

Abstract
The mechanical role of cardiac microtubules (MTs) has been a topic of some controversy. Early studies, which relied largely on pharmacological interventions that altered the MT cytoskeleton as a whole, presented no consistent role. Recent advances in the ability to observe and manipulate specific properties of the cytoskeleton have strengthened our understanding. Direct observation of MTs in working myocytes suggests a spring‐like function, one that is surprisingly tunable by post‐translational modification (PTM). Specifically, detyrosination of MTs facilitates an interaction with intermediate filaments that complex with the sarcomere, altering myocyte stiffness, contractility, and mechanosignalling. Such results support a paradigm of cytoskeletal regulation based on not only polymerization, but also associations with binding partners and PTMs that divide the MT cytoskeleton into functionally distinct subsets. The evolutionary costs and benefits of tuning cytoskeletal mechanics remain an open question, one that we discuss herein. Nevertheless, mechanically distinct MT subsets provide a rich new source of therapeutic targets for a variety of phenomena in the heart.
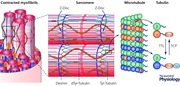
Keywords: cardiomyocyte, cytoskeleton, microtubules, muscle mechanics, myocytes
Abbreviations
- dTyr
detyrosination
- EC
excitation–contraction
- PTM
post‐translational modification
- MT
microtubule
- MAP
microtubule associated protein
- ROS
reactive oxygen species
Introduction
The purpose of the cardiomyocyte is to perform useful mechanical work; to convert chemical energy into a change in myocyte geometry that ultimately drives expulsion of blood from the heart. Within each myocyte work is performed on the molecular scale by the sarcomere, where myosin heads pull on actin filaments to shorten sarcomeres, and thus the myocyte. This shortening is dynamically regulated, and while contractile regulation is well studied at the level of the sarcomere – where much is known about the role of key players such as actin and myosin, the calcium sensor troponin, or the elastic spring titin – the role of the non‐sarcomeric cytoskeleton has remained more opaque, particularly regarding microtubules (MTs).
MTs consist of α/β tubulin heterodimers that polymerize to form stiff (3 orders of magnitude stiffer than actin), hollow tubes approximately 25 nm in diameter. MTs can be ‘dynamic’, undergoing continuous cycles of growth (polymerization) and shrinkage (catastrophe), but can also be ‘stabilized’, or protected from catastrophe. Stability is conferred by interactions between MTs and other cytoskeletal elements, microtubule associated proteins (MAPs) and motors, or even post‐translational modifications (PTMs) of tubulin itself (for recent review see Akhmanova & Steinmetz, 2015). MTs and their interacting partners have been remarkably well studied using reconstitution systems and single molecule approaches, yet uncovering their role in the myocyte, and their influence on myocyte mechanics, has proven to be a particularly rocky road.
In the early 1990s, Cooper and colleagues proposed that a proliferated and stabilized MT cytoskeleton could increase internal mechanical resistance to impede myocyte contraction (Tsutsui et al. 1993). The MT contribution to mechanics was particularly prominent after severe pressure overload, where MTs increased the viscosity and passive stiffness of the myocyte (Tagawa et al. 1997; Zile et al. 1998). Treatment of diseased myocardium with colchicine, which potently destabilizes the MT network, was found to decrease stiffness, improve contractility and boost cardiac output.
Over time these claims have proven controversial, confirmed by some (Wang et al. 1999; Shiels et al. 2007), and yet refuted by others (Bailey et al. 1997; de Tombe, 1998). A parallel body of work on the role of microtubules and free tubulin dimers in G‐protein coupled receptor signalling (Popova et al. 1997; Schappi et al. 2014) may provide some insight into the apparent unpredictability of results gleaned from gross microtubule manipulations, as excess free tubulin may alter cellular signalling on rapid time scales. This, combined with conflicting reports on the effects of colchicine on calcium fluxes and excitation–contraction (EC) coupling (Gomez et al. 2000; Calaghan et al. 2001), undermines a purely mechanical interpretation of studies where MTs are grossly disrupted.
