the basic cellular organization is preserved, moving forward visualization of the deeper muscle structure and whole-muscle connectome studies. As CLARITY and other clearing protocols are still being improved, the ultimate challenge will be to achieve maximum clearing while allowing molecular labeling of tissue components and retaining structural details –the classic challenge of all microscopy techniques.
Acknowledgments
We thank Gary Melvin for help with the CLARITY chambers, members of the lab for useful suggestions, and Kristien J. Zaal for critical reading of the manuscript. This project was supported by the Intramural Program of the National Institutes of Arthritis and Musculoskeletal and Skin Diseases.
Abbreviations
- CLARITY
Clear Lipid-exchanged Acrylamide-hybridized Rigid Imaging/Immunostaining/In situ hybridization-compatible Tissue-hYdrogel
- EDL
Extensor Digitorum Longus
- ETC
Electrophoretic Tissue Clearance
- FDB
Flexor Digitorum Brevis
- N.A.
numerical aperture
- NMJ
neuromuscular junction
- PFA
paraformaldehyde
References
- Calve S, Ready A, Huppenbauer C, Main R, Neu CP. Optical clearing in dense connective tissues to visualize cellular connectivity in situ. PLoS One. 2015;10:e0116662. doi: 10.1371/journal.pone.0116662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen F, Tillberg PW, Boyden ES. Optical imaging. Expansion microscopy. Science. 2015;347:543–548. doi: 10.1126/science.1260088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung K, Wallace J, Kim SY, Kalyanasundaram S, Andalman AS, Davidson TJ, Mirzabekov JJ, Zalocusky KA, Mattis J, Denisin AK, Pak S, Bernstein H, Ramakrishnan C, Grosenick L, Gradinaru V, Deisseroth K. Structural and molecular interrogation of intact biological systems. Nature. 2014;497:332–337. doi: 10.1038/nature12107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darabid H, Perez-Gonzalez AP, Robitaille R. Neuromuscular synaptogenesis: coordinating partners with multiple functions. Nature reviews Neuroscience. 2014;15:703–718. [PubMed] [Google Scholar]
- Freeman K, Tao W, Sun H, Soonpaa MH, Rubart M. In situ three-dimensional reconstruction of mouse heart sympathetic innervation by two-photon excitation fluorescence imaging. J Neurosci Methods. 2014;221:48–61. doi: 10.1016/j.jneumeth.2013.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hama H, Kurokawa H, Kawano H, Ando R, Shimogori T, Noda H, Fukami K, Sakaue-Sawano A, Miyawaki A. Scale: a chemical approach for fluorescence imaging and reconstruction of transparent mouse brain. Nature neuroscience. 2011;14:1481–1488. doi: 10.1038/nn.2928. [DOI] [PubMed] [Google Scholar]
- Ke MT, Fujimoto S, Imai T. SeeDB: a simple and morphology-preserving optical clearing agent for neuronal circuit reconstruction. Nature neuroscience. 2013;16:1154–1161. doi: 10.1038/nn.3447. [DOI] [PubMed] [Google Scholar]
- Kim N, Burden SJ. MuSK controls where motor axons grow and form synapses. Nature neuroscience. 2008;11:19–27. doi: 10.1038/nn2026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H, Park JH, Seo I, Park SH, Kim S. Improved application of the electrophoretic tissue clearing technology, CLARITY, to intact solid organs including brain, pancreas, liver, kidney, lung, and intestine. BMC Developmental Biology. 2014;14(1):48. doi: 10.1186/s12861-014-0048-3. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, Tapia JC, White OL, Lichtman JW. The interscutularis muscle connectome. PLoS biology. 2009;7:e32. doi: 10.1371/journal.pbio.1000032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer WM, Martin AP, Flynn JR, Reed SL, White RG, Furbank RT, Grof CP. PEA-CLARITY: 3D molecular imaging of whole plant organs. Scientific reports. 2015;5:13492. doi: 10.1038/srep13492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plotnikov S, Juneja V, Isaacson AB, Mohler WA, Campagnola PJ. Optical clearing for improved contrast in second harmonic generation imaging of skeletal muscle. Biophys J. 2006;90:328–339. doi: 10.1529/biophysj.105.066944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poguzhelskaya E, Artamonov D, Bolshakova A, Vlasova O, Bezprozvanny I. Simplified method to perform CLARITY imaging. Molecular neurodegeneration. 2014;9:19. doi: 10.1186/1750-1326-9-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ralston E, Lu Z, Biscocho N, Soumaka E, Mavroidis M, Prats C, Lomo T, Capetanaki Y, Ploug T. Blood vessels and desmin control the positioning of nuclei in skeletal muscle fibers. J Cell Physiol. 2006;209:874–882. doi: 10.1002/jcp.20780. [DOI] [PubMed] [Google Scholar]
- Ralston E, Swaim B, Czapiga M, Hwu WL, Chien YH, Pittis MG, Bembi B, Schwartz O, Plotz P, Raben N. Detection and imaging of non-contractile inclusions and sarcomeric anomalies in skeletal muscle by second harmonic generation combined with two-photon excited fluorescence. Journal of structural biology. 2008;162:500–508. doi: 10.1016/j.jsb.2008.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Susaki EA, Tainaka K, Perrin D, Kishino F, Tawara T, Watanabe TM, Yokoyama C, Onoe H, Eguchi M, Yamaguchi S, Abe T, Kiyonari H, Shimizu Y, Miyawaki A, Yokota H, Ueda HR. Whole-brain imaging with single-cell resolution using chemical cocktails and computational analysis. Cell. 2014;157:726–739. doi: 10.1016/j.cell.2014.03.042. [DOI] [PubMed] [Google Scholar]
- Szent-Gyorgyi A. Free-energy relations and contraction of actomyosin. Biol Bull. 1949;96:140–161. [PubMed] [Google Scholar]
- Tainaka K, Kubota SI, Suyama TQ, Susaki EA, Perrin D, Ukai-Tadenuma M, Ukai H, Ueda HR. Whole-body imaging with single-cell resolution by tissue decolorization. Cell. 2014;159:911–924. doi: 10.1016/j.cell.2014.10.034. [DOI] [PubMed] [Google Scholar]
- Tomer R, Ye L, Hsueh B, Deisseroth K. Advanced CLARITY for rapid and high-resolution imaging of intact tissues. Nature protocols. 2014;9:1682–1697. doi: 10.1038/nprot.2014.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verveer PJ, Swoger J, Pampaloni F, Greger K, Marcello M, Stelzer EH. High-resolution three-dimensional imaging of large specimens with light sheet-based microscopy. Nature methods. 2007;4:311–313. doi: 10.1038/nmeth1017. [DOI] [PubMed] [Google Scholar]
- Yang B, Treweek JB, Kulkarni RP, Deverman BE, Chen CK, Lubeck E, Shah S, Cai L, Gradinaru V. Single-cell phenotyping within transparent intact tissue through whole-body clearing. Cell. 2014;158:945–958. doi: 10.1016/j.cell.2014.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]