While careful reading of these studies reveals elegant science, the tools and knowledge of the time limited them. They relied heavily on blunt pharmacological approaches, and lacked specific tools to probe MTs in living cells. There have been tremendous advances in cytoskeletal biology over the last 10–15 years, and recent work empowered by such advances suggests that it is time to re‐evaluate the role of muscle MTs with a fresh perspective.
Recent results
Microtubule network architecture
One such advance is the use of fluorescent probes to track the growing ends of MTs (so called end‐binding proteins; for review see Jaworski et al. 2008). This approach has now been used to characterize the growth and organization of the MT network in striated muscle. In skeletal myofibres, the Ralston lab demonstrated how MTs nucleate from Golgi elements to form an orthogonal grid, with transverse elements running largely along the Z‐disk of the sarcomere, interspersed by perpendicular bundles of longitudinal tubules (Oddoux et al. 2013). In cardiac myocytes transverse MTs are rarer, and MTs predominantly grow along the long axis of the cell, interdigitating with the myofibrils. Despite the paucity of transverse elements, Z‐discs again feature prominently, as the Santana lab note that MTs tend to grow from Z‐disk to Z‐disk, often pausing or changing direction at this protein‐rich region, suggestive of protein–protein interactions that remain undefined (Drum et al. 2016). Work from the Song and Marty labs also supports interactions with the t‐tubule system and junctional sarcoplasmic reticulum, respectively, which may be important for the maintenance of EC coupling (Zhang et al. 2014; Osseni et al. 2016).
This muscle MT network can display considerable variety through post‐translational modifications (PTMs). Tubulin PTMs have garnered a flurry of attention of late, as the development of genetic tools to manipulate PTMs have revealed diverse structural and functional consequences (e.g. Barisic et al. 2015; Nirschl et al. 2016; Robison et al. 2016; Valenstein & Roll‐Mecak, 2016; for review see Janke & Bulinski, 2011). Muscle microtubules demonstrate an abundance of one such PTM called detyrosination (dTyr), the enzymatic cleavage of a C‐terminal tyrosine residue from α‐tubulin. This modification, first described over 40 years ago (Barra et al. 1974), has numerous functional roles, many of which have only come to light in the last 2 years. dTyr can mediate MT interactions with intermediate filaments (Gurland & Gundersen, 1995; Yoshiyama et al. 2003; Robison et al. 2016), alter the binding and processivity of motor proteins on MT tracks to regulate trafficking (Sirajuddin et al. 2014; Barisic et al. 2015; Nirschl et al. 2016), and stabilize MTs by protecting them from disassembly (Peris et al. 2009). Among the family of known PTMs, dTyr appears uniquely abundant in healthy muscle cells (Kerr et al. 2015). It is also pervasive in neuronal networks, and ablation of tubulin tyrosine ligase, the enzyme that reverses dTyr, results in lethality, likely to be due to neuronal derangement (Erck et al. 2005).
Microtubules in muscle mechanosignalling
Our group, in collaboration with the Ward lab, became interested in this PTM in the context of MT‐mediated mechanosignalling. In a heart or skeletal muscle cell, mechanical stress elicits an increase in the production of reactive oxygen species (ROS) and alterations in intracellular calcium homeostasis (Prosser et al. 2011; Khairallah et al. 2012; Pal et al. 2013). The stress‐dependent regulation of [Ca2+]i – termed X‐ROS signalling due to its reliance on NADPH oxidase 2 (NOX2) ROS production – requires an intact microtubule network (Iribe et al. 2009; Prosser et al. 2011; Khairallah et al. 2012). In the cardiomyocyte, X‐ROS signalling sensitizes ryanodine receptor calcium release channels (RyR2), increasing RyR2 activity in response to mechanical stress. The Chen Izu lab has utilized an elegant ‘cell‐in‐gel’ approach to show that along with NOX2, neuronal nitric oxide synthase (nNOS) and Ca2+/calmodulin‐dependent kinase II (CaMKII) are involved in the mechanoregulation of RyR2 and EC coupling (Jian et al. 2014), although it remains to be tested if MT integrity is required for the latter.
In certain myopathic conditions, such as that arising from Duchenne muscular dystrophy, myocytes demonstrate a hypersensitivity to mechanical stress and exacerbated X‐ROS signalling (Prosser et al. 2011; Khairallah et al. 2012; Kombairaju et al. 2014). We noted an elevation of detyrosinated MTs in these same conditions. Surprisingly, suppressing dTyr alone was sufficient to blunt aberrant mechanosignalling in dystrophic muscle cells, and to protect dystrophic mice from stress‐induced arrhythmias and contraction‐induced injury (Kerr et al. 2015). The Ervasti lab has also identified a link between altered MT networks and susceptibility to skeletal muscle injury in various murine models of muscular dystrophy (Belanto et al. 2014).
In review, the above findings suggest that (1) detyrosinated MTs can transmit mechanical signals in muscle cells; and (2) MTs can provide an internal resistance to myocyte contraction. Yet how MTs bear (and transmit) mechanical loads, and how dTyr may alter this load bearing and force transmission, remained conspicuously vague.
Detyrosinated microtubules in myocyte mechanics
Advanced imaging approaches and new molecular tools for manipulating MTs have begun to shed light on detyrosinated MTs in myocyte mechanics. Using a point spread function imaging approach capable of high speed acquisition, we were able to observe MT behaviour in beating adult myocytes for the first time (Robison et al. 2016). We found that MTs resemble springs, deforming into short‐wavelength sinusoidal buckles under compressive load (see Abstract Figure). As the energy required to buckle MTs into such unfavourable configurations detracts from the energy available for sarcomere shortening, this provided visible evidence of a mechanism for MTs to impede myocyte contraction. Buckles form over a broad range of wavelengths, but MTs most often buckled at a wavelength consistent with the distance of a sarcomere, suggesting that the buckling mode is constrained by some association with the underlying contractile apparatus.
Specifically suppressing dTyr decreased the likelihood that any given MT would buckle during a contraction, and MTs that did still buckle no longer preferred to buckle at sarcomeric wavelengths, indicating that the association between MTs and the sarcomere is at least partly mediated by dTyr. This association between detyrosinated MTs and the sarcomere appears important for the integration of MTs as resistance elements, as sarcomeres shortened farther and faster in myocytes where dTyr was suppressed.
Additional evidence indicates that the intermediate filament desmin, which wraps around the Z‐disc, is at least one component mediating an MT cross‐link to the sarcomere. When either desmin or dTyr was genetically suppressed, myocytes were less stiff and less viscous, consistent with the disruption of a dynamic cytoskeletal cross‐link. Disruption of both produced no additional effect, indicating that desmin and dTyr depend on each other for the effects they have on myocyte mechanics. Based on these results we propose that detyrosinated MTs and desmin form a dynamically cross‐linked network that mechanically couples the non‐sarcomeric cytoskeleton to the sarcomere, and which integrates MTs into the mechanical scheme of the myocyte (Robison et al. 2016).
The relevance of these results is highlighted by the fact that dTyr levels were increased in left ventricular samples from patients with cardiomyopathy, and increasing dTyr correlated with declining systolic function within patient groups. When dTyr was increased to a similar extent in murine myocytes, we found that sarcomere shortening was impaired and myocytes showed increases in both stiffness and viscosity, raising the possibility that elevated dTyr may limit contractility in cardiac disease. These increases in passive stiffness included both viscous (deformation rate dependent) and elastic (rate independent) components (Robison et al. 2016).
Discussion
The presence and apparent dynamic regulation of multiple subsets of MTs with divergent mechanical behaviours indicates that there are functionally important mechanical roles for microtubules. Yet the presence of internal, mechanically resistive elements ostensibly opposes the overall purpose of the myocyte of converting chemical energy into useful mechanical work. If certain subpopulations of MTs impair this basic function, their apparent conservation demands some benefit to offset this evolutionary cost. Here we briefly explore some putative benefits of a tunable cytoskeletal resistance network.
Microtubules as elastic rectifiers?
The observation of reversible sinusoidal buckling in myocytes with a high proportion of detyrosinated MTs immediately suggests the existence of some kind of spring mechanism, storing energy during the contractile phase from the action of myosin motors and then releasing it during relaxation to provide a restoring force and accelerate relaxation in preparation for the next beat.
Yet whether the unbuckling of MTs provides a sufficient restoring force to meaningfully facilitate relaxation remains to be determined. When buckling is decreased by suppressing dTyr, myocytes have a slightly slowed late relaxation, consistent with an impaired spring, yet paradoxically relax faster during early re‐extension (Robison et al. 2016). The interpretation of this finding is complicated, however, as: (1) when buckling is decreased myocytes also shorten further during contraction, potentially compressing other elastic elements that may then provide more restoring force than normal to speed early relaxation; (2) myocytes with reduced dTyr have decreased viscosity, which could independently speed the relaxation time of the system. The issue of MTs providing restoring force demands further experimentation and clarification, as it may (or may not) represent a mechanism of fundamental importance to contractile mechanics. We would caution against the use of colchicine for such studies, however, as the liberation of free tubulin is likely to have numerous confounding consequences.
Nevertheless, the presence of increased elastic stiffness in myocytes with abundant detyrosinated MTs suggests a role similar to the canonical role of titin springs. While such a role was initially dismissed based on studies of the mechanics of skinned muscle fibres, these conditions diverge from the in situ state in the dilution of free dimeric tubulin and GTP (Granzier & Irving, 1995). These conditions favour MT disassembly and therefore underestimation of MT contributions to mechanics. Innovative approaches will be required to tease out the relative contributions of titin, the non‐sarcomeric cytoskeleton, and the myofilaments (Sequeira et al. 2015) to the passive mechanical properties of the cardiomyocyte at physiological deformation rates and under different disease stressors where considerable cytoskeletal remodelling occurs.
Microtubules as viscous resistors?
If MTs provide some restoring force to the sarcomere, it must be from the elastic component; viscous forces oppose the direction of movement and so would resist relaxation, not assist it. Yet the proliferation or heavy detyrosination of MTs produces a substantial increase in the viscous properties of the myocyte along with the increase in elastic stiffness. Increased viscosity may be a simple cost of the augmentation to restoring force provided by microtubule buckling. Alternatively, the ability to tune viscosity both within the myocyte and perhaps differentially throughout the heart could provide independent benefit. MT dynamics and post‐translational modification are regulated on a time scale of seconds to minutes, and thus cytoskeletal cross‐linking represents a potential viscous rheostat with high‐spatiotemporal specificity.
Elevated viscosity could confer some protection to the heart. This line of thinking also allows us to speculate on whether detyrosinated MTs and intermediate filaments are increased in cardiomyopathy (Sato et al. 1997; Heling et al. 2000; Robison et al. 2016) for some selective advantage, or if cytoskeletal proliferation is merely a consequence of conserved cellular mechanisms. Cells under tension or elevated mechanical stress (for example a myocyte under pressure overload) often build and stabilize cytoskeletal structure (Flynn et al. 2010; Rehfeldt et al. 2012; Dingal & Discher, 2014). This is presumed to be a protective mechanism, perhaps to better match cellular stiffness to that of the surrounding tissue to protect against shear‐induced damage or other detriments of force. In the heart, tubulin expression begins to increase in patients with compensated hypertrophy, and dTyr progressively increases during decompensation along with the decline in left ventricular function (Robison et al. 2016). While this may simply reflect a consequence of the cell‐wide response to mechanical stress, limiting contractility could also be purposefully protective. Under pressure overload, myocytes have two means to normalize the stress on the ventricular wall: (1) thickening of the wall, which occurs at least partly via the addition of sarcomeres in parallel during the hypertrophic response, and (2) by limiting active pressure generation. If we assume that the hypertrophic capacity of the myocyte is limited, cytoskeletal stiffening may be eventually called upon to decrease wall stress by limiting pressure generation. In this case, elevated viscous resistance would reduce the risk of catastrophic rupture, a distinctly worse outcome than the obligate drop in cardiac efficiency.
A cost of rapid mechanical communication?
Finally, if we consider the heart in its dynamic role of adapting to changing external demand, there are clear benefits to coordinated mechanosignalling. We know that detyrosinated MTs are required for certain mechanosignalling pathways and also that detyrosinated MTs provide more viscoelastic resistance than their tyrosinated counterparts. A probable link between the two phenomena is the apparent interaction between detyrosinated MTs and desmin, which provides a direct mechanical connection to established mechanosensory complexes at the Z‐disk. It may simply be that the viscous losses associated with detyrosinated MTs are part of the energetic cost of maintaining an interconnected and rapidly activated tuning mechanism for adapting cardiac output to changing demand.
Mechanisms of mechanical divergence
Beyond speculation on why detyrosination pathways have been conserved, it may also be fruitful to ask how this modification produces such dramatically different mechanical behaviour. The modification of a single residue on the relatively unstructured C‐terminal tail of an otherwise tightly globular protein seems unlikely to induce intrinsic changes to the mechanics of microtubules. An alternative way in which dTyr could change structural properties is through the recruitment of MT binding partners. Indeed, post‐translational modifications of tubulin have been extensively investigated in non‐muscle tissues and provide precedent for this mechanism. In these contexts, tubulin modifications have been shown to change binding to MT motors (Liao & Gundersen, 1998; Kreitzer et al. 1999), MAPs (Kumar & Flavin, 1982; Chapin & Bulinski, 1994) and crosslinking to other cytoskeletal proteins (Gurland & Gundersen, 1995; Yoshiyama et al. 2003), resulting in changes in MT mechanical behaviour. Seen in the light of the more extensive body of literature covering MT modifications in other tissue types, specific interactions between subsets of the cardiac MTs and desmin have a clear functional precedent, representing a logical extension of MT behaviour into mechanically active cardiac tissue. This literature also makes clear that, exciting as we find recent results on detyrosinated MTs to be, they need not be at the centre of MT‐based regulation in the heart; a variety of MAPs could exert influence independent of dTyr as well, with or without other MT PTMs.
Conclusion
MT mechanics have been an esoteric realm of cardiac physiology for some time, with significant but unpredictable effects on the actual function of the myocyte. New advances in the basic science of MTs have lent clarity to the field, and tools now exist which can better tease out effects from the underlying complexity of the MT cytoskeleton. These tools have given us additional context in which to understand past results and unlocked new regulatory mechanisms. Considering the variety of particular MAPs and cytoskeletal crosslinks available to cardiac MTs, there is likely to be an extensive regulatory scheme governing MT behaviour of which the interactions of dTyr with the sarcomere are only one part. Regulation of the cardiac cytoskeleton represents a fruitful area of future investigations, both in terms of basic scientific investigation and potential therapeutic intervention.
Additional information
Competing interests
The authors have no competing interests.
Author contributions
B.P. and P.R. wrote the manuscript. Both authors have approved the final version of the manuscript and agree to be accountable for all aspects of the work. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
This work was supported by grants from the National Institutes of Health (NIH): NHLBI R01 HL133080 and R00 HL114879 to B.L.P, and by NIAMS T32 AR053461 to P.R.
Biographies
Dr. Robison completed his Ph.D. at the University of Maryland Medical School under the guidance of Dr. Martin Schneider. He joined the Prosser lab for a post‐doctoral fellowship when the lab opened in 2014. Dr. Robison specializes in advanced microscopy and image processing, which he applies to the study of the cytoskeleton and signaling within the muscle cell.

Dr. Prosser received his training at the University of Maryland Medical School under the tutelage of Drs. Martin Schneider (Ph.D.) and W. Jonathan Lederer (post‐doctoral fellowship). He established his own group as an Assistant Professor in the Department of Physiology at the University of Pennsylvania Perelman School of Medicine in 2014, as part of the Pennsylvania Muscle Institute. The Prosser lab studies the mechanobiology of the heart.
References
- Akhmanova A & Steinmetz MO (2015). Control of microtubule organization and dynamics: two ends in the limelight. Nat Rev Mol Cell Biol 16, 711–726. [DOI] [PubMed] [Google Scholar]
- Bailey BA, Dipla K, Li S & Houser SR (1997). Cellular basis of contractile derangements of hypertrophied feline ventricular myocytes. J Mol Cell Cardiol 29, 1823–1835. [DOI] [PubMed] [Google Scholar]
- Barisic M, Silva e Sousa R, Tripathy SK, Magiera MM, Zaytsev AV, Pereira AL, Janke C, Grishchuk EL & Maiato H (2015). Microtubule detyrosination guides chromosomes during mitosis. Science 348, 799–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barra HS, Arce CA, Rodríguez JA & Caputto R (1974). Some common properties of the protein that incorporates tyrosine as a single unit and the microtubule proteins. Biochem Biophys Res Commun 60, 1384–1390. [DOI] [PubMed] [Google Scholar]
- Belanto JJ, Mader TL, Eckhoff MD, Strandjord DM, Banks GB, Gardner MK, Lowe DA & Ervasti JM (2014). Microtubule binding distinguishes dystrophin from utrophin. Proc Natl Acad Sci USA 111, 5723–5728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calaghan S, White E & Le Guennec JY (2001). A unifying mechanism for the role of microtubules in the regulation of [Ca2+]i and contraction in the cardiac myocyte. Circ Res 89, E31. [PubMed] [Google Scholar]
- Chapin SJ & Bulinski JC (1994). Cellular microtubules heterogeneous in their content of microtubule‐associated protein 4 (MAP4). Cell Motil Cytoskeleton 27, 133–149. [DOI] [PubMed] [Google Scholar]
- de Tombe PP (1998). Altered contractile function in heart failure. Cardiovasc Res 37, 367–380. [DOI] [PubMed] [Google Scholar]
- Dingal PCDP & Discher DE (2014). Systems mechanobiology: tension‐inhibited protein turnover is sufficient to physically control gene circuits. Biophys J 107, 2734–2743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drum BML, Yuan C, Li L, Liu Q, Wordeman L & Santana LF (2016). Oxidative stress decreases microtubule growth and stability in ventricular myocytes. J Mol Cell Cardiol 93, 32–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erck C, Peris L, Andrieux A, Meissirel C, Gruber AD, Vernet M, Schweitzer A, Saoudi Y, Pointu H, Bosc C, Salin PA, Job D & Wehland J (2005). A vital role of tubulin‐tyrosine‐ligase for neuronal organization. Proc Natl Acad Sci USA 102, 7853–7858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flynn BP, Bhole AP, Saeidi N, Liles M, Dimarzio CA & Ruberti JW (2010). Mechanical strain stabilizes reconstituted collagen fibrils against enzymatic degradation by mammalian collagenase matrix metalloproteinase 8 (MMP‐8). PLoS One 5, e12337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez AM, Kerfant BG & Vassort G (2000). Microtubule disruption modulates Ca2+ signaling in rat cardiac myocytes. Circ Res 86, 30–36. [DOI] [PubMed] [Google Scholar]
- Granzier HL & Irving TC (1995). Passive tension in cardiac muscle: contribution of collagen, titin, microtubules, and intermediate filaments. Biophys J 68, 1027–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurland G & Gundersen GG (1995). Stable, detyrosinated microtubules function to localize vimentin intermediate filaments in fibroblasts. J Cell Biol 131, 1275–1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heling A, Zimmermann R, Kostin S, Maeno Y, Hein S, Devaux B, Bauer E, Klövekorn WP, Schlepper M, Schaper W & Schaper J (2000). Increased expression of cytoskeletal, linkage, and extracellular proteins in failing human myocardium. Circ Res 86, 846–853. [DOI] [PubMed] [Google Scholar]
- Iribe G, Ward CW, Camelliti P, Bollensdorff C, Mason F, Burton RAB, Garny A, Morphew MK, Hoenger A, Lederer WJ & Kohl P (2009). Axial stretch of rat single ventricular cardiomyocytes causes an acute and transient increase in Ca2+ spark rate. Circ Res 104, 787–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janke C & Bulinski JC (2011). Post‐translational regulation of the microtubule cytoskeleton: mechanisms and functions. Nat Rev Mol Cell Biol 12, 773–786. [DOI] [PubMed] [Google Scholar]
- Jaworski J, Hoogenraad CC & Akhmanova A (2008). Microtubule plus‐end tracking proteins in differentiated mammalian cells. Int J Biochem Cell Biol 40, 619–637. [DOI] [PubMed] [Google Scholar]
- Jian Z, Han H, Zhang T, Puglisi J, Izu LT, Shaw JA, Onofiok E, Erickson JR, Chen YJ, Horvath B, Shimkunas R, Xiao W, Li Y, Pan T, Chan J, Banyasz T, Tardiff JC, Chiamvimonvat N, Bers DM, Lam KS & Chen‐Izu Y (2014). Mechanochemotransduction during cardiomyocyte contraction is mediated by localized nitric oxide signaling. Sci Signal 7, ra27–ra27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr JP, Robison P, Shi G, Bogush AI, Kempema AM, Hexum JK, Becerra N, Harki DA, Martin SS, Ratieri R, Prosser BL & Ward CW (2015). Detyrosinated microtubules modulate mechanotransduction in heart and skeletal muscle. Nat Commun 6, 8526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khairallah RJ, Shi G, Sbrana F, Prosser BL, Borroto C, Mazaitis MJ, Hoffman EP, Mahurkar A, Sachs F, Sun Y, Chen Y‐W, Raiteri R, Lederer WJ, Dorsey SG & Ward CW (2012). Microtubules underlie dysfunction in duchenne muscular dystrophy. Sci Signal 5, ra56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kombairaju P, Kerr JP, Roche JA, Pratt SJP, Lovering RM, Sussan TE, Kim J‐H, Shi G, Biswal S & Ward CW (2014). Genetic silencing of Nrf2 enhances X‐ROS in dysferlin‐deficient muscle. Front Physiol 5, 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreitzer G, Liao G & Gundersen GG (1999). Detyrosination of tubulin regulates the interaction of intermediate filaments with microtubules in vivo via a kinesin‐dependent mechanism. Mol Biol Cell 10, 1105–1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar N & Flavin M (1982). Modulation of some parameters of assembly of microtubules in vitro by tyrosinolation of tubulin. Eur J Biochem 128, 215–222. [DOI] [PubMed] [Google Scholar]
- Liao G & Gundersen GG (1998). Kinesin is a candidate for cross‐bridging microtubules and intermediate filaments. Selective binding of kinesin to detyrosinated tubulin and vimentin. J Biol Chem 273, 9797–9803. [DOI] [PubMed] [Google Scholar]
- Nirschl JJ, Magiera MM, Lazarus JE, Janke C & Holzbaur ELF (2016). α‐Tubulin tyrosination and CLIP‐170 phosphorylation regulate the initiation of dynein‐driven transport in neurons. Cell Rep 14, 2637–2652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oddoux S, Zaal KJ, Tate V, Kenea A, Nandkeolyar SA, Reid E, Liu W & Ralston E (2013). Microtubules that form the stationary lattice of muscle fibers are dynamic and nucleated at Golgi elements. J Cell Biol 203, 205–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osseni A, Sébastien M, Sarrault O, Baudet M, Couté Y, Fauré J, Fourest‐Lieuvin A & Marty I (2016). Triadin and CLIMP‐63 form a link between triads and microtubules in muscle cells. J Cell Sci 129, 3744–3755. [DOI] [PubMed] [Google Scholar]
- Pal R, Basu Thakur P, Li S, Minard C & Rodney GG (2013). Real‐time imaging of NADPH oxidase activity in living cells using a novel fluorescent protein reporter. PLoS One 8, e63989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peris L, Wagenbach M, Lafanechère L, Brocard J, Moore AT, Kozielski F, Job D, Wordeman L & Andrieux A (2009). Motor‐dependent microtubule disassembly driven by tubulin tyrosination. J Cell Biol 185, 1159–1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popova JS, Garrison JC, Rhee SG & Rasenick MM (1997). Tubulin, Gq, and phosphatidylinositol 4,5‐bisphosphate interact to regulate phospholipase Cβ1 signaling. J Biol Chem 272, 6760–6765. [DOI] [PubMed] [Google Scholar]
- Prosser BL, Ward CW & Lederer WJ (2011). X‐ROS signaling: rapid mechano‐chemo transduction in heart. Science 333, 1440–1445. [DOI] [PubMed] [Google Scholar]
- Rehfeldt F, Brown AEX, Raab M, Cai S, Zajac AL, Zemel A & Discher DE (2012). Hyaluronic acid matrices show matrix stiffness in 2D and 3D dictates cytoskeletal order and myosin‐II phosphorylation within stem cells. Integr Biol (Camb) 4, 422–430. [DOI] [PubMed] [Google Scholar]
- Robison P, Caporizzo MA, Ahmadzadeh H, Bogush AI, Chen CY, Margulies KB, Shenoy VB & Prosser BL (2016). Detyrosinated microtubules buckle and bear load in contracting cardiomyocytes. Science 352, aaf0659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato H, Nagai T, Kuppuswamy D, Narishige T, Koide M, Menick DR & Cooper G (1997). Microtubule stabilization in pressure overload cardiac hypertrophy. J Cell Biol 139, 963–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schappi JM, Krbanjevic A & Rasenick MM (2014). Tubulin, actin and heterotrimeric G proteins: coordination of signaling and structure. Biochim Biophys Acta 1838, 674–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sequeira V, Najafi A, McConnell M, Fowler ED, Bollen IAE, Wüst RCI, dos Remedios C, Helmes M, White E, Stienen GJM, Tardiff J, Kuster DWD & van der Velden J (2015). Synergistic role of ADP and Ca2+ in diastolic myocardial stiffness. J Physiol 593, 3899–3916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiels H, O'Connell A, Qureshi MA, Howarth FC, White E & Calaghan S (2007). Stable microtubules contribute to cardiac dysfunction in the streptozotocin‐induced model of type 1 diabetes in the rat. Mol Cell Biochem 294, 173–180. [DOI] [PubMed] [Google Scholar]
- Sirajuddin M, Rice LM & Vale RD (2014). Regulation of microtubule motors by tubulin isotypes and post‐translational modifications. Nat Cell Biol 16, 335–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tagawa H, Wang N, Narishige T, Ingber DE, Zile MR & Cooper G (1997). Cytoskeletal mechanics in pressure‐overload cardiac hypertrophy. Circ Res 80, 281–289. [DOI] [PubMed] [Google Scholar]
- Tsutsui H, Ishihara K & Cooper G (1993). Cytoskeletal role in the contractile dysfunction of hypertrophied myocardium. Science 260, 682–687. [DOI] [PubMed] [Google Scholar]
- Valenstein ML & Roll‐Mecak A (2016). Graded control of microtubule severing by tubulin glutamylation. Cell 164, 911–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Li F, Campbell SE & Gerdes AM (1999). Chronic pressure overload cardiac hypertrophy and failure in guinea pigs: II. Cytoskeletal remodeling. J Mol Cell Cardiol 31, 319–331. [DOI] [PubMed] [Google Scholar]
- Yoshiyama Y, Zhang B, Bruce J, Trojanowski JQ & Lee VM‐Y (2003). Reduction of detyrosinated microtubules and Golgi fragmentation are linked to tau‐induced degeneration in astrocytes. J Neurosci 23, 10662–10671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Chen B, Guo A, Zhu Y, Miller JD, Gao S, Yuan C, Kutschke W, Zimmerman K, Weiss RM, Wehrens XHT, Hong J, Johnson FL, Santana LF, Anderson ME & Song L‐S (2014). Microtubule‐mediated defects in junctophilin‐2 trafficking contribute to myocyte T‐tubule remodeling and Ca2+ handling dysfunction in heart failure. Circulation 129, 1742–1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zile MR, Richardson K, Cowles MK, Buckley JM, Koide M, Cowles BA, Gharpuray V & Cooper G (1998). Constitutive properties of adult mammalian cardiac muscle cells. Circulation 98, 567–579. [DOI] [PubMed] [Google Scholar]